
Abstract
Cardiomyocyte apoptosis is a major process in pathogenesis of a number of heart diseases, including ischemic heart diseases and cardiac failure. Ensuring survival of cardiac cells by blocking apoptotic events is an important strategy to improve cardiac function. Although the role of ER disruption in inducing apoptosis has been demonstrated, we do not yet fully understand how it influences the mitochondrial apoptotic machinery in cardiac cell models. Recent investigations have provided evidences that the prosurvival protein HCLS1-associated protein X-1 (Hax1) protein is intimately associated with the pathogenesis of heart disease, mitochondrial biology, and protection from apoptotic cell death. To study the role of Hax1 upon ER stress induction, Hax1 was overexpressed in cardiac cells subjected to ER stress, and cell death parameters as well as mitochondrial alterations were examined. Our results demonstrated that the Hax1 is significantly downregulated in cardiac cells upon ER stress induction. Moreover, overexpression of Hax1 protected from apoptotic events triggered by Tunicamycin-induced ER stress. Upon treatment with Tunicamycin, Hax1 protected from mitochondrial fission, downregulation of mitofusins 1 and 2 (MFN1 and MFN2), loss of mitochondrial membrane potential (∆Ψm), production of reactive oxygen species (ROS) and apoptotic cell death. Taken together, our results suggest that Hax1 inhibits ER stress-induced apoptosis at both the pre- and post-mitochondrial levels. These findings may offer an opportunity to develop new agents that inhibit cell death in the diseased heart.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pathogenesis of cardiac diseases involves a critical stress condition that can lead to a high number of apoptotic cardiomyocytes [1–5]. Mitochondria play an essential role in mediating programmed cell death and their alterations result in rapid and extensive cell loss [1, 6–10]. The endoplasmic reticulum (ER) is an essential organelle that regulates the quality control of secretory, transmembrane, and ER-resident proteins which mediate proper protein folding and protein transport [11, 12]. ER stress contributes to the pathogenesis of a number cardiovascular disorders and can lead to increased disease progression [13]. ER protein folding can be disturbed by diverse arrays of biological conditions such as nutrient deprivation, ROS, calcium ion disequilibrium, and improper posttranslational modifications. These conditions may lead to presence of numerous unfolded or misfolded proteins in the ER, leading to ER stress [14]. This stress on the ER elicits special signaling pathways, known as ER stress responses [11, 15, 16]. The latter events allow the cells to inhibit the unfolded or misfolded protein in the ER, and thus ensure cell survival. However, exaggerated ER stress can cause apoptosis via mitochondria-dependent or -independent pathway [17, 18], a process that has not been fully characterized in cardiac cells. The cross-talk between the ER and mitochondria is an important cellular phenomenon that is accompanied by dynamic interorganellar structural and functional events leading to cell death [19–21]. A number of signaling molecules were suggested to mediate ER stress–induced apoptosis, including the mitochondrial permeability regulators Bcl-2 family proteins, kinases, proteases, and transcription factors [22].
Hax1 was originally identified as a protein that forms a complex with HS-1 (hematopoietic lineage cell-specific protein-1) in lymphocytes [23]. Recently, it has been shown that Hax1 protects from cardiac ischemic injury via enhancing cell survival against ischemia/reperfusion [24]. Interestingly, Hax1 has been shown to inhibit cardiomyocyte cell death induced by hydrogen peroxide [25]. Moreover, global deletion of Hax1 leads to a short lifespan due to progressive death of neuronal cells [26]. Furthermore, Hax1 gene mutations in humans lead to severe neutropenia and symptoms of immunodeficiency, neurological disease and neurodevelopment abnormalities [27–29]. In addition, Hax1 has multifunctional roles in other key cellular processes like calcium homeostasis and cell migration [27, 30]. Despite the reported prosurvival role of Hax1 various models [27], its function in cardiac cell apoptosis needs further elucidation. Moreover, Hax1 was reported to be localized to cardiac mitochondria and sarcoplasmic reticulum [31]. Unlike mouse Hax1, the human and rat genes have been shown to be heavily spliced, giving rise to variants that display differences in the NH2 terminus [32]. Interestingly, Koontz and Kontrogianni-Konstantopoulos have reported that Hax1 is a family of anti- and proapoptotic proteins that may regulate cell fate via homo- or heterodimerization [30].
In this study, the role of Hax1 in regulating ER stress-induced mitochondrial alteration and apoptosis was examined using the stable cell model H9C2. These cells have several features of cardiac muscle cells with the advantage of being immortalized. Hax1 overexpression protected cardiac cells from apoptosis induced by ER stress (Tunicamycin) and further that Hax1 preserved mitochondrial morphology, restored the mRNA expression of the mitochondrial fusion regulators MFN1 and MFN2, blocked mitochondrial membrane depolarization, and reduced ROS level following Tunicamycin-induced ER stress. Collectively, these data demonstrate that Hax1 protects from mitochondrial alterations during ER stress-induced apoptosis.
