
Abstract
Pulmonary fibrosis (PF) is a disease in which excessive extracellular matrix (ECM) accumulation occurs in pulmonary mesenchyme, which induces the destruction of alveolar structures and poor prognosis. Macrophage death is responsible for ECM accumulation after alveolar epithelial injury in PF. Depending on the local micro-environments, macrophages can be polarized to either classically activated (M1) or alternatively activated (M2) macrophage phenotypes. In general, M1 macrophages can promote inflammation and sterilization, stop the continuous damage process and prevent excessive repair, while M2 macrophages are anti-inflammatory and promote tissue repair, and excessive M2 macrophage activity may inhibit the absorption and degradation of ECM. Emerging evidence has revealed that death forms such as pyroptosis mediated by inflammasome affect polarization direction and ultimately lead to the development of PF. Pharmacological manipulation of macrophages death signals may serve as a logical therapeutic strategy for PF. This review will focus on the current state of knowledge regarding the regulation and underlying mechanisms of macrophages and their mediators in the influence of macrophage death on the development of PF. We expect to provide help in developing effective therapeutic strategies in clinical settings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Pulmonary fibrosis (PF) is a progressive disease that can raise the mortality and the rate of disability of patients with lung disease. The clear mechanism of the pathological procedure is still unknown, while there lots of advances in past decades. Macrophage, with the ability to polarize into different phenotypes, is an innate immunological cell that plays a contradictory but connected role in pulmonary fibrosis. How the dead form of cells participates in fibrosis by effect macrophages has attracted researchers a lot whereas the association between macrophages and cell death is in a mess yet. This review aims to figure out the underlying interrelationship between macrophage and cell death form and expounds a possible mechanism of pulmonary fibrosis from a novel sight.
Introduction
Pulmonary fibrosis (PF) is the most common fibrosing lung disease with poor prognosis and no effective treatment [1]. It is characterized by the development of excessive ECM deposition, leading to decreased static lung compliance, disrupted gas exchange and respiratory failure and death [2]. The median survival time for patients with idiopathic pulmonary fibrosis (IPF), the most classical disease of PF, is approximately 3 years after initial diagnosis due to unsatisfactory effect of current anti-fibrosis drugs [3].
Previous studies have confirmed that excessive deposition of ECM is an important factor in the progression of PF [4]. The formation of ECM is a pathological process of abnormal repair after alveolar epithelial cells (AECs) injury [5]. Therefore, it is very important to control the inflammatory response and carry out normal repair of the damaged tissue. In normal tissue repair, macrophages are important cells to degrade and absorb ECM [6]. Macrophages are mainly involved in tissue damage repair in the immune regulatory pathway of the human body [7]. Bone marrow monocytes migrate into tissues and become tissue macrophages [8]. At this time, the macrophages are in a dormant state, which is called resting-state (M0) macrophages [9]. When AECs are injured, monocytes gather in the lung interstitium [10]. Under the influence of pro-inflammatory factors, M0 macrophages differentiate into classically activated (M1) macrophages [7]. Once activated, M1 macrophages will produce tumor necrosis factor-α (TNF-α), L-1β and oxygen free radicals to fight infection or remove foreign substances, thus terminating the damage repair process and preventing excessive repair [9]. However, the differentiation of M0 into alternatively activated(M2) macrophages can be over-anti-inflammatory and promote abnormal tissue repair [11]. This excessive activity of M2 macrophages may lead to the occurrence of various fibrosis diseases [9]. In conclusion, macrophages play an important role in maintaining the homeostasis of lung tissue, and the maladjustment of the polarization direction of macrophages can lead to the occurrence of PF.
The direction of macrophage polarization is controlled by different cell death forms such as apoptosis, pyroptosis and autophagy. These death forms are also closely related to PF [12,13,14]. Pyroptosis is a process of programmed cell death mediated by classical inflammasome pathway [15]. The activation of inflammasome pathway will induce macrophages to gather and produce cascade inflammatory reaction [16]. The lysed macrophages will not only promote the secretion of cytokines such as IL-1 and IL-18, but also lead to TGF-β1 secreted by M2 macrophages, which promotes the ECM deposition and the inflammatory process in PF [9]. Autophagy can also regulate the polarization direction of macrophages and participate in the regulation of inflammatory response [12]. Autophagy is an evolutionarily conserved and genetically regulated pathway that serves to degrade and clear subcellular components [12, 17]. Excessive macrophage autophagy will increase the occurrence of apoptosis, promote the transformation from M1 to M2, and degrade the activation of NF-κB (nuclear factor-kappa B) inflammatory signal pathway, inhibiting the production and release of proinflammatory factor TNF-α, IL-6, IL-1β and IL-12 [18]. Finally, the above process affected the degradation of ECM and promoted the abnormal repair [12]. Therefore, autophagy is involved in PF by regulating the direction of polarization of macrophages and regulating cytokine secretion.
Emerging evidence has revealed that macrophages death plays important roles in influencing the progression of PF [10]. There is increasing recognition that inflammation and cell death reciprocally affect each other and form an auto-amplification loop of these two factors, which in turn exaggerates fibrosis [19]. Therefore, pharmacological manipulation of macrophages death signals may potentially serve as a logical therapeutic strategy for PF. This review will focus on recent advances in the regulation of AM death and underlying mechanisms on the development of PF.
Analysis of macrophages in human PF lungs
The distinction of the composition of macrophages in human fibrosis pulmonary lungs, compared with normal people, has been reported [20,21,22]. Researchers analyzed the composition of macrophages in fibrosis lung, finding that resident alveolar macrophages and monocytes-derived macrophages, which however now can be further classified into 3 or 4 populations via single-cell RNA-sequencing(scRNA-seq), comprise the major macrophage population [21]. In 2018, researchers first described that there were at least 2 distinct macrophage populations associated with the process of PF, which is both highly expressing genes SPP1 and CHI3L1 [21]. Furthermore, a study suggested that only the SPPS1himacrophages which notably express MERTK were increased in the lung tissues which indicated this subpopulation of macrophages would be the potential highlight of a therapeutic field in PF [23].
Scholars applied a self-organizing map algorithm (FlowSOM) to identify which subsets of myeloid cells participate in fibrosis, revealing that the myeloid lineage cells in IPF can be divided into functional six subsets: monocyte-like cells, monocyte-derived macrophages, monocyte-derived dendritic cells, interstitial macrophages, dendritic cells and alveolar macrophages [24]. Amid them, monocyte-like cells, monocyte-derived macrophages (CD206-subsets) and interstitial macrophages (CD206-subsets) are decreased in IPF lungs. In addition, researchers observed that the AMs from patients with IPF over-expressed a range of regulative genes like ITGB1, ITGB2, S100A8, and FN1 [24]. Collectively, by analysis, the diversity of macrophages in IPF, and specific pathological mechanisms of PF would be deeply clarified in the future.
Role of macrophages in PF
Macrophage-derived secretory proteins and PF
In any phase of PF, macrophages mediate different immunocytes to participate in the process of fibrosis via releasing a variety of proteins, like growth factors, chemokines, and enzymes [25]. TGF-β, some of which is released by macrophages, as the most major effective profibrotic factor is widely researched [26]. It is positive feedback that cells exposed to TGF-β secrete more of this cytokine, interestingly [25]. Besides, macrophages secrete platelet derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and insulin like growth factor-1 (IGF-1) to advance the proliferation of fibroblasts and the synthesis of collagen [27]. Nintedanib, an intracellular inhibitor of tyrosine kinase receptor, has been demonstrated to hinder fibrosis by targeting PDGF, VEGF and FGF (fibroblast growth factor) and is considered as a novel pharmacological treatment [28, 29]. Macrophages can release chemokine (C-C motif) ligand-2 (CCL2) and chemokine (C-C motif) ligand-18 (CCL18) to recruit circulation-derived monocyte to aggravate fibrosis, which is similar to IL-1β [30]. Nevertheless, macrophages, with the capability of secreting MMPs (matrix metalloproteinases) to promote the degradation of ECM, can abate fibrosis [31, 32]. Quite a few researches prove that the expression of MMP-3 augments in the lung tissue of patients with PF, which induces fibroblast activation and epithelial interstitial transformation [31, 33, 34]. In contrast, MMP-3 can not only activate TGF-β to exacerbate fibrosis but show the ability to encumber IGF-1 to execute the role of anti-fibrosis [31]. Macrophages rely on those secretory proteins, showing distinct functions, some of which even are contradictory [7]. This phenomenon may relate to different micro-environment to which macrophages exposure and to the different subpopulations of macrophages which would be further elucidated following.
