
Abstract
Pelodiscus sinensis is the most common turtle species raised in East and Southeast Asia. The Japanese strain and the Qingxi black strain are important aquatic breeds of P. sinensis in China with unique characteristics in terms of production performance and appearance, including skin color. In this study, melanin content measure and histological analysis on skin samples of these two strains were carried out to compare their color characteristics. The results showed that Qingxi black turtles clearly have a greater ability to deposit melanin than the Japanese strain especially in the abdomen. Then, de novo transcriptome assembly and differential expression profiling analyses on the ventral skin from the two strains were performed to identify the genes responsible for the differences in skin color using the Illumina RNA-Seq system with three biological replicates. A total of 19,331 annotated unigenes were found by aligning to the reference genome of P. sinensis using TopHat v2.0.12. Differential expression analysis revealed that 670 genes were expressed differently, including 185 upregulated genes and 485 downregulated genes in Qingxi black strain using the DESeq R package (|log2FoldChange| ≥ 1, padj < 0.05). Sixteen differentially expressed genes (DEGs), which were randomly selected, were confirmed by quantitative real-time PCR (qRT-PCR). GO and KEGG analyses revealed four DEGs (agouti signaling protein, frizzled family receptor 1, phospholipase C, and protein kinase C) were related to melanogenesis pathway. Gene expression levels of the four DEGs as well as three genes from the tyrosinase gene family were measured by qRT-PCR. The results indicated that agouti signaling protein, tyrosinase-related protein, and dopachrome tautomerase could be the main genes responsible for the difference in abdominal skin color between the two turtle strains. This study provided valuable information for further analysis of the melanogenesis mechanisms in different varieties of P. sinensis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Chinese soft-shelled turtle, Pelodiscus sinensis, is a commercially important species raised in East and Southeast Asia for its remarkable medicinal and nutritional properties (Lu et al. 2010). It has become one of the most important species in the Chinese aquaculture industry. In 2014, production of this species was 355,000 t, with a total value of 250 million Chinese yuan (He et al. 2015), and the annual production has remained above 320,000 t in recent years. In addition, the Japanese strain and Qingxi black strain of P. sinensis have become two new important aquatic breeds through selection and breeding over the past 20 years in China (Zhang et al. 2017). These two varieties each have unique characteristics in terms of appearance and production performance. For example, the Japanese strain has a cyan-colored carapace, an almost white abdomen, with the important feature of fast growth, large body size, and it is the most widely bred variety in China. In contrast, the whole body of the Qingxi black turtle is black, and its value mainly depends on its rich melanin content (Jimbow 1999). The Qingxi black turtle is a body color variation of P. sinensis with descendible and characteristic black belly color. It is a good material for genetic and breeding research. And it is rich in melanin with edible and medicinal value, which gives it high economic value. However, little is known about the mechanisms that underlie the color differences in the skin of different P. sinensis strains.
Skin pigmentation of mammals is due to melanin, which is a biopolymer produced by melanocytes. Skin color patterns are determined by the quantity and distribution of two types of organic pigment: eumelanin (black to brown) and pheomelanin (yellow to red) (Barsh 2006; Ito 2003). Both are produced by melanocytes in the basal epidermis. They are synthesized and accumulated in specialized organelles called melanosomes, and are then transferred to keratinocytes of the epidermis (Jimbow et al. 2000). Melanin synthesis involves a complex regulatory system and signaling network; this synthesis depends on the sequential expression of a series of genes or loci, and many signal molecules are involved in regulation of this process. Melanogenesis mainly includes four signal transduction pathways: cyclic adenosine monophosphate (cAMP), wingless/integrated, mitogen-activated protein kinase (MAPK), and phospholipase C (PLC), of which cAMP pathway is the dominant pathway involved in regulation of melanin synthesis in mammals.
The cAMP pathway is initiated by the binding of α-melanocyte-stimulating hormone (α-MSH) to the melanocortin-1 receptor (MC1R), which elevates intracellular cAMP levels and increases expression of tyrosinase (TYR) (Hunt et al. 1994). Synthesis of both eumelanin and pheomelanin starts from tyrosine oxidation catalyzed by TYR (Ito 2003). The resulting dopaquinone is a precursor for eumelanin synthesis, promoted by tyrosinase-related protein 1 (TYRP1) and dopachrome tautomerase (DCT). Dopaquinone can also transform into pheomelanin in the presence of high cysteine concentrations and/or low tyrosinase activity (Ito 2003; Land et al. 2003). Furthermore, the agouti signaling protein (ASP) normally acts as a competitive antagonist of melanocortin peptides at the MC1R to block cAMP synthesis, leading to downregulation of eumelanogenesis and upregulation of pheomelanogenesis (Bultman et al. 1992; Lu et al. 1994; Millar et al. 1995). Many studies of the genes involved in the formation of melanin in humans and mice have been conducted, but little is known about the process in turtles.