Materials and methods
Cell culture, overexpression construct and QRT PCR
H9C2 rat cardiomyoblasts (ATCC) were grown in high glucose DMEM containing 10 % FBS in 5 % CO2, 95 % air at 37 °C. The cells were washed with PBS and kept in serum free DMEM medium before use. ER stress was induced by Tunicamycin (Sigma) 3 µM or with DMSO (vehicle control) and incubated for 24 and 48 h. pcDNA3 vector was used to express the mouse Hax1 protein in H9C2 cells. Cells were transfected with empty vector or a constructs expressing Hax1-HA-CT (kind gift from Dr. Luca Pellegrini, Faculty of Medicine, Université Laval, Quebec, QC, Canada) as described earlier [33], and analyzed after 24 and 48 h. The broad caspase inhibitor Z-VAD-FMK (Calbiochem) was used at the final concentration of 20 µM. To analyze gene expression, total RNA isolation was performed by using with TRIzol (Invitrogen). Total RNA purification, reverse transcription into cDNA (with random hexamer primers), and Real-time PCR analysis were performed as previously described [34]. Data were normalized to the endogenous reference 18S expression and reported as fold change relative to control. Quantitative RT-PCR was performed by using specific primers against the following target genes Chop (forward: 5′-GAAAGCAGAAACCGGTCCAAT-3′, reverse: 5′-GGATGAGATATAGGTGCCCCC-3′), Grp78 (forward: 5′-CACGTCCAACCCGGAGAA-3′, reverse: 5′-ATTCCAAGTGCGTCCGATG-3′), MFN1 (forward: 5′-CCATCACTGCGATCTTCGGCCA-3′, reverse: 5′-CAGCGAGCTTGTTTCTGTAGCCCT-3′), MFN2 (forward: 5′-AGCGTCCTCTCCCTCTGACA-3′, reverse: 5′-TTCCACACCACTCCTCCGAC-3′), Hax1 exogenous (forward: 5′-CTCTCAGAGATCTTCAGCTTTGG-3′, reverse: 5′-TGGGACGTCGTATGGGTATAG-3′), Hax1 endogenous (forward: 5′-TAAACTGGTGGTCTCGTGTTG-3′, reverse: 5′-AGATCAAAGACGCTCATTCCC-3′),18S (forward: 5′-AGTCCCTGCCCTTTGTACACA-3′, reverse: 5′-CGATCCGAGGGCCTCACTA-3′).
Analysis of cell viability, apoptosis and caspase activity
Apoptosis was detected by TUNEL assay, using the In Situ Cell Death Detection Kit, Fluorescein (Roche) according to manufacturer’s instructions. Cell viability was assessed by the MTS/PMS assay (Promega) as described before [35]. DEVDase assay was performed to detect caspase activity, as described by the manufacturer (Calbiochem). Immunostaining of active caspase-3 cells was made by fixing the cells in 4 % paraformaldehyde for 10 min at room temperature, washing them three times in PBS, blocking with 1 % normal goat serum in PBS for 1 h at room temperature, and labeling with anti-cleaved caspase-3 primary antibody (Cell Signaling) (1:100 in blocking buffer) overnight at 4 °C. Cells were incubated with anti-mouse secondary antibody Alexa Fluor 568 (Molecular Probes) for 1 h at room temperature and counterstained using DAPI. Cell apoptosis was also assessed by Annexin V-FITC/PI staining. The cells were stained with Annexin V-FITC/PI, in the dark for 30 min at room temperature, and analyzed by flow cytometry (FACSCalibur; BD Biosciences).
Western blot analysis
Total cells were extracted using NP-40 lysis buffer (KeyGEN). Proteins were resolved on sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Immobilon; Millipore). Membranes were probed with anti-Hax1 (BD Biosciences), anti-cleaved caspase-3 (Cell Signaling Technology), anti-GAPDH (Santa Cruz). Protein bands were incubated with HRP-conjugated antibodies and visualized using electrochemiluminescence by a chemiluminescence instrument.
Assessment of mitochondrial alterations and ROS production
Mitochondria were observed after incubating the cells with 100 nM of MitoTracker Green FM (Invitrogen) for 30 min and then washing three times with media. Cells which exhibited mitochondrial fission were readily identified as fragmented small punctate mitochondria as described earlier [36–39]. These cells were quantified in each experimental group by using fluorescence microscopy. Loss of ∆Ψm was assessed using tetramethylrhodamine ethyl ester (TMRE, Molecular Probes) (20 nM) for 30 min in DMEM medium. At the end of the incubation, cells were washed in DMEM medium containing TMRE (20 nM) and allowed to equilibrate. The mitochondrial membrane depolarization was recognized by the decline of mitochondria-localized intensity of fluorescence. Mitochondrial membrane potential was also measured using the fluorescent probe JC-1. At low membrane potential, JC-1 is a green-fluorescent monomer, whereas at higher potential, JC-1 emits red fluorescence. The ratio of JC-1 green to red fluorescence reflects the ratio of apoptosis. Cells were incubated with JC-1 dye diluted in culture medium at 37 °C for 30 min. Cells were then immediately analyzed by flow cytometry (FACSCalibur; BD Biosciences, USA). To assess ROS generation, 2 μm of dihydroethidium (DHE) (Invitrogen) was used. The cells were incubated for 15 min as described [40]. The quantification of fluorescence intensity was performed with ImageJ software (NIH).
Statistical analysis
Statistical significance was determined by Student’s t test. The number of samples varied from 3 to 6 independent experiments. Data are presented as mean ± standard deviation (SD) or standard error of the mean (SEM). P value below 0.05 was considered statistically significant.