Macrophage polarization in PF
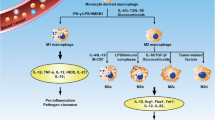
Macrophage activation is periodical which indicates the plastic feature of macrophages [35]. In the process of macrophage activation, macrophages can differentiate into two functionally distinct sub-phenotypes which differ in inducing factors and expression of surface markers (Fig. 1). Firstly, the M1 macrophage is involved in PF mainly in the early stages of injury to mediate the inflammatory response, basically induced by lipopolysaccharide (LPS), granulocyte-macrophage colony stimulating factor (GM-CSF), IFN-γ, TNF-α [36]. M2 macrophages can release several profibrotic cytokines and newly study points out that M2 can show phenotype of M1 by giving LPS and IFN-γ in vitro culture, additionally finding that the fibrosis of model mice was alleviated [35, 37]. That may be a potential way of treating PF. However, researchers tended to consider both M1 and M2 in the pathogenesis of PF in recent years [10]. Functionally, M1 macrophages, which show an excellent ability to mediate tissue injury, contribute to boosting the immunoreaction of the host by releasing intracellular and pro-inflammatory cytokines and chemokines, like TNF-α, IL-1, IL-6 and IL-12 and the removal of pathogens through generating the ROS (active oxygen species) [38]. At the early stage of inflammation, M0 can transform into M1 induced by inducers like LPS, IFN-γ and GM-CSF [9, 39]. Normally, M1 macrophages promote an inflammatory response in lung air space and then re-transform into M2 macrophages which play an important role in wound healing or anti-inflammation [7]. However, without appropriate termination, M1 will cause excessive inflammation and exacerbate the injury of the AECs via the cytotoxic and pro-inflammatory effects, finally causing aberrant fibroblast proliferation and hypernomic ECM deposition [9]. M2 macrophages are used to be considered the main effect cell in the repair or fibrosis of tissue injury and can release pro-fibrotic cytokines such as TGF-β, IL-4, IL-10 and PDGF, which promote transformation and proliferation of fibroblast and myofibroblast [40]. Researchers used to establish an early PF model via bleomycin, finding that the depletion of M2 will abate PF [41]. Collectively, the unbalance of the proportion of M1 with M2 play an important role in the development of PF.

M0 can polarity into M1 and M2 by distinct stimuli. M1 plays an inflammatory role by releasing ROS, IL-1, IL-6, and IL-12 while M2 has the profibrotic potential with the releasing of TGF-β, IL-4, IL-10 and PDGF in PF
Pyroptosis in macrophage drived PF
Pyroptosis is a form of regulated cell death
Pyroptosis is defined as inflammatory programmed cell necrosis which induces DNA damage and chromatin condensation, having the ability to protect the host from the infection of pathogenic bacteria or other non-infection damage by inducing local inflammation [15]. However, because of the fleetly numerous pro-inflammatory cytokines released, and the inflammasome dependency, pyroptosis is mechanistically distinct from apoptosis and any other forms of cell death [42]. When cells are stimulated by inflammatory or injury factors, intracellular Caspase can be activated by the corresponding pathway and cleavage pro-gasdermin, pro-IL-1β and pro-IL-18, ultimately lead to cytomemobrane rupture and release of IL-1β and IL-18 [15, 43]. The assembly of inflammasome plays an extraordinarily important role in this progression [44]. Inflammasomes are a kind of cytosolic multi-protein platform and are constituted by at least three main components, including nod-like receptors (NLRs), pro-caspase-1, and apoptosis-associated spot-like proteins (ACS) [16]. The NLRs family, which is prominent basis of the classification of the inflammasome, is a cytoplasmic group of pattern recognition receptors (PRRs) and characteristic proteins contained at the N-termini of NLRs subdivide these receptors into at least four subfamilies: NLRAs (with a trans-activating domain at the end), NAIPS (with apoptosis inhibitory repeat at the N terminal), NLRPs (with pyrin binding domain at the N terminal) and NLRCs (with Caspase recruitment domain protein, CARD, at the end) [45].
Depending on the distinction between the activated enzyme and the main procedure, pyroptosis can be divided into two pathways [15]. Firstly, the canonical inflammasome pathway characteristically requires the activation of the enzyme of caspase-1 [42, 43, 46, 47]. However, the activation of capcase-1 depends on the assembly of inflammasome which is activated by different upstream pathways [45]. Different types of stimuli, including double-stranded DNA, and bacterial LPS act on NLRs (like the NOD-like receptor thermal protein domain associated protein 3, NLRP3) and spark downstream procedure, those phosphorylated NLRP3 recruit CARD-containing ACSs, which further recruit pro-caspase-1 through their CARDs and the three of them assemble to form the inflammasome, which converts the pro-caspase-1 into caspase-1 [43]. Converted caspase-1 can simultaneously cleavage pro-IL1β, IL-18 and gasdermin D (GSDMD), producing bioactive IL1β, IL-18 and two snippets of GSDMD (N-terminal and T-terminal GSDMD) [47]. And the NT-GSDMD oligomerizes and forms plasma-membrane pores which leads to the fatal release of proinflammatory intracellular contents in the end [15, 42, 47]. Differing from the canonical pathway of pyroptosis, the upstream PRRs are unnecessary for the non-canonical pathway [15, 48]. Recent studies further indicated that both caspase-4 and caspase-5 in humans can be directly triggered via binding to cytosolic LPS [49, 50]. Caspase-11/4/5 cannot directly mature pro-IL-1β and IL-18, unexpectedly, they can mediate the secretion of IL-1β/ IL-18 via NLRP3/caspase-1 pathway and can cleave GSDMD, resulting in pyroptosis eventually [51, 52].
Pyroptosis-derived cytokines in macrophage
Pyroptosis happening in macrophages can cause the release of intracellular cytokines which have an extraordinary ability on leading to inflammation [43]. Those cytokines including IL-1β and IL-18 both exist in macrophages in precursor form and will be cleaved once the caspase is mature [53].
IL-1β, which shows robust relevance to inflammation, participates in a variety of immune phenomena like the migration of monocyte, fever of a host and expression of various chemokines [53, 54]. As to IL-18, it plays an important role in the expression of IFN-γ and activation of Th cells and other immune cells [55]. Dimeric caspase on the inflammasome can cleave the pro-IL-1β, pro-IL-18 and gasdermin meanwhile, and the N-terminal from the cleaved gasdermin form a pore on the cell membrane which leads to k + efflux and cell swelling and result in the releasing of IL-1β and IL-18 [47]. New research has suggested that LPS can bind with and directly activate caspase-4/5/11 without the need for PPRs, while caspase-4/5/11 cannot cleave the pro-IL-1β and pro-IL-18 but via the NLRP3 pathway to activate the caspase-1 to mature those cytokines [47, 56]. Additionally, researchers reported that LPS activates caspase-1-dependent pyroptosis by binding to toll-like receptor-4 (TLR4) expression on the macrophage surface, subsequently upregulating IL-1RI expression on macrophages through NF- kB signaling pathway, increasing macrophage sensitivity to IL-1β and promoting assemble of the inflammasome, thus further promoting pyroptosis [57]. Differing from those pathways mentioned above, high mobility group box 1 (HMGB1), which is released from cells in the injury area, binds to the receptor for advanced glycation end products the receptor for advanced glycation end products (RAGE), inducing the endocytosis of HMGB1 and formation of ACS which activates the caspase-1 later [58,59,60]. Significantly, some studies showed that the assembly of inflammasome and activation of caspase not consequentially induce pyroptosis but cause necroptosis and the mechanism of this phenomenon is still in the mist [61, 62].