Transcriptome profiling analysis is a rapid and effective method for conducting genome surveys and identifying functional genes and molecular markers due to its comprehensive profiling of gene expression. It has been widely used for functional gene screening in studies of aquatic animals (Wu et al. 2010; Xiao et al. 2015; Yadetie et al. 2013; Zhu et al. 2016). In this study, we performed transcriptome analysis on skin samples from the Japanese and Qingxi black strains of P. sinensis, which exhibit distinct color differences. The goal of this study was to identify melanin biosynthesis–related genes that are differentially expressed in the skin of the two turtle strains. Results of this study will help elucidate the melanogenesis mechanisms that underlie skin pigmentation in P. sinensis.
Materials and methods
Sample collection
All Chinese soft-shelled turtles (Qingxi black strain and Japanese strain) used in this study were obtained from the national turtle breeding center (Deqing, Huzhou, China). Healthy soft-shelled turtles were selected as experimental animals (mean weight 312.2 ± 10.3 g). Tissue samples of three Qingxi black turtles and three Japanese turtles were collected from dorsal and ventral skin, frozen immediately in liquid nitrogen, and stored at − 80 °C until use. As for histological analysis, approximately 0.5 × 0.5-cm skin tissue samples from these turtles were fixed in a 4% paraformaldehyde solution for 6 h, respectively.
Measurement of melanin content
The melanin content in the skin samples was determined following the procedure described by Hosoi (Hosoi et al. 1985) with some modifications. The frozen skin samples were ground with liquid nitrogen, suspended in 1 M NaOH, and heated for 1 h at 80 °C. The lysates then were centrifuged at 12000×g at 4 °C for 10 min, and the supernatants were collected. The absorbance of extracted melanin was read at 405 nm using a SpectraMax 190 ELISA microplate reader (Molecular Devices, Sunnyvale, USA). All measurements were repeated three times.
TYR activity assay
Skin samples (0.2 g) described in the previous section ground with liquid nitrogen were mixed with 2 mL of protein extraction lysate (0.1 M sodium phosphate buffer, 0.5% TritonX-100, 5 mM EDTA, 1 mM PMSF, pH 6.9) and lysed on a shaking table for 24 h at 4 °C. The lysates then were centrifuged at 12000×g at 4 °C for 10 min, and the supernatants were collected for the TYR activity and protein concentration assays. The substrate was 0.1% 3,4-dihydroxyphenylalanine (L-DOPA) in 0.1 M sodium phosphate buffer, pH 6.9. The substrate volume was 0.7 mL, and 0.3 mL of enzyme solution was added to the substrate system for 5 h at 37 °C. Absorption was recorded at 475 nm using a UV-1800 spectrophotometer (Shimadzu, Tokyo, Japan). A unit of enzyme activity was defined as an increase of 0.001 per minute of absorbance at 475 nm. The protein concentration was determined using the bicinchoninic acid method with the total protein assay kit from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All measurements were repeated three times.
Histological analysis
The fixed tissues were dehydrated by placing each tissue in a graded series of ethyl alcohol. After dehydration, the tissues were embedded in paraffin for histological studies. The paraffin sections were serially cut at 5-μm thickness with a RM2235 microtome (Leica, NuBloch, Germany) and stained with hematoxylin and eosin (HE) to show the melanin deposits. To visualize melanin more clearly as black deposits, Fontana-Masson staining of 5-μm thickness serial paraffin sections was used (Yoon et al. 2003). To visualize TYR activity, the tissues were cleaned to remove polyformaldehyde and then incubated in 0.1% L-DOPA in 0.01 M sodium phosphate buffer, pH 6.9. Subsequently, the samples were refixed in 4% paraformaldehyde solution, dehydrated to make 5-μm thickness paraffin sections, and stained using nuclear fast red. Through this method, tyrosinase could show up in a black color. Morphological structures were measured using a Nikon 80i (Nikon, Tokyo, Japan) equipped with a biological color microscopy image analysis system.