Results
Tunicamycin downregulates Hax1 expression and induces efficient ER stress
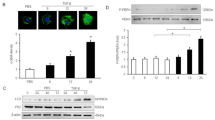
To investigate the potential role Hax1 in ER stress-induced apoptotic cell death, we first analyzed if ER stress modulates Hax1 level. We treated H9C2 cells with the potent ER stress activator Tunicamycin, an inhibitor of N-linked glycosylation, and checked the mRNA and protein levels of Hax1 at 24 and 48 h after treatment. Tunicamycin was chosen because it has been used efficiently to induce ER stress in various cellular and animal models including cardiac tissues [41–43]. Moreover, Tunicamycin treatment allows careful assessment of ER-mitochondrial crosstalk during the apoptotic process [44]. Interestingly, Tunicamycin treatment induced significant decrease in the expression level of endogenous Hax1 mRNA at both 24 and 48 h after treatment (Fig. 1a). Moreover, induction of ER stress by Tunicamycin for 24 and 48 h resulted in remarkable reduction of Hax1 protein compared to the control (Fig. 1b). To verify efficient ER stress induction by Tunicamycin, we analyzed the expression of the ER stress markers GRP78 and Chop. Our results revealed significant induction of the mRNA level of both GRP78 and Chop at 24 and 48 h after treatment (Fig. 1c, d).

Tunicamycin downregulates Hax1 expression and induces efficient ER stress. a Tunicamycin induced significant reduction of the endogenous Hax1 mRNA level at 24 and 48 h after treatment. (T) indicates Tunicamycin treatment. b Representative western blot showing decreased Hax1 protein level in lysates from cells treated with Tunicamycin for 24 and 48 h. GAPDH is shown as a loading control. c, d Tunicamycin induced remarkable ER stress indicated by the increased mRNA level of the ER stress markers GRP78 and Chop at 24 and 48 h after treatment. UT in a, c, d and (−) in b are untreated control cells. (+) in b indicates Tunicamycin treatment. mRNA data are shown as mean ± SEM, normalized to controls. The asterisk indicates a significant difference (P < 0.05)
Effect of Hax1 overexpression on Tunicamycin-induced upregulation of ER stress markers
Then we extended our studies to address the question if Hax1 overexpression affects the expression of ER stress markers induced by Tunicamycin. To verify significant overexpression of Hax1 in H9C2 cells, we assessed its mRNA and protein levels. Increased exogenous Hax1 mRNA and protein expression levels were significantly seen at both 24 and 48 h after Hax1 plasmid transfection (Fig. 2a, b). To investigate if overexpressed Hax1 could affect Tunicamycin-induced ER stress, we checked mRNA level of the ER stress markers GRP78 and chop in cells treated with Tunicamycin and/or overexpressing Hax1 at 24 and 48 h after transfection. Hax1 overexpression did not change Tunicamycin-induced elevation of chop or GRP78 mRNA expression (Fig. 2c, d).

Hax1 overexpression and its effect on ER stress markers. a Exogenous Hax1 mRNA level in cells overexpressing Hax1 and transfected with empty vector (EV) or untreated at 24 and 48 h after transfection. Increased exogenous Hax1 mRNA expression is significantly seen at both 24 and 48 h after transfection. b Hax1 protein level in cells overexpressing Hax1 for 24 and 48 h. GAPDH is shown as a loading control. c, d mRNA analysis of the ER stress markers GRP78 and chop in cells treated with Tunicamycin and/or overexpressing Hax1 at 24 and 48 h after transfection. Hax1 overexpression did not change Tunicamycin-induced elevation of chop or GRP78 mRNA expression. Data are shown as mean ± SEM, normalized to controls. The asterisk indicates a significant difference (P < 0.05). NS indicates that there is no significant change. UT cells and EV transfected cells were used as controls
Tunicamycin-induced apoptosis is inhibited by Hax1 overexpression
In the present model, we examined the role of Hax1 in the downstream apoptotic events triggered by the potent ER stress inducer Tunicamycin in H9C2 cells. We first tested the ability of Hax1 overexpression to improve cell viability and to inhibit cell apoptosis when ER stress is induced. We performed time course experiments and examined apoptosis parameters in H9C2 cells using various experimental procedures including TUNEL labeling of apoptotic DNA fragmentation, cell viability assay, caspase activation assay, active caspase-3 staining, western blot detection of cleaved caspase-3 and simultaneous flow cytometric quantification of both viable and apoptotic cells with an Annexin V-FITC/PI staining. Tunicamycin treatment caused significant decrease in cell viability, increase in apoptotic DNA fragmentation, elevated general caspase activation, increased caspase-3 positive cells and increased protein level of cleaved caspase-3 (Fig. 3a–g). Overexpression of Hax1 blocked Tunicamycin-induced apoptotic events and increased cell viability (Fig. 3a–g). All these findings clearly demonstrate that Tunicamycin induces apoptotic cell death in a Hax1-dependent manner.