Macrophage pyroptosis in PF
The fact that macrophage polarization does have a great effect on PF is generally proven [6]. Similarly, macrophage pyroptosis, because of the release of pro-inflammatory cytokines and regulating polarization, is considered to relate to PF (Fig. 2). CCL2, the classical inflammatory cell chemokines, not only plays an important role in boosting inflammation in the early period of PF but has a profibrotic capacity [63]. Growth evidence proved the notion that CCL2 can rise the expression of ECM and promote the TGF-β released by fibroblasts in the pulmonary, stimulating the deposition of collagen [63]. Additionally, CCL2, by activating the ERK1/2 pathway, contributes to the release of IL-6 which efficaciously restrains the death of fibroblasts via the IL-6/STAT3 pathway [30, 64]. The conception that the NLRP3-inflammasome which participates in macrophage pyroptosis is associated with PF is widely accepted [16, 65,66,67]. On the one hand, NLRP3-inflammasome, as the most classical inflammasome of pyroptosis, induce the inflammatory death of macrophages. On the other hand, it has been demonstrated that this inflammasome promoted the epithelial interstitial transformation in bleomycin-induced fibrosis by regulating levels of TGF-β [68,69,70,71,72]. NOD-like receptors are widely expressed in the cytoplasm of immune cells such as monocytes, lymphocytes and NK cells and have the function of identifying damage associated molecular patterns (DAMPs) and pathogen associated molecular patterns (PAMPs) and participating in the pyroptosis and the polarization of macrophages [73, 74]. Research in 2022 reported that the expression level of M1 macrophage and the NLRP-related protein casp-1 are decreased by applying discoidin domain receptor-1 inhibitor (DDR1-I) in Raw264.7 macrophages in vitro [75]. Additionally, researchers found that oleamide shows the capacity of activation of NLRP3-inflammasome, consequently promoting polarization of M0 into M1 [76]. And the activation of NLRP3-inflammasome is one of the most important procedures of pyroptosis [44]. Also, the TLR4/NK-κB pathway working as a classical inflammatory pathway is not only associated with macrophage polarization but participating in macrophage pyroptosis [57]. TLR4 which is classically activated by DAMPs recruits downstream myeloid differentiation factor (88MyD88) by homophile interaction [77]. MyD88 is a type of cytosolic-soluble protein, which has the ability to phosphorylate IKKβ and further phosphorylate I-κB leading to the forfeit of bioactivity of suppression κB [78]. The final effect of this course is the release of the P65 / P50 NF- κ B dimer and the promotion of the transcription of NLRP3-inflammasome as well as other inflammatory factor precursors [79]. After completion of transcription of NLRP3-inflammasome, it will mediate the pyroptosis of macrophages [80]. Pyroptotic macrophages will release inflammatory cytokines IL-1β which is an important regulatory factor in polarization [53, 74]. With pretreated with low concentrate of Pirfenidone for 24 h, researchers found that the polarization of M2 was inhibited in Raw264.7 cells because of downregulation of NF-κB p50 [81].
A conclusion that can be drawn from those researches is that there is a potential association between macrophage polarization and pyroptosis, and both of their effect on fibrosis of pulmonary. A deeper mechanistic understanding of this relationship which needs to be further researched would offer an innovative and more precise targeted therapy strategy for PF.

Pyroptosis has two main pathways: canonical pathway and non-canonical pathway. The canonical pathway is regulated by inflammasome assembly which is mainly activated by PAMPs and DAMPs. Active NLRP3 binds pro-caspase-1 via ACS to form the NLRP3-inflammasome, further hydrolyzing and activating pro-caspase-1. Caspase-1 of dimerization cleaves GSDMD and pro-IL-1β and pro-IL-18, forming an unselective pore on the cytomembrane, secreting IL-1β and IL-18 and influx of water, causing cell death. Non-canonical pathway initialed by caspase-4/5/11 which is activated by LPS. Active caspase-4/5/11 activates caspase-1 and cleaves GSDMD directly, leading to pyrotosis
The macrophage of other death forms in PF
Macrophage apoptosis in PF
Macrophage apoptosis is associated with PF (Fig. 3). Compared with pyroptosis, apoptosis, which is mediated by two certain pathways: the intrinsic and the extrinsic pathway, is traditionally considered as a non-immunologically linked form of cell death [19]. Research in 2016 reported that Grp78 as the major unfolded protein response regulator shows the ability to suppress macrophage apoptosis, consequently exacerbating bleomycin-induced PF [82]. Moreover, after being disposed of by LPS, researchers found that PF in a mouse model of silicosis was aggravated because of promoted apoptosis and inflammation in AMs [83]. Of note, apoptosis-relating caspase-3/-7 can hinder pyroptosis in the way of cleaving the non-inflammatory site Asp87 of GSDMD [46]. Postponed apoptosis of macrophages conversely induces pathological inflammation and continuously boosts pro-inflammatory cytokines release [45, 84]. TREM-1 knockout mice show diminishing inflammation in the LPS-induced pulmonary injury model [85]. Mice are protected from bleomycin-induced PF once c-FLIP, an anti-apoptotic protein, is deleted from CD11bhi macrophages [86]. In conclusion, excessive infiltration of macrophages promotes PF and suitable repression of this effect would be a novel research direction in treating PF in the future.

Apoptosis is mediated by intrinsic and extrinsic pathways. a Intrinsic pathway; b Extrinsic pathway
Macrophage autophagy in PF
Autophagy, which is mediated by the mechanistic target of Rapamycin (mTOR), the ER stress, the insulin pathway, and a variety of other pathways, features as a form of catabolic cellular components that is highly conserved and autophagosome-dependent (Fig. 4.) [87, 88]. In addition, to contribute in sustain the metabolic balance of cells, autophagy has also been shown a relationship with macrophage pyroptosis in PF [89,90,91]. Recent research reported that Resolvin D2 (RvD2), an anti-innate-immune mediator, promoted the degradation of NLRP3-inflammasome with the indeterminate mechanism, however [92]. Moreover, the loss of autophagy-related proteins was observed in the capacity of augmenting the release of IL-1β and pyroptosis in 2008 [93]. PAMPs and DAMPs activated autophagy by PRRs signaling pathway downregulates pyroptosis via eliminating the cleaved GSDMD produced by caspase which may be related to the AMPK-eEF-2 K pathway [18]. DAMPs, like HMGB1 and interleukin, initial and promote local inflammation in the early stage of fibrosis and ROS, reactive nitrogen species (RNS), and mitochondrial DNA (mtDNA) have the ability to induce autophagy by causing mitochondria damage [94, 95]. Damaged or dysfunctional mitochondria can release ROS and mtDNA, forming a cascade of pro-pyroptotic factors, resulting in NLRP3-inflammasome excessive activation. However, those factors initial mitophagy meanwhile, activating the mitochondrial autophagosome, following decomposing the damaged mitochondrial, leading inhibition of pyroptosis. RNS, similar to ROS, is responsible for the promotion of pyroptosis, whose clearance may abate macrophage pyroptosis via autophagy [94, 96,97,98]. After the knockout of the autophagy-related protein-7 (Atg7) gene, the activity of the inflammasome of macrophage and the level of serum IL-1β are raised in mice models of pseudomonas aeruginosa-induced sepsis [99]. All these findings indicate that autophagy has an excellent ability to eliminate inflammation. which may link with suppressing macrophage pyroptotic death.