RNA isolation
Total RNA was extracted from frozen samples using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Extracted RNA was resuspended in diethy pyrocarbonate–treated ddH2O. RNA degradation and contamination were assessed using a 1% agarose gel. RNA purity was checked using the NanoPhotometer® spectrophotometer (Implen, Inc., Westlake Village, CA, USA). RNA concentration was measured using the Qubit® RNA Assay Kit and a Qubit® 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, La Jolla, CA, USA).
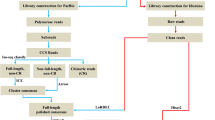
RNA-Seq library preparation and Illumina sequencing
RNA-Seq library construction and Illumina sequencing were performed by Novogene Biological Information Technology Company (Beijing, China). First, 3 μg of total RNA from each sample were used as input material for RNA-Seq library preparation and sequencing. The RNA samples were from the ventral skin of three Japanese and three Qingxi black P. sinensis, respectively. cDNA libraries were generated using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA) following the manufacturer’s instructions after mRNA was purified and fragmented. Clustering of the index-coded samples was performed on a cBot Cluster Generation System using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq platform, and 125 base pair/150 base pair paired-end reads were generated.
RNA-Seq data analysis
RNA-Seq reads were assessed for quality using FastQC (version 0.10.1; http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). Raw sequence data were transformed by base calling into sequence data, stored in fastq format, and cleaned by removing reads containing adapters and poly-N and low-quality reads. At the same time, the phred quality scores Q20 and Q30, and GC content of the clean data were calculated. All downstream analyses were based on the clean data with high quality. Reference genome and gene model annotation files were downloaded from the reference genome of P. sinensis website (GenBank: AGCU00000000.1, ftp://ftp.ensembl.org/pub/release-81/fasta/pelodiscus_sinensis/dna/) directly. Index of the reference genome was built using Bowtie v2.2.3 and paired-end clean reads were aligned to the reference genome using TopHat v2.0.12 (Trapnell et al. 2009). The value of mismatch was set as 2, and the other parameters were the default of the software. Gene sequence, gene ID, gene description, and other information were directly extracted from R package according to the annotation information of reference genome. The Cufflinks v2.1.1 reference annotation–based transcript (RABT) assembly method was used to construct and identify both known and novel transcripts from TopHat alignment results. GATK2 v3.2 software was used to perform SNP and Indel calling.
Differential expression analysis
HTSeq v0.6.1 was used to count the read numbers mapped to each gene. Fragments per kilobase of exon per million fragments mapped of each gene was calculated based on the length of the gene and the read count mapped to the gene (Trapnell et al. 2010). Differential expression analysis of the two species of P. sinensis was performed using the DESeq R package (1.18.0) (Wang et al. 2009). The resulting P values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with an absolute value of log2FoldChange ratio ≥ 1 and an adjusted P value (padj) < 0.05 identified by DESeq were designated as being differentially expressed, which was divided into upregulated and downregulated lists of transcripts.
Functional enrichment analysis
Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) was conducted to identify biological process, molecular function, and cellular component terms. The GO seq R package was used for this analysis, and gene length bias was corrected (Young et al. 2010). GO terms with corrected P values < 0.05 were considered to be significantly enriched by DEGs. The Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to assign and predict putative functions and pathways associated with the DEGs. KOBAS software was used to test the statistical enrichment of DEGs in KEGG pathways (Mao et al. 2005).
Quantitative real-time polymerase chain reaction (qRT-PCR)
qRT-PCR was carried out using the CFX96™ Real-Time System (Bio-Rad, Hercules, CA, USA) to analyze the expression profiles of the primary genes. The GAPDH gene was used as the internal reference. Table 1 shows the primer sequences and related information. The RNA samples used for qRT-PCR amplifications were the same as those used to construct the RNA-Seq library. First-strand cDNA was synthesized from total RNA by M-MLV reverse transcriptase (Transgen, Beijing, China) for 1 h at 42 °C with oligo dT primers following the manufacturer’s protocol. qRT-PCR was performed using SYBR Green I real-time PCR Master Mix (Takara, Dalian, China). It was carried out in a 12 μL reaction mixture containing 6.25 μL of SYBR Premix Ex Taq, 1 μL of 1:5 diluted cDNA template, 0.25 μL of each of the primers, and 4.25 μL of ddH2O. The thermal cycling profile consisted of initial denaturation at 95 °C for 30 s and 40 cycles at 95 °C for 15 s, 55 °C for 30 s, and 72 °C for 30 s. The relative expression level was calculated using the 2−ΔΔCt method, where ΔΔCt = ΔCttest – ΔCtcalibrator and data were normalized to the endogenous GAPDH or RPS18.