Hax1 inhibits ER stress-induced cell death and caspase activation. a Tunicamycin induced significant apoptotic cell death detected by TUNEL. The percent of Tunicamycin-induced apoptosis was significantly decreased upon Hax1 overexpression. b Tunicamycin reduced cell viability analyzed by MTS assay. The number of viable cells in the Tunicamycin treated group was significantly decreased upon Hax1 overexpression. c Tunicamycin induced significant apoptotic cell death detected by active caspase-3 positivity. The number of Tunicamycin-induced increase in caspase-3 positive cells was significantly decreased upon Hax1 overexpression. d Tunicamycin induced caspase activation detected by DEVDase assay. The high caspase activity induced by Tunicamycin was significantly decreased upon Hax1 overexpression. e Western blot analysis showing Tunicamycin-induced apoptotic cell death detected by cleaved caspase-3. The increased caspase-3 cleavage induced by Tunicamycin was partly reversed by Hax1 overexpression. GAPDH is shown as a loading control. f, g Increased apoptosis rate in the Tunicamycin treated group, detected by Annexin V-FITC/PI staining, while the trend was partly reversed by Hax1 overexpression. EV transfected cells were used as controls in all experiments of this figure. The effect of Hax1 overexpression on Tunicamycin-induced apoptosis was analyzed at 24 and 48 h after transfection. Data are shown as the mean ± standard deviation (SD), normalized to controls. The asterisk indicates a significant difference (P < 0.05)
Hax1 inhibits Tunicamycin-induced mitochondrial fission and mitochondrial depolarization
Changes in mitochondrial dynamics are tightly associated with the ER–mitochondria crosstalk and are involved in a number of molecular pathways crucial for cell survival [45–48]. We hypothesized that Hax1 would protect from ER stress-induced apoptosis upstream of the apoptotic mitochondrial fission. To investigate if Hax1 inhibits ER stress-induced mitochondrial fission in H9C2 cells, we treated the cells with Tunicamycin and assessed mitochondrial morphology, using MitoTracker Green. Under these conditions, the cells displayed extensive Tunicamycin-induced mitochondrial fission which was remarkably prevented by Hax1 overexpression (Fig. 4a–d). We then wanted to assess changes in ∆Ψm in cells treated with Tunicamycin or/and subjected to Hax1 overexpression. We used TMRE dye which labels active mitochondria (Fig. 4e) and the fluorescent dye JC-1 which indicates changes in the electrochemical membrane potential of the mitochondria (Fig. 4f–i). Tunicamycin treatment caused significant loss of ∆Ψm in H9C2 cells. Upon Hax1 overexpression, Tunicamycin-induced mitochondrial loss of ∆Ψm was significantly inhibited. Taken together, these results demonstrate that Hax1 is required for protecting mitochondrial morphology and integrity during ER stress-induced apoptosis.

Hax1 inhibits Tunicamycin-induced mitochondrial fission and mitochondrial depolarization. a–c Mitochondrial morphology of single H9C2 cells, assessed by Mitotracker green, showing normal mitochondrial morphology in a control cell transfected with EV, remarkable mitochondrial fission in a cell treated with Tunicamycin and transfected with EV, and a cell exhibiting inhibition of Tunicamycin-induced mitochondrial fission by Hax1 overexpression, respectively. Scale bar 20 µm. d There was increased percent of cells displaying mitochondrial fission in the Tunicamycin treated group. The number of cells with mitochondrial fission in the Tunicamycin treated group was significantly decreased upon Hax1 overexpression. e Tunicamycin induced significant depolarization of the mitochondria detected by reduction in the intensity of TMRE dye. Hax1 blocked mitochondrial depolarization induced by Tunicamycin. f–h Cells were stained with the ∆Ψm-sensitive dye JC-1 and Hoechst 33342 (fluorescence DNA dye). JC-1 analysis confirmed increased collapse of mitochondrial membrane potential in the Tunicamycin treated group, while the trend was partly reversed by Hax1 overexpression. Scale bar200 µm. i quantitative analysis of JC-1 staining experiments. Data are shown as the mean ± SD, normalized to controls. The asterisks indicate the significant differences (P < 0.05)
Hax1 restores MFN1 and MFN2 levels upon ER stress induction
MFN1 and MFN2 are known to protect from cardiomyocyte apoptosis via preventing mitochondrial fission [49]. The increased mitochondrial fission detected in H9C2 cells subjected to ER stress prompted us to analyze the effect of ER stress on mRNA expression of the key mitochondrial morphology regulators MFN1 and MFN2, as well as to examine the role of Hax1 overexpression in modulating these factors. While Tunicamycin-induced ER stress caused late reduction in MFN1 mRNA (at 48 h after Tunicamycin treatment), it led to both early and late downregulation of MFN2 mRNA i.e. at 24 and 48 h after treatment (Fig. 5a, b). Interestingly, overexpression of Hax1 prevented reduction of MFN1 and MFN2 mRNA in Tunicamycin-treated cells (Fig. 5a, b). The same treatment conditions used for MFN1 and MFN2 mRNA experiments were applied for cells analyzed microscopically and revealed significant block of ER stress-induced mitochondrial fission in cells overexpressing Hax1 (Fig. 4a–d).