Autophagy mediates PF via multi-pathway on the other hand. In the acute inflammatory phase of PF, autophagy is widely activated, to clear the invading pathogenic substances [100, 101]. However, it had been reported that, in the lung tissue of patients with PF, the autophagosomes were remarkably decreased [102, 103] and ubiquitinated proteins were accumulated in cells [12], which indicated that autophagy is inhibited in the fibrotic phase of this disease which leads to disability of removing ECM and returning fibrosis of pathological pulmonary. Chemokine (C-X3-C motif) receptor 1(CX3CR1), expressed on the macrophage, NK cells and T lymphocytes, is the receptor for CX3CL1 and is important to induce the generation of ROS and macrophage autophagy [104]. The overexpression of CX3CL1 promoted fibrosis in a mouse model of hyperoxic lung injury, which is related to the activation of Akt1-mediated autophagy of macrophage. By further using CX3CL1 inhibitor, 3-methyladenine (3-MA), macrophage autophagy, and fibrosis of pulmonary were reduced [105]. The classical hypoglycemics metformin is recently found to be able to activate the AMPK-mTOR pathway, thereby attenuating PF in silicosis [106]. Besides, autophagy exacerbates fibrosis via promoting M2 macrophage polarization. DHA (docosahexaenoic acid) enhances the transformation of M2 through autophagy and the p38-MAPK pathway [107]. Ubiquitin-specific protease 19 (USP19), as a type of deubiquitinating enzyme, positively effect M2-like macrophage polarization by the autophagy-related response to NLRP3 [108]. However, interestingly, researchers found that isoprenaline can down-regulate autophagy by activating ROS-ERK and mTOR signaling pathways, enhancing M2 macrophage polarization [109].
In summary, a growing number of researches demonstrate the robust relationship between autophagy and pyroptosis in PF. It is clear that autophagy participates in process of PF in a pyroptosis-dependent or M2 macrophage-dependent way. Nevertheless, the question of whether are there more interaction between autophagy and fibrosis is open yet.

The main process of autophagy is the formation of autolysosomes
The crosstalk between the signaling pathway of pyroptosis, apoptosis and autophagy
Growth number of researches have been established a more complex mode of cell death and as crosstalk among them [18, 46]. To make a deep understanding of those forms of cell death in PF, it is necessary to elucidate some essential signal pathways and the crosstalk of them (Fig. 5). The term inflammasome was first described in 2002 [110]. It had been widely proven in past decades that the nod-like receptor family and pyrin and HIN domain family were involved in the formation of an inflammasome [111,112,113]. The NLRP3 is greatly important for human immune defenses, whose activation may relate to multifactor. The NLRP3 is mainly composed of 3 domains: C-terminal leucine-rich repeats (LRR), a nucleotide-binding oligomerization domain (NACHT) domain and the N-terminal pyrin domain (PYD). The NLRP3 recruits and activates caspase-1 by binding ASC [114]. The activation of NLRP3 which includes initial up-regulation of NLRP3 and the level of pro-IL-1β is essential for this procedure [115]. To date, it was thought that k+ outflowing, ROS and lysosomal damage are the three major hypotheses in the field of activation of NLRP3, however, the precise mechanism is in controversial yet [116,117,118]. In fact, some of researches suggest that NK-κB plays a specific role in the activation of NLRP3 [119, 120]. Once activated by DAMPs or PAMPs, TLRs recruit downstream MyD88 which phosphorylates IKKβ via the IRAK-TRAF6-NIK pathway and then further phosphorylates I-κB, leading to the deprivation of bio-activity of inhibiting κB, which final causing the transcription of NLRP3-inflammasome and the precursor of other inflammatory cytokines [120, 121]. The inflammasomes need to undergo several other post-translational modifications including ubiquitination [122] and sumoylation [123] before it is activated. After the assembly of NLRP3-inflammasome is finished, caspase-1 would be activated in certain vitro conditions [71, 124]. However, new researches also indicated that TLR4/MyD88 signaling pathway can initial NLRP3-inflammasome through a non-transcriptional mechanism which may relate to the production of mtROS [125].
Distinct from pyroptosis, caspases that participate in apoptosis can be functionally classified as initiator caspases, including caspase-8 and -9 and executioner caspases, including caspase-3 and -7 [126,127,128]. Tumor necrosis factor receptor 1 (TNFR1) activated by TNF-α recruits the RIPK1 which is ubiquitinated after the complex-I is formed, and RIPK1 will be deubiquitinated if the inhibitor of apoptosis proteins are lacking and deubiquitinated RIPK1 forms complex-II, then activating pro-apoptotic caspase-8, one component of complex-II, finally causing apoptosis [19]. Caspase-8 is the major part of the connection between pyroptosis and apoptosis. Caspase-8 not only has the ability of proteolysis to process caspase-1 but also plays an integral role in the transcription of NLRP3 and IL-1β [129]. Furthermore, caspase-8 can directly activate GSDMD whose cleavage participates in the formation of NLRP3 and production of IL-1β during the infection of Yersinia, following the blockade of TAK1 [130]. Caspase-1 also is the evidence for the relativity between pyroptosis and apoptosis, in 2008 when it was observed that caspase-1 can cleave caspase-7 in macrophages [131]. Researchers found that caspase-1 can activate caspase-3 in the condition of deficiency of GSDMD [132].
Similar to apoptosis, it has been found that signaling pathways regulating autophagy are involved in regulating pyroptosis [18]. The mTOR pathway plays a more crucial role in the autophagy of cells. Insulin and IGF activate the RTKs and activate the PI3K-AKT signaling pathway, the activated AKT phosphorylates TSC1/2 which dissociates from lysosome and subsequently activates Rheb, the activator of mTORC1 [133, 134]. Interestingly, AKT also participates in the modulation of the mTORC2 pathway of which mechanism is still not completely clear [135]. The overproduction of ROS can not only activate NLRP3 inflammasome which has been mentioned above but induces autophagy also [136]. Researchers found that the pyroptosis of Leydig cells was alleviated after being processed by adrenomedullin by promoting autophagy via the ROS/AMPK/mTOR pathway [137]. SESN2, a stress-derived protein, inhibits activation of NLRP3-inflammasome in macrophage, by inducing mitophagy which is a form of autophagy [138]. Cytosolic DNA acts as the activator of stimulator of interferon genes (STING) via the GMP/cGAMP pathway which is the classical pathway of inducing autophagy [139].
Collectively, the crosstalk between apoptosis, autophagy and pyroptosis has been further studied in recent years but some of the mechanisms still are unclear. More researches are needed to conduct on the crosstalk between pyroptosis and the other two cell death forms, especially in the field of PF. In the past, we often take the impact of only one mode of cell death into consideration in PF, while simultaneously neglecting the potential effect of other forms of cell death. An all-inclusive understanding of cell death happening in the process of PF will provide a novel direction of research in PF which possibly contributes to the treatment and quality of life of patients with PF.

The crosstalk between the signaling pathway of pyroptosis, apoptosis, and autophagy. DAMPs and PAMPs can activate the corresponding receptor, recruiting cytosolic MyD88, and starting the NK-κB pathway. In the process of post-transcription of NLRP3, pro-caspase-1 and pro-IL-1β/18, the apoptotic caspase-8 can promote the activation of GSDMD and induce the proteolysis of NLRP3 and contribute to the transcription of NLRP3, pro-caspase-1 and pro-IL-1β/18. SESN2 can induce mitophagy which releases the mtROS which acts as a PAMP. Adrenomedullin promotes autophagy via ROS/AMPL/mTOR pathway, abating pyroptosis of the cell
Conclusion
Lung M1 and M2 macrophages are distinct cell subtypes and are both involved in the pathogenesis of PF. M1 and M2 macrophages play different roles in the pathogenesis of PF. Generally, M1 macrophages are responsible for wound healing after alveolar epithelial injury, while M2 macrophages are determined to over repair the damaged tissue and terminate the degradation of ECM in the lung [9]. A variety of regulatory cytokines, chemokines, mediators and immune-regulatory cells affect macrophage polarization in the lung [9]. Studies have provided evidence for a connection between cell death and macrophage polarization and understanding of the impact of macrophage death on ECM accumulation is critical in fully elucidating the mechanisms underlying PF. Following an initial event of chronic persistent injury, cell death and inflammation can induce each other and drive a release of regulatory cytokines, chemokines, mediators that lead to exaggerated fibrosis effects [10, 102]. Macrophage pyroptosis can activate the release of signaling pathways caspase-1 and IL-1 and promote the secretion of TGF-β1 [43], thus promoting the proliferation and differentiation of myofibroblasts and inflammatory response. Apoptosis and autophagy were thought to be the form of cell death during homeostasis and development and has been heavily studied and discussed in numerous pieces of literature on PF [19, 101, 102]. The gaps in our knowledge of cell death include whether different types of cell death signaling developed separately as responses to specific triggers or whether they represent parts of a signaling network that follow common regulatory mechanisms.