Statistical analyses
The results of data analyses are presented as mean ± standard deviation of three replicate experiments. The data were analyzed statistically using one-way analysis of variance followed by post-hoc comparison of means (least significant difference test) using IBM SPSS Statistics 20 software. Differences were considered to be significant at P < 0.05 or P < 0.01.
Results
Melanin deposition comparison between the two strains of P. sinensis
Figure 1a shows the Japanese and Qingxi black strains of P. sinensis. The dorsal skin of the Qingxi black turtle is relatively deeper than that of the Japanese strain, and the color difference is more pronounced on the ventral skin, where it is close to black for the Qingxi black turtle and almost white for the Japanese strain. The melanin content in the dorsal and ventral skin of the Qingxi black turtle was significantly higher than that in the Japanese strain (P < 0.01; Fig. 1b). Additionally, TYR activity in the dorsal and ventral skin of the Qingxi black turtle was significantly higher than that in the Japanese strain (P < 0.05; Fig. 1c). HE staining showed that more melanin was present in the stratum spinosum and stratum basale of the epidermis of the Qingxi black turtle than that of the Japanese strain (Fig. 2a). HE stain results were confirmed by the specific Fontana-Masson stain for melanin. Melanin deposition was clearly present in the ventral epidermis of the Qingxi black turtle (distributed in the stratum spinosum and the melanocytes of the stratum basale), whereas no obvious melanin deposits were detected in the epidermis of the ventral skin of the Japanese strain (Fig. 2b). Distribution of the L-DOPA stain for melanocytes in the stratum basale also showed that Qingxi black turtles may have a greater ability to deposit melanin than the Japanese strain (Fig. 2c).

The Japanese strain and Qingxi black strain of P. sinensis: a color comparison of dorsal and ventral skin, b melanin content comparison, and c tyrosinase activity comparison. JN PS represents the Japanese strain, and QX PS represents the Qingxi black strain. **P < 0.01 and *P < 0.05 represent significant differences

Histological comparison of melanin deposition in the ventral skin of the two P. sinensis strains: (a) HE staining, (b) Fontana-Masson staining, and (c) L-DOPA staining of ventral skin sections. I: the Japanese strain, II: the Qingxi black strain. SC, stratum corneum; SG, stratum granulosum; SS, stratum spinosum; SB, stratum basale; D, dermis
Transcriptome sequence assembly
The total number of raw reads per library ranged from 48.17 to 65.40 million from the Japanese and Qingxi black turtle strains (Table 2). The quality assessment of sequencing data showed that the distribution of quality Q30 was higher than 89.45% in each sample and that the GC and AT content was almost equal in each sequencing cycle (Table 2). After filtering, 61.25–65.31% clean reads with a length range of 125 to 150 base pairs were successfully matched to the P. sinensis genome using TopHat software, and 58.94–63.00% reads were uniquely mapped to the genome for the two turtle strains (Table 2). The total number of known genes identified was 19,331, and there were 3469 novel genes predicted in addition. The details are listed in Supplemental file 1. These results demonstrated that the sequencing quality was quite good and therefore the subsequent transcriptome analysis results were reliable.
Identification of differential expression analysis
In total, 670 DEGs were detected between the Qingxi black and Japanese strains of P. sinensis. Among them, 185 DEGs were upregulated and 485 were downregulated in the Qingxi black strain (Fig. 3). The details are listed in Supplemental file 2. To further investigate the biological importance of the DEGs, GO enrichment analysis was performed to determine the functional categories of each gene. The DEGs were assigned to various GO terms to determine their functional classifications (Supplemental file 3). Figure 4 shows the 30 most enriched GO terms in the Qingxi black versus Japanese strain comparison. These DEGs were classified into the following three functional categories: biological process (18 DEGs), cellular component (6 DEGs), and molecular function (6 DEGs). The majority of the DEGs in the biological process category were involved in regulation of vasculature development, regulation of angiogenesis, and gastrulation. Within the cellular component category, virion and virion parts were the dominant annotated DEGs. In the molecular function category, the most of the DEGs were related to catalytic activity and hydrolase activity. However, the GO term for melanin biosynthetic process was not significantly enriched among the DEGs.