Hax1 inhibits downregulation of MFN1 and MFN2 mRNA caused by Tunicamycin treatment. a Tunicamycin treatment did not affect MFN1 mRNA in cells transfected with EV for 24 h. Hax1 overexpression increased MFN1 mRNA expression in cells treated with Tunicamycin at 48 h after transfection. b Tunicamycin treatment inhibited MFN2 mRNA expression level in cells transfected with EV for 24 and 48 h. EV transfected cells were used as controls in all experiments of this figure. Data are shown as the mean ± SEM, normalized to controls. The asterisk indicates a significant difference (P < 0.05). NS indicates that there is no significant change
Tunicamycin-induced mitochondrial alteration is partially caspase dependent
Hax1 has been previously reported to inhibit apoptosis by physically interacting with caspase-9 and blocking its processing and activation [25]; however, it is not yet clear if caspase inhibition by Hax1 contributes to mitochondrial protection during apoptosis. Caspases were previously suggested to precede the occurrence of mitochondrial initiation of apoptosis but this role remained controversial [50, 51]. In this study, we have shown that Hax1 overexpression suppresses caspase activity in Tunicamycin-treated H9C2 cells. In order to determine if the effect of ER stress on mitochondrial events is caspase-dependent, we co-treated the cells with Tunicamycin and the broad caspase inhibitor Z-VAD-FMK. Tunicamycin treatment induced significant mitochondrial fission which was partially reversed by caspase inhibition (Fig. 6a). Consistently, Tunicamycin caused mitochondrial depolarization and this was decreased, in part, by caspase inhibition (Fig. 6b). Thus, both ER stress-induced mitochondrial fission and loss of ∆Ψm were partially caspase-dependent events. These findings are supported by previous findings that revealed a partial contribution of caspases to mitochondrial alterations upon apoptosis induced by different factors [7]. Determination of the role of Hax1 in the caspase-dependent mitochondrial alterations of cardiomyocytes subjected to ER stress requires further studies.

Tunicamycin-induced mitochondrial alteration is partially caspase dependent. a Tunicamycin induced significant mitochondrial fission which was partially inhibited by the caspase inhibitor Z-VAD-FMK. b Mitochondrial depolarization induced by Tunicamycin was also partially inhibited by the caspase blocker Z-VAD-FMK. Treatment with Z-VAD-FMK without Tunicamycin addition did not affect mitochondrial polarization in EV transfected control cells. EV transfected cells were used as controls in all experiments of this figure. Data are shown as the mean ± SD, normalized to controls. The asterisk indicates a significant difference (P < 0.05)

Hax1 blocks Tunicamycin-induced ROS. Tunicamycin induced significant ROS production in cells transfected with EV (detected by DHE). Hax1 overexpression inhibited Tunicamycin-induced ROS production. EV transfected cells were used as controls. Data are shown as the mean ± SD, normalized to controls. The asterisk indicates a significant difference (P < 0.05)
Hax1 blocks Tunicamycin-induced ROS
To investigate if Hax1 protects from ER stress-induced ROS, we assessed ROS production in H9C2 overexpressing Hax1 and/or treated with the ER-stressor Tunicamycin. Tunicamycin resulted in increased accumulation of ROS detected by DHE dye labeling (Fig. 7). Overexpression of Hax1 protected from accumulation of intracellular ROS induced by Tunicamycin (Fig. 7). These data demonstrated that Tunicamycin-induced ROS production is Hax1-dependent.
Discussion
In the present study, we have demonstrated a significant role for Hax1 in protecting against apoptotic events caused by Tunicamycin-induced ER stress in H9C2 cardiac cells. These apoptotic features include mitochondrial fission, downregulation of MFN1 and MFN2 mRNA, mitochondrial depolarization, and ROS production. Cardiomyocyte apoptosis is tightly linked with the pathology of heart diseases such as hypertrophy, hypoxia, ischemia/reperfusion injury, developmental abnormalities and heart failure [24, 52–54]. Previous studies have demonstrated that ER stress plays an essential role in cardiomyocyte apoptosis triggered by Ischemia/Reperfusion [24]. In addition, extensive ER stress induced by ischemia or hypoxia/reoxygenation usually leads to the upregulation of specific ER stress response markers [24, 55, 56]. The effect of Hax1 on apoptosis continues to emerge. Hax1 has been recently shown to suppress apoptosis in various models including cardiac cells, both in vitro and in vivo [24, 25, 27, 31, 57, 58]. Moreover, Hax1 may regulate many unrelated molecules, indicating its contribution to diverse signaling mechanisms within the cell [23, 57–60]. Given the crucial pro-death role of ER stress in the diseased heart, it is essential to dissect different preferential apoptotic pathways triggered by this mechanism in different cardiac cell models. Identification of factors that modulate Hax1 expression level and its downstream targets may lead to new therapeutic interventions.
Recently, in a different experimental model than the one presented here, Hax1 has been shown to confer protective effects in primary cardiac cells via by inhibiting IRE-1 signaling [24]. Our results are in agreement with the anti-apoptotic role of Hax1 demonstrated in the latter study; however we detected no effect of Hax1 on the expression of cardiac ER stress markers GRP78 and Chop. These differences might be due to variations in Hax1 subcellular localization, expression levels or the differentiation stage in different cardiac cell types. On the other hand, cell death inducers could also have different effects on pathways leading to apoptosis. Thus, it is possible that Hax1 exerts the cardioprotective effects through diverse mechanisms in different cardiac cell models.
Alterations in mitochondrial morphology were demonstrated to contribute to different aspects of cardiac biology, including heart development, cardiac cell response to ischemia/reperfusion injury, diabetic cardiomyopathy, heart failure, and programmed cell death [61, 62]. Mitochondrial fission is known to be a major mechanism that mediates post-mitochondrial events of apoptosis [5, 7, 63, 64]. Understanding how the mitochondrial dynamic machinery influences cell injury may offer new therapeutic targets for treating heart disease. Although Hax1 was demonstrated to have mitochondrial protective effects, its role in mitochondrial fission of cardiac cells has not been clarified. In this study, we showed that Tunicamycin significantly promoted ER stress-induced mitochondrial fission in H9c2 cardiac cells in a Hax1-dependent manner.