Although therapies for PF included a variety of drugs and non-pharmacological interventions remain unsolved regarding the exact mechanisms of manipulating the balance of M1/M2 phenotype in PF pathogenesis and are unable to effectively attenuate PF [3]. Comprehensive understanding of the molecular mechanisms that regulate cell death will allow the development of strategies that control cell death, thereby developing novel interventions for PF.
Data availability
Not applicable.
Abbreviations
- PF:
-
Pulmonary fibrosis
- ECM:
-
Extracellular matrix
- AECs:
-
Alveolar epithelial cell
- TNF-α:
-
Tumor necrosis factor-α
- NF-κb:
-
Nuclear factor-kappa B
- ScRNA-seq:
-
Single-cell RNA-sequencing
- PDGF:
-
Single-cell RNA-sequencing
- VEGF:
-
Vascular endothelial growth factor
- IGF-1:
-
Insulin like growth factor
- FGF:
-
Fibroblast growth factor
- CCL:
-
Chemokine (C-C motif) ligand
- MMP:
-
Matrix metalloproteinases
- IPF:
-
Idiopathic PF
- GM-CSF:
-
Granulocyte-macrophage colony stimulating factor
- ROS:
-
Reactive oxygen species
- LPS:
-
Lipopolysaccharide
- ACS:
-
Apoptosis-associated speck-like protein
- NLRs:
-
Nod-like receptor
- PRR:
-
Pattern recognition receptor
- CARD:
-
Caspase recruitment domain protein
- GSDM:
-
Gasdermin
- TLR:
-
Toll-like receptor
- HMGB1:
-
High mobility group box 1
- RAGE:
-
Receptor for advanced glycation end products
- STAT:
-
Signal transducer and activator of transcription
- DAMP:
-
Damage associated molecular pattern
- PAMP:
-
Pathogen associated molecular pattern
- MyD88:
-
Myeloid differentiation factor 88
- IKKB:
-
I-kappa-B kinase B
- BCL-xL:
-
B-cell lymphoma extra large
- BCL-2:
-
B-cell lymphoma 2
- BAK:
-
BCL-2 antagonist/killer 1
- MOMP:
-
Mitochondrial outer membrane permeabilization
- SMAC:
-
Second mitochondria–derived activator of caspase
- APAF1:
-
Apoptotic protease activating factor 1
- Asp:
-
Aspartic acid
- Fas:
-
CD95
- FasL:
-
Fas ligand
- FADD:
-
Fas-associated death domain
- cFLP:
-
Cellular FLICE-inhibitory protein
- IAP:
-
Inhibitor of apoptosis protein
- DISC:
-
Death-inducing signaling complex
- DNase:
-
Deoxyribonuclease
- RIPK1:
-
Receptor-interacting protein kinase 1
- TRADD:
-
TNFR1-associated death domain
- mTOR:
-
Mechanistic target of rapamycin
- RVD2:
-
Resolvin D2
- Atg:
-
Autophagy-related protein
- AMPK:
-
AMP-activated protein kinase
- MAPK:
-
Mitogen-activated protein kinase
- RNS:
-
Reactive nitrogen species
- mtDNA:
-
Mitochondrial DNA
- CX3CR1:
-
Chemokine (C-X3-C motif) receptor 1
- 3-MA:
-
3-Methyladenine
- DHA:
-
Docosahexaenoic acid
- USP19:
-
Ubiquitin-specific protease 19
- TNFR1:
-
Tumor necrosis factor receptor 1
- STING:
-
Stimulator of interferon genes
- LRR:
-
Leucine-rich repeats
- NACHT:
-
Nucleotide-binding oligomerization domain
References
Wakwaya Y, Brown KK (2019) Idiopathic pulmonary fibrosis: epidemiology, diagnosis andOutcomes. Am J Med Sci 357(5):359–369. https://doi.org/10.1016/j.amjms.2019.02.013
Moss BJ, Ryter SW, Rosas IO (2022) Pathogenic mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev Pathol. https://doi.org/10.1146/annurev-pathol-042320-030240. .17:515 – 46
Noble PW, Barkauskas CE, Jiang D (2012) Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 122(8):2756–2762. https://doi.org/10.1172/JCI60323
Thannickal VJ, Toews GB, White ES, Lynch JP III, Martinez FJ (2004) Mechanisms of pulmonary fibrosis. Annu Rev Med 55:395–417. https://doi.org/10.1146/annurev.med.55.091902.103810
Deng Z, Fear MW, Suk Choi Y, Wood FM, Allahham A, Mutsaers SE et al (2020) The extracellular matrix and mechanotransduction in pulmonary fibrosis. Int J Biochem Cell Biol 126:105802. https://doi.org/10.1016/j.biocel.2020.105802
Witherel CE, Sao K, Brisson BK, Han B, Volk SW, Petrie RJ et al (2021) Regulation of extracellular matrix assembly and structure by hybrid M1/M2 macrophages. Biomaterials 269:120667. https://doi.org/10.1016/j.biomaterials.2021.120667
Arora S, Dev K, Agarwal B, Das P, Syed MA (2018) Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology 223(4–5):383–96. https://doi.org/10.1016/j.imbio.2017.11.001
Tan SY, Krasnow MA (2016) Developmental origin of lung macrophage diversity. Development 143(8):1318–1327. https://doi.org/10.1242/dev.129122
Yunna C, Mengru H, Lei W, Weidong C (2020) Macrophage M1/M2 polarization. Eur J Pharmacol. https://doi.org/10.1016/j.ejphar.2020.173090. .877:173090
Cheng P, Li S, Chen H (2021) Macrophages in lung Injury, repair and fibrosis. Cells 10(2):436
Roszer T (2015) Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. https://doi.org/10.1155/2015/816460
Lv X, Li K, Hu Z (2020) Autophagy and pulmonary fibrosis. Adv Exp Med Biol. https://doi.org/10.1007/978-981-15-4272-5_40. .1207:569 – 79
Hong Q, Zhang Y, Lin W, Wang W, Yuan Y, Lin J et al (2022) Negative feedback of the cAMP/PKA pathway regulates the effects of endoplasmic reticulum stress-induced NLRP3 inflammasome activation on type II alveolar epithelial cell pyroptosis as a novel mechanism of BLM-induced pulmonary fibrosis. J Immunol Res 2022:2291877. https://doi.org/10.1155/2022/2291877
Sharma P, Alizadeh J, Juarez M, Samali A, Halayko AJ, Kenyon NJ et al (2021) Autophagy, apoptosis the unfolded protein response and lung function in idiopathic pulmonary fibrosis. Cells 10(7):1642
Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X (2021) Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther 6(1):128. https://doi.org/10.1038/s41392-021-00507-5
Pinkerton JW, Kim RY, Robertson AAB, Hirota JA, Wood LG, Knight DA et al (2017) Inflammasomes in the lung. Mol Immunol 86:44–55. https://doi.org/10.1016/j.molimm.2017.01.014
Wu MY, Lu JH (2019) Autophagy and macrophage functions: inflammatory response and phagocytosis. Cells 9(1):70
Guo R, Wang H, Cui N (2021) Autophagy regulation on pyroptosis: mechanism and medical implication in sepsis. Mediators Inflamm. https://doi.org/10.1155/2021/9925059
Sauler M, Bazan IS, Lee PJ (2019) Cell death in the lung: the apoptosis-necroptosis axis. Annu Rev Physiol 81:375–402. https://doi.org/10.1146/annurev-physiol-020518-114320
Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F et al (2020) Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 6(28):eaba1983. https://doi.org/10.1126/sciadv.aba1983
Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S et al (2019) Single-cell transcriptomic analysis of human lung provides insights into the Pathobiology of Pulmonary Fibrosis. Am J Respir Crit Care Med 199(12):1517–1536. https://doi.org/10.1164/rccm.201712-2410OC
Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A et al (2019) Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20(2):163–172. https://doi.org/10.1038/s41590-018-0276-y
Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E et al (2019) Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. https://doi.org/10.1183/13993003.02441-2018
Serezani APM, Pascoalino BD, Bazzano JMR, Vowell KN, Tanjore H, Taylor CJ et al (2022) Multiplatform single-cell analysis identifies Immune cell types enhanced in pulmonary fibrosis. Am J Respir Cell Mol Biol 67(1):50–60. https://doi.org/10.1165/rcmb.2021-0418OC
Zhang L, Wang Y, Wu G, Xiong W, Gu W, Wang CY (2018) Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir Res 19(1):170. https://doi.org/10.1186/s12931-018-0864-2
Boutanquoi PM, Burgy O, Beltramo G, Bellaye PS, Dondaine L, Marcion G et al (2020) TRIM33 prevents pulmonary fibrosis by impairing TGF-beta1 signalling. Eur Respir J. https://doi.org/10.1183/13993003.01346-2019