Algorithm to identify genes differentially expressed between the two strains. The genes were classified into three classes. Red color indicates those genes that were upregulated (meaning that gene expression in the Qingxi black strain was greater than that in the Japanese strain). Green color indicates those genes that were downregulated. Blue color indicates those genes that were not differentially expressed

GO classifications of the DEGs. All of the DEGs were assigned to three categories: cellular component, molecular function, and biological process
To confirm the reliability of the RNA-Seq data, 16 of the 670 DEGs were randomly selected for qRT-PCR analysis. The results showed a high correlation between the RNA-Seq and qRT-PCR data, and the expression patterns of the selected DEGs determined by qRT-PCR were largely identical to those determined by RNA-Seq (Fig. 5). These results indicate that expression profiling of DEGs as determined by RNA-Seq was reliable and accurate.


Verification of the selected DEGs by qRT-PCR: (a) relative expression of the selected DEGs by qRT-PCR between the Qingxi black strain and the Japanese strain, (b) comparison of RNA-Seq data with qRT-PCR data. (c) Correlation of the expression ratio of selected genes measured by RNA-Seq and qRT-PCR. **P < 0.01 and *P < 0.05 represent significant difference. ASNS, asparagine synthase; MAPK7, mitogen-activated protein kinase 7; POLL, DNA polymerase lambda; SMAD3, SMAD family member 3; CCNB2, cyclin B2; DDX5, DEAD-box helicase 5; KER, keratin; ASP, agouti signaling protein; GTF2B, general transcription factor IIB; RHI, ribonuclease H1; POLD2, DNA polymerase alpha subunit 2; GHITM, growth hormone–inducible transmembrane protein; SLC25A17, solute carrier family 25 member 17; PKC, protein kinase C, beta; NUP88, nucleoporin 88; EXOSC7, exosome component 7. Data were normalized to the reference gene GAPDH (I) and RPS18 (II)
Identification of critical pathways and DEGs involved in KEGG analysis
One hundred eighteen pathways were identified with a KEGG pathway annotation (Supplemental file 4). The top 20 enriched KEGG pathways of DEGs between the Japanese strain and Qingxi black strain of P. sinensis were showed in Table 3. Pathways in lysosome (12 genes), base excision repair (5 genes), nicotinate and nicotinamide metabolism (5 genes), and pantothenate and CoA biosynthesis (3 genes) were the most significantly enriched KEGG pathways. We believed that these pathways were significant during the growth of the two Chinese soft-shelled turtles, in particular metabolic pathways. In addition, the MAPK pathway (9 genes) was known to play an important role in regulating gene expression, it could change the activation of transcription factors via phosphorylation and influence the gene expression pattern of the two strains.
By KEGG pathway enrichment, we also screened a series of significant DEGs in critical pathways: DNA polymerase delta subunit (POLD, − 1.9739) involved in the regulation of DNA replication, repair, and homologous recombination; voltage-dependent calcium channel alpha-1 subunit (CACNA1, − 5.6307), protein kinase C beta type (PRKCB, − 3.082), tumor protein p53 (TP53, − 1.2639), vascular endothelial growth factor B (VEGFB, − 1.5594), and cytosolic phospholipase A2 (cPLA2, − 1.8395) involved in MAPK pathway and other important signaling pathways; mitotic-specific cyclin-B (CCNB, 1.4924) involved in cellular senescence, cell cycle, oocyte meiosis and p53 signaling pathway; interferon regulatory factor-3 (SMAD3, 1.3983) and phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform (PIK3CA, − 3.7891) involved in cellular senescence too; 3′5′-cyclic nucleotide phosphodiesterase (PDE1B, − 2.4478) and phosphatidylinositol phospholipase (PLCB3, − 1.2187) involved in calcium signaling pathway and other metabolic pathways. These DEGs played diverse roles in a number of different and important pathways, and they might be the main functional genes for the difference of various traits between the two strains.
Identification of melanin synthesis–related genes
To identify the DEGs involved in melanin biosynthetic pathway in the two strains of P. sinensis, the KEGG for the melanogenesis pathway was studied in detail (Fig. 6). The four DEGs involved in the melanogenesis pathway were agouti signaling protein (ASP), frizzled family receptor 1 (Frizzled), phospholipase C (PLC), and protein kinase C (PKC). They were all downregulated in the Qingxi black turtle strain compared with the Japanese strain (padj < 0.05) (Table 4). Table 3 also shows the changes in expression of TYR, TYRP1, and DCT, which were all involved in the synthesis of melanin. The read counts for these three melanin synthesis genes were all greater in the Qingxi black turtle samples than in the Japanese strain samples (padj < 0.1), but they were not DEGs screened by RNA-Seq .