The two mitofusins, MFN1 and MFN2 are known to localize to the outer mitochondrial membrane to ensure adequate cell viability [65]. MFN1 and MFN2 are known to protect from cardiomyocyte apoptosis, at least via preventing mitochondrial fission [49]. Importantly, defects in mitochondrial fusion by combined ablation of MFN1 and MFN2 have been shown to result in lethal heart failure [66, 67]. Furthermore, properly fused mitochondria have been proposed to facilitate the subcellular delivery of energy and to maintain mitochondrial membrane potential [68]. Mitochondrial membrane depolarization is considered to be a prerequisite for the release of cytochrome c from the intermembrane space mitochondria into the cytoplasm [69]. We have shown here an evidence that Hax1 inhibits ER stress-induced downregulation of MFN1 and MFN2 mRNA. Moreover, this study is in agreement with previous findings which demonstrated that Hax1 protects from mitochondrial membrane depolarization [57]. This is important because loss of MFN1 or MFN2 could render the mitochondria more susceptible to lose their membrane potential [65]. Depletion of MFN2 alone from cardiac cells has been shown to decrease the resistance to mitochondrial membrane depolarization downstream of H2O2, and correlated with high levels of apoptosis markers [70]. While the detailed mechanism by which Hax1 affects MFN1 and MFN2 mRNA remains unclear, our data offer a possible explanation for why mitochondrial fission and mitochondrial membrane depolarization are prevented in cells protected from damaging effects of ER stress.
In the present study, we found that both ER stress-induced mitochondrial fission and loss of mitochondrial membrane potential were partially caspase dependent events. Therefore, although caspase activity appears as a later event in cardiomyocyte apoptosis [71, 72], it also contributes to mitochondrial alterations caused by ER stress. It is possible that caspases act upstream of the mitochondria to sensitize cells to ER stress-induced apoptosis. This concept is supported by analyses performed on isolated mitochondria and revealed that caspase-2 acts as an upstream mediator of the mitochondrial events upon apoptosis induction [73, 74]. Earlier in vitro experiments showed that Hax1 inhibits cardiomyocyte apoptosis, by blocking caspase-9 activation [25]. Determination of the role of Hax1 in caspase–mediated mitochondrial alterations caused by ER stress requires further investigations.
In summary, the present study has uncovered roles for Hax1 in protection from ER stress-induced apoptosis. Our findings are consistent with Hax1 acting as mitochondrial damage antagonist in cardiac cells; when Hax1 is downregulated, cells exhibit enhanced mitochondrial apoptotic alterations and decreased survival in response to ER stress (Fig. 8). Further research may characterize other signaling components by which Hax1 exerts its cardioprotective effects in cardiac stress conditions.

Proposed model: ER stress triggers apoptosis through mitochondrial events in a Hax1-dependent manner. Schematic representation illustrating the role of Hax1 in protecting from mitochondrial alterations and apoptosis induced by ER stress in H9C2 cardiac cell line. Induction of ER stress triggers damaging events including improper mitochondrial fission and fragmentation, mitochondrial depolarization, ROS production, caspase activation, and apoptotic cell killing. The mitochondrial alterations induced by ER stress are caspase dependent. Hax1 appears to block ER stress-induced mitochondrial alterations and apoptosis downstream of the ER, possibly by upregulating MFN1 and MFN2 mRNA. A comprehensive analysis on these events may shed new lights in the ER stress/mitochondrial pathways, assist in a better clarification of anti-apoptotic roles of Hax1 and, in the long run, could allow developing novel cardioprotection strategies
References
van Empel VP, Bertrand AT, Hofstra L, Crijns HJ, Doevendans PA, De Windt LJ (2005) Myocyte apoptosis in heart failure. Cardiovasc Res 67:21–29
Wang Y, Huang S, Sah VP, Ross J Jr, Brown JH, Han J et al (1998) Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem 273:2161–2168
Mel’nikova NP, Timoshin SS, Jivotova EY, Pelliniemi LJ, Jokinen E, Abdelwahid E (2006) Angiotensin-II activates apoptosis, proliferation and protein synthesis in the left heart ventricle of newborn albino rats. Int J Cardiol 112:219–222
Abdelwahid E, Smith G (2007) Apoptosis in chronic heart failure. Int J Cardiol 114:375
Abdelwahid E, Petrovic D, Feng Q, Mistiaen WP (2007) Molecular mechanisms and new developments in the regulation of programmed cell death (apoptosis) and its role in pathogenesis of heart diseases. Nova Science Publishers, Inc, New York
Abdelwahid E, Rolland S, Teng X, Conradt B, Hardwick JM, White K (2011) Mitochondrial involvement in cell death of non-mammalian eukaryotes. Biochim Biophys Acta 1813:597–607
Abdelwahid E, Yokokura T, Krieser RJ, Balasundaram S, Fowle WH, White K (2007) Mitochondrial disruption in Drosophila apoptosis. Dev Cell 12:793–806
Yu T, Sheu SS, Robotham JL, Yoon Y (2008) Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res 79:341–351
Youle RJ, Karbowski M (2005) Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol 6:657–663
Su B, Wang X, Bonda D, Perry G, Smith M, Zhu X (2010) Abnormal mitochondrial dynamics–a novel therapeutic target for Alzheimer’s disease? Mol Neurobiol 41:87–96