Khalil N, Bereznay O, Sporn M, Greenberg AH (1989) Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med 170(3):727–737. https://doi.org/10.1084/jem.170.3.727
Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S et al (2015) Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J 45(5):1434–1445. https://doi.org/10.1183/09031936.00174914
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y et al (2019) Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 381(18):1718–1727. https://doi.org/10.1056/NEJMoa1908681
Liu X, Das AM, Seideman J, Griswold D, Afuh CN, Kobayashi T et al (2007) The CC chemokine ligand 2 (CCL2) mediates fibroblast survival through IL-6. Am J Respir Cell Mol Biol 37(1):121–128. https://doi.org/10.1165/rcmb.2005-0253OC
Yamashita CM, Dolgonos L, Zemans RL, Young SK, Robertson J, Briones N et al (2011) Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol 179(4):1733–1745. https://doi.org/10.1016/j.ajpath.2011.06.041
Madala SK, Pesce JT, Ramalingam TR, Wilson MS, Minnicozzi S, Cheever AW et al (2010) Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13-dependent fibrosis. J Immunol 184(7):3955–3963. https://doi.org/10.4049/jimmunol.0903008
Robert S, Gicquel T, Victoni T, Valenca S, Barreto E, Bailly-Maitre B et al (2016) Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci Rep. https://doi.org/10.1042/BSR20160107
Radisky DC, Przybylo JA (2008) Matrix metalloproteinase-induced fibrosis and malignancy in breast and lung. Proc Am Thorac Soc 5(3):316–322. https://doi.org/10.1513/pats.200711-166DR
Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E (2015) Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol 53(5):676–688. https://doi.org/10.1165/rcmb.2015-0012OC
Aggarwal NR, King LS, D’Alessio FR (2014) Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol 306(8):L709–L725. https://doi.org/10.1152/ajplung.00341.2013
Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM (2012) Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol 47(4):417–426. https://doi.org/10.1165/rcmb.2012-0090OC
Murray PJ, Wynn TA (2011) Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol 89(4):557–563. https://doi.org/10.1189/jlb.0710409
Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A (2010) Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 215(9–10):788–795. https://doi.org/10.1016/j.imbio.2010.05.020
Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M et al (2016) Chop Deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther 24(5):915–925. https://doi.org/10.1038/mt.2016.36
Wang Y, Zhang L, Wu GR, Zhou Q, Yue H, Rao LZ et al (2021) MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci Adv. https://doi.org/10.1126/sciadv.abb6075
Frank D, Vince JE (2019) Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ 26(1):99–114. https://doi.org/10.1038/s41418-018-0212-6
Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7(2):99–109. https://doi.org/10.1038/nrmicro2070
Tsuchiya K (2020) Inflammasome-associated cell death: pyroptosis, apoptosis, and physiological implications. Microbiol Immunol 64(4):252–269. https://doi.org/10.1111/1348-0421.12771
Martinon F, Tschopp J (2007) Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ 14(1):10–22. https://doi.org/10.1038/sj.cdd.4402038
Taabazuing CY, Okondo MC, Bachovchin DA (2017) Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol 24(4):507–514. https://doi.org/10.1016/j.chembiol.2017.03.009. (e4)
Shi J, Gao W, Shao F (2017) Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 42(4):245–254. https://doi.org/10.1016/j.tibs.2016.10.004
Platnich JM, Muruve DA (2019) NOD-like receptors and inflammasomes: a review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys 670:4–14. https://doi.org/10.1016/j.abb.2019.02.008
Fernandez-Duran I, Quintanilla A, Tarrats N, Birch J, Hari P, Millar FR et al (2022) Cytoplasmic innate immune sensing by the caspase-4 non-canonical inflammasome promotes cellular senescence. Cell Death Differ 29(6):1267–1282. https://doi.org/10.1038/s41418-021-00917-6
An J, Kim SH, Hwang D, Lee KE, Kim MJ, Yang EG et al (2019) Caspase-4 disaggregates lipopolysaccharide micelles via LPS-CARD interaction. Sci Rep 9(1):826. https://doi.org/10.1038/s41598-018-36811-4
Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB et al (2015) NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol 45(10):2918–2926. https://doi.org/10.1002/eji.201545655
Ruhl S, Broz P (2015) Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol 45(10):2927–2936. https://doi.org/10.1002/eji.201545772
Arend WP, Palmer G, Gabay C (2008) IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223:20–38. https://doi.org/10.1111/j.1600-065X.2008.00624.x
Weber A, Wasiliew P, Kracht M (2010) Interleukin-1beta (IL-1beta) processing pathway. Sci Signal 3(105):cm2. https://doi.org/10.1126/scisignal.3105cm2
Dinarello CA (1999) IL-18: a TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J Allergy Clin Immunol 103(1 Pt 1):11–24. https://doi.org/10.1016/s0091-6749(99)70518-x
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526(7575):660–665. https://doi.org/10.1038/nature15514
He X, Qian Y, Li Z, Fan EK, Li Y, Wu L et al (2016) TLR4-Upregulated IL-1beta and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci Rep 6:31663. https://doi.org/10.1038/srep31663
Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5(4):331–342. https://doi.org/10.1038/nri1594
Vijayakumar EC, Bhatt LK, Prabhavalkar KS (2019) High mobility Group Box-1 (HMGB1): a potential target in therapeutics. Curr Drug Targets 20(14):1474–1485. https://doi.org/10.2174/1389450120666190618125100
Yang H, Wang H, Andersson U (2020) Targeting inflammation driven by HMGB1. Front Immunol 11:484. https://doi.org/10.3389/fimmu.2020.00484
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440(7081):237–241. https://doi.org/10.1038/nature04516
Mollen KP, Anand RJ, Tsung A, Prince JM, Levy RM, Billiar TR (2006) Emerging paradigm: toll-like receptor 4-sentinel for the detection of tissue damage. Shock 26(5):430–437
Deng X, Xu M, Yuan C, Yin L, Chen X, Zhou X et al (2013) Transcriptional regulation of increased CCL2 expression in pulmonary fibrosis involves nuclear factor-kappab and activator protein-1. Int J Biochem Cell Biol 45(7):1366–1376. https://doi.org/10.1016/j.biocel.2013.04.003
Murray LA, Argentieri RL, Farrell FX, Bracht M, Sheng H, Whitaker B et al (2008) Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int J Biochem Cell Biol 40(10):2174–2182. https://doi.org/10.1016/j.biocel.2008.02.016
Toldo S, Mauro AG, Cutter Z, Abbate A (2018) Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 315(6):H1553–H68. https://doi.org/10.1152/ajpheart.00158.2018
Zhang WJ, Chen SJ, Zhou SC, Wu SZ, Wang H (2021) Inflammasomes and fibrosis. Front Immunol 12:643149. https://doi.org/10.3389/fimmu.2021.643149
Brusselle GG, Provoost S, Bracke KR, Kuchmiy A, Lamkanfi M (2014) Inflammasomes in respiratory disease: from bench to bedside. Chest 145(5):1121–1133. https://doi.org/10.1378/chest.13-1885
Liu W, Han X, Li Q, Sun L, Wang J (2022) Iguratimod ameliorates bleomycin-induced pulmonary fibrosis by inhibiting the EMT process and NLRP3 inflammasome activation. Biomed Pharmacother 153:113460. https://doi.org/10.1016/j.biopha.2022.113460