Melanogenesis pathway in the skin of P. sinensis. Downregulated genes in samples from the Qingxi black strain are marked with a green outline
To confirm the reliability of the RNA-Seq data for the melanogenesis pathway, these seven genes were analyzed using qRT-PCR. The expression patterns of the selected genes determined by qRT-PCR were largely identical to those determined by RNA-Seq (Fig. 7). Expression levels of the four DEGs (ASP, Frizzled, PLC, and PKC) screened using RNA-Seq exhibited downregulation in the Qingxi black turtle samples. TYRP1 expression was upregulated and TYR expression did not change significantly in the Qingxi black turtle skin samples compared with the Japanese strain. DCT expression levels could not be compared between the two strains because the expression level in the Japanese strain was too low to test, and it was in accordance with the result that the RNA-Seq read count of DCT in this strain was 0 (Table 3).

Expression quantity verification of the selected genes in the melanogenesis pathway by qRT-PCR. **P < 0.01 and *P < 0.05 represent significant differences. ASP, agouti signaling protein; Frizzled, frizzled family receptor 1; PLC, phospholipase C, beta 3; PKC, protein kinase C, beta; TYR, tyrosinase; TYRP1, tyrosinase-related protein 1
Discussion
In this study, we investigated the differences in melanin content and gene expression in the skin of the two important strains of P. sinensis that have different skin pigmentation patterns. Histological analysis and melanin content assay showed that Qingxi black turtles may have a greater ability to deposit melanin in the epidermis of the ventral skin than the Japanese strain. Melanin was distributed in the stratum spinosum and the melanocytes of the stratum basale. A total of 670 DEGs from the ventral skin were detected by RNA-Seq between the two strains, among which 185 were upregulated and 485 were downregulated in the Qingxi black strain, respectively. Four DEGs (ASP, Frizzled, PLC, and PKC) in the melanogenesis pathway were identified in the two strains of P. sinensis by KEGG analysis, and they exhibited downregulation of expression in the Qingxi black turtle samples confirmed by qRT-PCR.
According to previous reports, ASP is the reverse regulator of the melanin synthesis pathway, and changes in ASP gene sequence, mRNA splicing form, and expression quantity can directly regulate melanin synthesis at cellular and organism levels (Fontanesi et al. 2012; Nakayama et al. 2010; Virador et al. 2000; Yoshihara et al. 2012). The effect of ASP on pigment type switching is responsible for a variety of coat color patterns across a broad range of mammalian species. Our results indicated that ASP may function in a similar way in reptiles. The expression level of ASP in the ventral skin of Qingxi black turtles was significantly lower than that in the Japanese strain. Thus, the ability of ASP to antagonize α-MSH decreased, which may have promoted the formation and deposition of eumelanin and melanin in Qingxi black turtles. Combined with KEGG results regarding the melanogenesis pathway, these results suggest that the decreased expression of ASP mRNA in the skin of Qingxi black turtles might be responsible for melanin deposition in the skin. However, the effect of the expression level decreases detected for the Frizzled, PLC, and PKC genes was not clear in this study.
In addition, the tyrosinase gene family also plays a major role in the two species’ melanin deposition. Synthesis of melanin starts from tyrosine oxidation catalyzed by TYR, and the subsequent eumelanin synthesis mainly promoted by TYRP1 and DCT (Ito 2003; Land et al. 2003). In our result, TYR activity in the skin of the Qingxi black turtle was significantly higher than that in the Japanese strain, and it indicated that tyrosinase could be a major factor for the melanin deposition difference. The tyrosinase family genes (TYR, TYRP1, and DCT) were analyzed for expression by qRT-PCR to verify the active genes involved in melanin biosynthetic pathways and different skin color depositions in Chinese soft-shelled turtle. TYR expression showed no significant expression change. The upregulated expression of TYRP1 in Qingxi black turtles likely is beneficial for synthesis of eumelanin. In the Japanese strain, DCT expression was not detected by RNA-Seq or qRT-PCR, and this lack and the lower expression of TYRP1 may explain why the ventral skin was almost white in the Japanese strain. These results are in agreement with previous reports stating that TYRP1 and DCT are essential for eumelanin synthesis (Kobayashi et al. 1994; Tsukamoto et al. 1992; Winder et al. 1994).