Kaufman RJ (2002) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110:1389–1398
Ruddock LW, Molinari M (2006) N-glycan processing in ER quality control. J Cell Sci 119:4373–4380
Ono Y, Shimazawa M, Ishisaka M, Oyagi A, Tsuruma K, Hara H (2012) Imipramine protects mouse hippocampus against Tunicamycin-induced cell death. Eur J Pharmacol 696:83–88
Gorlach A, Klappa P, Kietzmann T (2006) The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal 8:1391–1418
Sidrauski C, Chapman R, Walter P (1998) The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol 8:245–249
Urano F, Bertolotti A, Ron D (2000) IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci 113(Pt 21):3697–3702
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA et al (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403:98–103
Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND et al (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129:1337–1349
Kornmann B, Walter P (2010) ERMES-mediated ER-mitochondria contacts: molecular hubs for the regulation of mitochondrial biology. J Cell Sci 123:1389–1393
Pizzo P, Pozzan T (2007) Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics. Trends Cell Biol 17:511–517
Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG et al (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39:121–132
Pirot P, Ortis F, Cnop M, Ma Y, Hendershot LM, Eizirik DL et al (2007) Transcriptional regulation of the endoplasmic reticulum stress gene chop in pancreatic insulin-producing cells. Diabetes 56:1069–1077
Suzuki Y, Demoliere C, Kitamura D, Takeshita H, Deuschle U, Watanabe T (1997) HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J Immunol 158:2736–2744
Lam CK, Zhao W, Cai W, Vafiadaki E, Florea SM, Ren X et al (2013) Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90. Circ Res 112:79–89
Han Y, Chen YS, Liu Z, Bodyak N, Rigor D, Bisping E et al (2006) Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ Res 99:415–423
Chao JR, Parganas E, Boyd K, Hong CY, Opferman JT, Ihle JN (2008) Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature 452:98–102
Fadeel B, Grzybowska E (2009) HAX-1: a multifunctional protein with emerging roles in human disease. Biochim Biophys Acta 1790:1139–1148
Boztug K, Ding XQ, Hartmann H, Ziesenitz L, Schaffer AA, Diestelhorst J et al (2010) HAX1 mutations causing severe congenital neuropenia and neurological disease lead to cerebral microstructural abnormalities documented by quantitative MRI. Am J Med Genet A 152 A:3157–3163
Lanciotti M, Indaco S, Bonanomi S, Coliva T, Mastrodicasa E, Caridi G et al (2010) Novel HAX1 gene mutations associated to neurodevelopment abnormalities in two Italian patients with severe congenital neutropenia. Haematologica 95:168–169
Koontz J, Kontrogianni-Konstantopoulos A (2014) Competition through dimerization between antiapoptotic and proapoptotic HS-1-associated protein X-1 (Hax-1). J Biol Chem 289:3468–3477
Zhao W, Waggoner JR, Zhang ZG, Lam CK, Han P, Qian J et al (2009) The anti-apoptotic protein HAX-1 is a regulator of cardiac function. Proc Natl Acad Sci USA 106:20776–20781
Grzybowska EA, Sarnowska E, Konopinski R, Wilczynska A, Sarnowski TJ, Siedlecki JA (2006) Identification and expression analysis of alternative splice variants of the rat Hax-1 gene. Gene 371:84–92
Jeyaraju DV, Cisbani G, De Brito OM, Koonin EV, Pellegrini L (2009) Hax1 lacks BH modules and is peripherally associated to heavy membranes: implications for Omi/HtrA2 and PARL activity in the regulation of mitochondrial stress and apoptosis. Cell Death Differ 16:1622–1629
Gurgen D, Hegner B, Kusch A, Catar R, Chaykovska L, Hoff U et al (2011) Estrogen receptor-beta signals left ventricular hypertrophy sex differences in normotensive deoxycorticosterone acetate-salt mice. Hypertension 57:648–654
Santra M, Skorski T, Calabretta B, Lattime EC, Iozzo RV (1995) De novo decorin gene expression suppresses the malignant phenotype in human colon cancer cells. Proc Natl Acad Sci USA 92:7016–7020
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F et al (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1:515–525
Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E (2009) Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ 16:899–909
Chou CH, Lin CC, Yang MC, Wei CC, Liao HD, Lin RC et al (2012) GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS One 7:e49112
Eura Y, Ishihara N, Yokota S, Mihara K (2003) Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem 134:333–344
Yaglom JA, Ekhterae D, Gabai VL, Sherman MY (2003) Regulation of necrosis of H9c2 myogenic cells upon transient energy deprivation. Rapid deenergization of mitochondria precedes necrosis and is controlled by reactive oxygen species, stress kinase JNK, HSP72 and ARC. J Biol Chem 278:50483–50496
Shen M, Wang L, Wang B, Wang T, Yang G, Shen L et al (2014) Activation of volume-sensitive outwardly rectifying chloride channel by ROS contributes to ER stress and cardiac contractile dysfunction: involvement of CHOP through Wnt. Cell Death Dis 5:e1528
Li G, Scull C, Ozcan L, Tabas I (2010) NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol 191:1113–1125
Tanjore H, Lawson WE, Blackwell TS (2013) Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta 1832:940–947