Alyaseer AAA, de Lima MHS, Braga TT (2020) The role of NLRP3 inflammasome activation in the epithelial to mesenchymal transition process during the fibrosis. Front Immunol 11:883. https://doi.org/10.3389/fimmu.2020.00883
Min L, Shu-Li Z, Feng Y, Han H, Shao-Jun L, Sheng-Xiong T et al (2022) NecroX-5 ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3-mediated epithelial-mesenchymal transition. Respir Res 23(1):128. https://doi.org/10.1186/s12931-022-02044-3
Tian R, Zhu Y, Yao J, Meng X, Wang J, Xie H et al (2017) NLRP3 participates in the regulation of EMT in bleomycin-induced pulmonary fibrosis. Exp Cell Res 357(2):328–334. https://doi.org/10.1016/j.yexcr.2017.05.028
Liang Q, Cai W, Zhao Y, Xu H, Tang H, Chen D et al (2020) Lycorine ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3 inflammasome activation and pyroptosis. Pharmacol Res 158:104884. https://doi.org/10.1016/j.phrs.2020.104884
Wang L, Zhao M (2022) Suppression of NOD-like receptor protein 3 inflammasome activation and macrophage M1 polarization by hederagenin contributes to attenuation of sepsis-induced acute lung injury in rats. Bioengineered 13(3):7262–7276. https://doi.org/10.1080/21655979.2022.2047406
Zhang J, Liu X, Wan C, Liu Y, Wang Y, Meng C et al (2020) NLRP3 inflammasome mediates M1 macrophage polarization and IL-1beta production in inflammatory root resorption. J Clin Periodontol 47(4):451–460. https://doi.org/10.1111/jcpe.13258
Wang H, Wen Y, Wang L, Wang J, Chen H, Chen J et al (2022) DDR1 activation in macrophage promotes IPF by regulating NLRP3 inflammasome and macrophage reaction. Int Immunopharmacol 113. https://doi.org/10.1016/j.intimp.2022.109294
Wisitpongpun P, Potup P, Usuwanthim K (2022) Oleamide-mediated polarization of M1 macrophages and IL-1β production by regulating NLRP3-Inflammasome activation in primary human monocyte-derived macrophages. Front Immunol. https://doi.org/10.3389/fimmu.2022.856296
Li C, Yu Y, Li W, Liu B, Jiao X, Song X et al (2017) Phycocyanin attenuates pulmonary fibrosis via the TLR2-MyD88-NF-kappaB signaling pathway. Sci Rep 7(1):5843. https://doi.org/10.1038/s41598-017-06021-5
Yan F, Guan J, Peng Y, Zheng X (2017) MyD88 NEDDylation negatively regulates MyD88-dependent NF-kappaB signaling through antagonizing its ubiquitination. Biochem Biophys Res Commun 482(4):632–637. https://doi.org/10.1016/j.bbrc.2016.11.084
Yang Y, Wang H, Kouadir M, Song H, Shi F (2019) Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 10(2):128. https://doi.org/10.1038/s41419-019-1413-8
Zheng M, Kanneganti TD (2020) The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev 297(1):26–38. https://doi.org/10.1111/imr.12909
Ying H, Fang M, Hang QQ, Chen Y, Qian X, Chen M (2021) Pirfenidone modulates macrophage polarization and ameliorates radiation-induced lung fibrosis by inhibiting the TGF‐β1/Smad3 pathway. J Cell Mol Med 25(18):8662–8675. https://doi.org/10.1111/jcmm.16821
Ayaub EA, Kolb PS, Mohammed-Ali Z, Tat V, Murphy J, Bellaye PS et al (2016) GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J Pathol 239(4):411–425. https://doi.org/10.1002/path.4738
Tan S, Yang S, Chen M, Wang Y, Zhu L, Sun Z et al (2020) Lipopolysaccharides promote pulmonary fibrosis in silicosis through the aggravation of apoptosis and inflammation in alveolar macrophages. Open Life Sci 15(1):598–605. https://doi.org/10.1515/biol-2020-0061
Wang Y, Gao W, Shi X, Ding J, Liu W, He H et al (2017) Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547(7661):99–103. https://doi.org/10.1038/nature22393
Yuan Z, Syed M, Panchal D, Joo M, Bedi C, Lim S et al (2016) TREM-1-accentuated lung injury via miR-155 is inhibited by LP17 nanomedicine. Am J Physiol Lung Cell Mol Physiol 310(5):L426–L438. https://doi.org/10.1152/ajplung.00195.2015
McCubbrey AL, Barthel L, Mohning MP, Redente EF, Mould KJ, Thomas SM et al (2018) Deletion of c-FLIP from CD11b(hi) macrophages prevents development of bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol 58(1):66–78. https://doi.org/10.1165/rcmb.2017-0154OC
Yu L, Chen Y, Tooze SA (2018) Autophagy pathway: cellular and molecular mechanisms. Autophagy 14(2):207–215. https://doi.org/10.1080/15548627.2017.1378838
Yang J, Carra S, Zhu WG, Kampinga HH (2013) The regulation of the autophagic network and its implications for human disease. Int J Biol Sci 9(10):1121–1133. https://doi.org/10.7150/ijbs.6666
Kim JY, Paton JC, Briles DE, Rhee DK, Pyo S (2015) Streptococcus pneumoniae induces pyroptosis through the regulation of autophagy in murine microglia. Oncotarget 6(42):44161–44178. https://doi.org/10.18632/oncotarget.6592
Liu M, Lu J, Yang S, Chen Y, Yu J, Guan S (2022) Alliin alleviates LPS-induced pyroptosis via promoting mitophagy in THP-1 macrophages and mice. Food Chem Toxicol 160:112811. https://doi.org/10.1016/j.fct.2022.112811
Zhuo L, Chen X, Sun Y, Wang Y, Shi Y, Bu L et al (2020) Rapamycin Inhibited pyroptosis and reduced the release of IL-1beta and IL-18 in the septic response. Biomed Res Int 2020:5960375. https://doi.org/10.1155/2020/5960375
Cao L, Wang Y, Wang Y, Lv F, Liu L, Li Z (2021) Resolvin D2 suppresses NLRP3 inflammasome by promoting autophagy in macrophages. Exp Ther Med 22(5):1222. https://doi.org/10.3892/etm.2021.10656
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T et al (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456(7219):264–268. https://doi.org/10.1038/nature07383
Denning NL, Aziz M, Gurien SD, Wang P (2019) DAMPs and NETs in Sepsis. Front Immunol 10:2536. https://doi.org/10.3389/fimmu.2019.02536
Zindel J, Kubes P (2020) DAMPs, PAMPs, and LAMPs in immunity and sterile inflammation. Annu Rev Pathol 15:493–518. https://doi.org/10.1146/annurev-pathmechdis-012419-032847
Jiang C, Jiang L, Li Q, Liu X, Zhang T, Dong L et al (2018) Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology 410:26–40. https://doi.org/10.1016/j.tox.2018.09.002
Teng JF, Mei QB, Zhou XG, Tang Y, Xiong R, Qiu WQ et al (2020) Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-kappaB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers 12(1):193
Liu Q, Zhang D, Hu D, Zhou X, Zhou Y (2018) The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol. https://doi.org/10.1016/j.molimm.2018.09.010. .103:115 – 24
Pu Q, Gan C, Li R, Li Y, Tan S, Li X et al (2017) Atg7 deficiency intensifies inflammasome activation and pyroptosis in pseudomonas sepsis. J Immunol 198(8):3205–3213. https://doi.org/10.4049/jimmunol.1601196
Deretic V (2021) Autophagy in inflammation, infection, and immunometabolism. Immunity 54(3):437–453. https://doi.org/10.1016/j.immuni.2021.01.018
Clarke AJ, Simon AK (2019) Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol 19(3):170–183. https://doi.org/10.1038/s41577-018-0095-2
Kota A, Deshpande DA, Haghi M, Oliver B, Sharma P (2017) Autophagy and airway fibrosis: is there a link? F1000Res. https://doi.org/10.12688/f1000research.11236.1
Zhang J, Wang H, Chen H, Li H, Xu P, Liu B et al (2022) ATF3 -activated accelerating effect of LINC00941/lncIAPF on fibroblast-to-myofibroblast differentiation by blocking autophagy depending on ELAVL1/HuR in pulmonary fibrosis. Autophagy 18(11):2636–2655. https://doi.org/10.1080/15548627.2022.2046448
Helmke A, Nordlohne J, Balzer MS, Dong L, Rong S, Hiss M et al (2019) CX3CL1-CX3CR1 interaction mediates macrophage-mesothelial cross talk and promotes peritoneal fibrosis. Kidney Int 95(6):1405–1417. https://doi.org/10.1016/j.kint.2018.12.030