In conclusion, we carried out histological and transcriptome analyses to compare differences in melanin deposition and gene expression between two breeds of P. sinensis. Our results suggest that ASP, TYRP1, and DCT may play important roles in the different melanin deposition patterns via the cAMP signaling pathway. Additionally, the major pathways and main factors involved in melanin synthesis in P. sinensis are similar to those of mammals. Results of this study provide valuable information for further analysis of the role of melanin synthesis–related DEGs and the melanogenesis mechanisms at work in different varieties of P. sinensis.
References
Barsh GS (2006) Regulation of pigment type switching by agouti, melanocortin signaling, attractin and mahoganoid. Pigmentary Syst: Physiol Pathophysiol:395–409. https://doi.org/10.1002/9780470987100.ch19
Bultman SJ, Michaud EJ, Woychik RP (1992) Molecular characterization of the mouse agouti locus. Cell 71:1195–1204. https://doi.org/10.1016/S0092-8674(05)80067-4
Fontanesi L, Rustempasic A, Brka M, Russo V (2012) Analysis of polymorphisms in the agouti signalling protein (ASIP) and melanocortin 1 receptor (MC1R) genes and association with coat colours in two Pramenka sheep types. Small Rumin Res 105:89–96. https://doi.org/10.1016/j.smallrumres.2012.02.008
He ZY, Zhang HQ, Cai YW (2015) High-efficiency culture models strategy for softshelled turtle. China Agriculture Press, Beijing, pp 1–5(15):17,19–17,80
Hosoi J, Abe E, Suda T, Kuroki T (1985) Regulation of melanin synthesis of B16mouse melanoma cells by 1R, 25-dihydroxy vitamin D3 and retinoic acid. Cancer Res 45:1474–1478
Hunt G, Donatien P, Lunec J, Todd C, Kyne S, Thody A (1994) Cultured human melanocytes respond to MSH peptides and ACTH. Pigm Cell Melanoma R 7:217–221. https://doi.org/10.1111/j.1600-0749.1994.tb00052.x
Ito S (2003) The IFPCS presidental lecture: a chemist’s view of melanogenesis. Pigm Cell Melanoma R 16:230–236. https://doi.org/10.1034/j.1600-0749.2003.00037.x
Jimbow K (1999) Biological role of tyrosinase-related protein and its relevance to pigmentary disorders (vitiligo vulgaris). J Dermatol 26(11):734–737. https://doi.org/10.1111/j.1346-8138.1999.tb02084.x
Jimbow K, Hua C, Gomez PF, Hirosaki K, Shinoda K, Salopek TG, Matsusaka H, Jin HY, Yamashita T (2000) Intracellular vesicular trafficking of tyrosinase gene family protein in eu- and pheomelanosome biogenesis. Pigm Cell Melanoma R 13(Suppl 8):110–117. https://doi.org/10.1034/j.1600-0749.13.s8.20.x
Kobayashi T, Urabe K, Winder AJ, Jime’nez-Cervantes C, Imokawa G, Brewington T, Solano F, Garcı’a-Borro’n JC, Hearing VJ (1994) Tyrosinase related protein 1 (TRP1) functions as a DHICA oxidase in melanin biosynthesis. EMBO J 13:5818–5825. https://doi.org/10.1002/j.1460-2075.1994.tb06925.x
Land EJ, Ito S, Wakamatsu K, Riley PA (2003) Rate constants for the first two chemical steps of eumelanogenesis. Pigm Cell Melanoma R 16:487–493. https://doi.org/10.1034/j.1600-0749.2003.00082.x
Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, Cone RD (1994) Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature 371:799–802. https://doi.org/10.1038/371799a0
Lu JF, Wan Q, Yin ZM, Lin L, Weng SB, Ye YW, Hang ST (2010) Extraction and characterization of collagen from calipash of Chinese soft-shelled turtle (Pelodiscus sinensis). J Fish China 34(6):801–808. https://doi.org/10.3724/SP.J.1231.2010.06800
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21(19):3787–3793. https://doi.org/10.1093/bioinformatics/bti430
Millar SE, Miller MW, Stevens ME, Barsh GS (1995) Expression and transgenic studies of the mouse agouti gene provide insight into the mechanisms by which mammalian coat color patterns are generated. Development 121(10):3223–3232
Nakayama K, Shotake T, Takeneka O, Ishida T (2010) Variations in the coding region of the agouti signaling protein gene do not explain agouti/non-agouti phenotypes in macaques. J Mamm Evol 17(3):211–214. https://doi.org/10.1007/s10914-010-9137-7
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9):1105–1111. https://doi.org/10.1093/bioinformatics/btp120