Han C, Nam MK, Park HJ, Seong YM, Kang S, Rhim H (2008) Tunicamycin-induced ER stress upregulates the expression of mitochondrial HtrA2 and promotes apoptosis through the cytosolic release of HtrA2. J Microbiol Biotechnol 18:1197–1202
Ngoh GA, Papanicolaou KN, Walsh K (2012) Loss of mitofusin 2 promotes endoplasmic reticulum stress. J Biol Chem 287:20321–20332
Korobova F, Ramabhadran V, Higgs HN (2013) An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339:464–467
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334:358–362
Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S (2011) Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 30:556–568
Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–2022
Grossmann J, Walther K, Artinger M, Kiessling S, Scholmerich J (2001) Apoptotic signaling during initiation of detachment-induced apoptosis (“anoikis”) of primary human intestinal epithelial cells. Cell Growth Differ 12:147–155
Grossmann J (2002) Molecular mechanisms of “detachment-induced apoptosis–Anoikis”. Apoptosis 7:247–260
Sun J, Sun G, Meng X, Wang H, Wang M, Qin M et al (2013) Ginsenoside RK3 Prevents Hypoxia-Reoxygenation Induced Apoptosis in H9c2 Cardiomyocytes via AKT and MAPK Pathway. Evid Based Complement Alternat Med 2013:690190
Abdelwahid E, Kalvelyte A, Stulpinas A, de Carvalho KA, Guarita-Souza LC, Foldes G (2016) Stem cell death and survival in heart regeneration and repair. Apoptosis 21:252–268
Abdelwahid E, Rice D, Pelliniemi LJ, Jokinen E (2001) Overlapping and differential localization of Bmp-2, Bmp-4, Msx-2 and apoptosis in the endocardial cushion and adjacent tissues of the developing mouse heart. Cell Tissue Res 305:67–78
Wang C, Li YZ, Wang XR, Lu ZR, Shi DZ, Liu XH (2012) Panax quinquefolium saponins reduce myocardial hypoxia-reoxygenation injury by inhibiting excessive endoplasmic reticulum stress. Shock 37:228–233
Wu XD, Zhang ZY, Sun S, Li YZ, Wang XR, Zhu XQ et al (2013) Hypoxic preconditioning protects microvascular endothelial cells against hypoxia/reoxygenation injury by attenuating endoplasmic reticulum stress. Apoptosis 18:85–98
Lam CK, Zhao W, Liu GS, Cai WF, Gardner G, Adly G et al (2015) HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc Natl Acad Sci USA 112:E6466–E6475
Lam AK, Galione A, Lai FA, Zissimopoulos S (2013) Hax-1 identified as a two-pore channel (TPC)-binding protein. FEBS Lett 587:3782–3786
Radhika V, Onesime D, Ha JH, Dhanasekaran N (2004) Galpha13 stimulates cell migration through cortactin-interacting protein Hax-1. J Biol Chem 279:49406–49413
Kawaguchi Y, Nakajima K, Igarashi M, Morita T, Tanaka M, Suzuki M et al (2000) Interaction of Epstein-Barr virus nuclear antigen leader protein (EBNA-LP) with HS1-associated protein X-1: implication of cytoplasmic function of EBNA-LP. J Virol 74:10104–10111
Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA et al (2016) Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol 594:509–525
Kane LA, Youle RJ (2010) Mitochondrial fission and fusion and their roles in the heart. J Mol Med (Berl) 88:971–979
Hom J, Sheu SS (2009) Morphological dynamics of mitochondria–a special emphasis on cardiac muscle cells. J Mol Cell Cardiol 46:811–820
Ong SB, Hausenloy DJ (2010) Mitochondrial morphology and cardiovascular disease. Cardiovasc Res 88:16–29
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200
Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF Jr, Stanley WC et al (2012) Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol 302:H167–H179
Chen Y, Liu Y, Dorn GW 2nd (2011) Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 109:1327–1331
Skulachev VP (2001) Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci 26:23–29
Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD et al (1999) Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc Natl Acad Sci USA 96:8144–8149
Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM et al (2011) Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol 31:1309–1328
Shi Y (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 9:459–470
Olsson M, Zhivotovsky B (2011) Caspases and cancer. Cell Death Differ 18:1441–1449
Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES (2002) Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem 277:13430–13437
Enoksson M, Robertson JD, Gogvadze V, Bu P, Kropotov A, Zhivotovsky B et al (2004) Caspase-2 permeabilizes the outer mitochondrial membrane and disrupts the binding of cytochrome c to anionic phospholipids. J Biol Chem 279:49575–49578
Acknowledgments
We thank Dr. Luca Pellegrini (Faculty of Medicine, Université Laval, Quebec, QC, Canada) for Hax1 overexpression construct, Dr. Thomas Simmen for discussion and Dr. Aikaterini Kontrogianni-Konstantopoulos for kindly sharing information on Hax1 expression. We are grateful to Mr. Denislam Zaripov for art drawing. E.A. was supported by the National Heart, Lung, and Blood Institute (NIH/NHLBI), Grant SP0012613. X.L. was supported by the National Natural Science Foundation of China (81272278). K.A.T.C was supported by Coordination for the Improvement of Higher Education Personnel (CAPES) of Brazil, Grant PE 1711.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Abdelwahid, E., Li, H., Wu, J. et al. Endoplasmic reticulum (ER) stress triggers Hax1-dependent mitochondrial apoptotic events in cardiac cells. Apoptosis 21, 1227–1239 (2016). https://doi.org/10.1007/s10495-016-1286-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-016-1286-6