Chen Y, Zhang H, Li F, Wang X (2020) Inhibition of CX3C receptor 1-mediated autophagy in macrophages alleviates pulmonary fibrosis in hyperoxic lung injury. Life Sci 259:118286. https://doi.org/10.1016/j.lfs.2020.118286
Cheng D, Xu Q, Wang Y, Li G, Sun W, Ma D et al (2021) Metformin attenuates silica-induced pulmonary fibrosis via AMPK signaling. J Transl Med 19(1):349. https://doi.org/10.1186/s12967-021-03036-5
Kawano A, Ariyoshi W, Yoshioka Y, Hikiji H, Nishihara T, Okinaga T (2019) Docosahexaenoic acid enhances M2 macrophage polarization via the p38 signaling pathway and autophagy. J Cell Biochem 120(8):12604–12617. https://doi.org/10.1002/jcb.28527
Liu T, Wang L, Liang P, Wang X, Liu Y, Cai J et al (2021) USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol 18(10):2431–2442. https://doi.org/10.1038/s41423-020-00567-7
Shan M, Qin J, Jin F, Han X, Guan H, Li X et al (2017) Autophagy suppresses isoprenaline-induced M2 macrophage polarization via the ROS/ERK and mTOR signaling pathway. Free Radic Biol Med. https://doi.org/10.1016/j.freeradbiomed.2017.05.021. .110:432 – 43
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10(2):417–426. https://doi.org/10.1016/s1097-2765(02)00599-3
Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L et al (2012) Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36(4):561–571. https://doi.org/10.1016/j.immuni.2012.02.014
Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I (2019) The pyrin inflammasome in health and disease. Front Immunol 10:1745. https://doi.org/10.3389/fimmu.2019.01745
Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W et al (2020) HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science. https://doi.org/10.1126/science.aas8995
Elliott EI, Sutterwala FS (2015) Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 265(1):35–52. https://doi.org/10.1111/imr.12286
Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. https://doi.org/10.3390/ijms20133328
Koumangoye R (2022) The role of Cl(-) and K(+) efflux in NLRP3 inflammasome and innate immune response activation. Am J Physiol Cell Physiol 322(4):C645–C52. https://doi.org/10.1152/ajpcell.00421.2021
Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V et al (2016) ROS-Mediated NLRP3 inflammasome activation in brain, heart, kidney and testis ischemia/reperfusion injury. Oxid Med Cell Longev 2016:2183026. https://doi.org/10.1155/2016/2183026
Lauterbach MA, Saavedra V, Mangan MSJ, Penno A, Thiele C, Latz E et al (2021) 1-Deoxysphingolipids cause autophagosome and lysosome accumulation and trigger NLRP3 inflammasome activation. Autophagy 17(8):1947–1961. https://doi.org/10.1080/15548627.2020.1804677
Li LL, Dai B, Sun YH, Zhang TT (2020) The activation of IL-17 signaling pathway promotes pyroptosis in pneumonia-induced sepsis. Ann Transl Med 8(11):674. https://doi.org/10.21037/atm-19-1739
Baker RG, Hayden MS, Ghosh S (2011) NF-kappaB, inflammation, and metabolic disease. Cell Metab 13(1):11–22. https://doi.org/10.1016/j.cmet.2010.12.008
Sun SC (2017) The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol 17(9):545–558. https://doi.org/10.1038/nri.2017.52
Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49(2):331–338. https://doi.org/10.1016/j.molcel.2012.11.009
Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH et al (2018) SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun 9(1):3001. https://doi.org/10.1038/s41467-018-05321-2
Jager B, Seeliger B, Terwolbeck O, Warnecke G, Welte T, Muller M et al (2021) The NLRP3-Inflammasome-Caspase-1 pathway is upregulated in idiopathic pulmonary fibrosis and acute exacerbations and is inducible by apoptotic A549 cells. Front Immunol 12:642855. https://doi.org/10.3389/fimmu.2021.642855
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES (2012) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287(43):36617–36622. https://doi.org/10.1074/jbc.M112.407130
Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP et al (2019) Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575(7784):683–687. https://doi.org/10.1038/s41586-019-1770-6
Song JS, Kang CM, Rhee CK, Yoon HK, Kim YK, Moon HS et al (2009) Effects of elastase inhibitor on the epithelial cell apoptosis in bleomycin-induced pulmonary fibrosis. Exp Lung Res 35(10):817–829. https://doi.org/10.3109/01902140902912527
Wang L, Scabilloni JF, Antonini JM, Rojanasakul Y, Castranova V, Mercer RR (2006) Induction of secondary apoptosis, inflammation, and lung fibrosis after intratracheal instillation of apoptotic cells in rats. Am J Physiol Lung Cell Mol Physiol 290(4):L695–L702. https://doi.org/10.1152/ajplung.00245.2005
Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP et al (2014) FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192(4):1835–1846. https://doi.org/10.4049/jimmunol.1302839
Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B et al (2018) Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362(6418):1064–1069. https://doi.org/10.1126/science.aau2818
Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J et al (2008) Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 7(12):2350–2363. https://doi.org/10.1074/mcp.M800132-MCP200
Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T et al (2019) Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun 10(1):2091. https://doi.org/10.1038/s41467-019-09753-2
DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW (2008) Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 22(2):239–251. https://doi.org/10.1101/gad.1617608
Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H et al (2014) Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156(4):771–785. https://doi.org/10.1016/j.cell.2013.11.049
Ballesteros-Alvarez J, Andersen JK (2021) mTORC2: the other mTOR in autophagy regulation. Aging Cell 20(8):e13431. https://doi.org/10.1111/acel.13431
Li L, Tan J, Miao Y, Lei P, Zhang Q (2015) ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol 35(5):615–621. https://doi.org/10.1007/s10571-015-0166-x
Li MY, Zhu XL, Zhao BX, Shi L, Wang W, Hu W et al (2019) Adrenomedullin alleviates the pyroptosis of Leydig cells by promoting autophagy via the ROS-AMPK-mTOR axis. Cell Death Dis 10(7):489. https://doi.org/10.1038/s41419-019-1728-5
Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J et al (2016) SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 12(8):1272–91
Gui X, Yang H, Li T, Tan X, Shi P, Li M et al (2019) Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567(7747):262–266. https://doi.org/10.1038/s41586-019-1006-9
Acknowledgements
We apologize to colleagues whose work was not cited here because of space limitations. This work was supported by University of Electronic Science and Technology of China.
Funding
This research was supported by the Key R&D projects of Sichuan Provincial Department of Science and Technology [grant number 2022YF50265].
Author information
Authors and Affiliations
Contributions
GY wrote the manuscript. YY performed the literature search, YL critically revised the manuscript. XL devised the conceptual ideas, revised the manuscript, and supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, G., Yang, Y., Liu, Y. et al. Regulation of alveolar macrophage death in pulmonary fibrosis: a review. Apoptosis 28, 1505–1519 (2023). https://doi.org/10.1007/s10495-023-01888-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-023-01888-4