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzber SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28(5):511–515. https://doi.org/10.1038/nbt.1621
Tsukamoto K, Jackson IJ, Urabe K, Montague PM, Hearing VJ (1992) A second tyrosinase-related protein, TRP2, is a melanogenic enzyme termed DOPAchrome tautomerase. EMBO J 11:519–526. https://doi.org/10.1002/j.1460-2075.1992.tb05082.x
Virador VM, Santis C, Furumura M, Kalbacher H, Hearing VJ (2000) Bioactive motifs of agouti signal protein. Exp Cell Res 259(1):54–63. https://doi.org/10.1006/excr.2000.4975
Wang L, Feng Z, Wang X, Zhang X (2009) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26(1):136–138. https://doi.org/10.1093/bioinformatics/btp612
Winder AJ, Kobayashi T, Tsukamoto K, Urabe K, Aroca P, Kameyama K, Hearing VJ (1994) The tyrosinase gene family: interactions of melanogenic proteins to regulate melanogenesis. Cell Mol Biol Res 40:613–626
Wu Z, Zhang W, Lu Y, Lu C (2010) Transcriptome profiling of zebrafish infected with Streptococcus suis. Microb Pathog 48:178–187. https://doi.org/10.1016/j.micpath.2010.02.007
Xiao J, Zhong H, Liu Z, Yu F, Luo Y, Gan X, Zhou Y (2015) Transcriptome analysis revealed positive selection of immune-related genes in tilapia. Fish Shellfish Immun 44:60–65. https://doi.org/10.1016/j.fsi.2015.01.022
Yadetie F, Karlsen OA, Lanzen A, Berg K, Olsvik P, Hogstrand C, Goksøyr A (2013) Global transcriptome analysis of Atlantic cod (Gadus morhua) liver after in vivo methylmercury exposure suggests effects on energy metabolism pathways. Aquat Toxicol 126:314–325. https://doi.org/10.1016/j.aquatox.2012.09.013
Yoon TJ, Lei TC, Yamaguchi Y, Batzer J, Wolber R, Hearing VJ (2003) Reconsti-tuted 3-dimensional human skin of various ethnic origins as an in vitro modelfor studies of pigmentation. Anal Biochem 318:260–269. https://doi.org/10.1016/S0003-2697(03)00172-6
Yoshihara C, Fukao A, Ando K, Tashiro Y, Taniuchi S, Takahashi S, Takeuchi S (2012) Elaborate color patterns of individual chicken feathers may be formed by the agouti signaling protein. Gen Comp Endocrinol 175(3):495–499. https://doi.org/10.1016/j.ygcen.2011.12.009
Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11(2):R14. https://doi.org/10.1186/gb-2010-11-2-r14
Zhang H, Xu X, Zhongyang He Z, Tianlun Zheng T, Jianzhong Shao J (2017) De novo transcriptome analysis reveals insights into different mechanisms of growth and immunity in a Chinese soft-shelled turtle hybrid and the parental varieties. Gene 605:54–62. https://doi.org/10.1016/j.gene.2016.12.003
Zhu Q, Zhang L, Li L, Que H, Zhang G (2016) Expression characterization of stress genes under high and low temperature stresses in the pacific oyster, Crassostrea gigas. Mar Biotechnol 18:176–188. https://doi.org/10.1007/s10126-015-9678-0
Funding
This study was financially supported by the National Natural Science Foundation of China (31602138) and the Zhejiang Province Public Welfare Technology Application Research Project (2017C32053). Dr. Guoying Qian was financially supported by the Zhejiang Provincial Project of Selective Breeding of Aquatic New Varieties (2016C02055-4-4). Yuexiu Si was financially supported by the scientific research fund of Zhejiang Chinese Medical University (2019ZR08). Linmeng Zhang was financially supported by the Zhejiang Provincial Undergraduates’ Scientific and Technical Innovation Project (2017R420024) and the Zhejiang Provincial Top Key Discipline of Biological Engineering (CX2017019). Lili Zhang was financially supported by the Zhejiang Provincial Top Key Discipline of Biological Engineering (CX2018022).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Matthias Hammerschmidt
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Si, Y., Zhang, L., Zhang, L. et al. Transcriptome analysis provides insight into the role of the melanin pathway in two differently pigmented strains of the turtle Pelodiscus sinensis. Dev Genes Evol 229, 183–195 (2019). https://doi.org/10.1007/s00427-019-00639-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00427-019-00639-3