
Abstract
Mitochondria are dynamic organelles that usually exist in extensive and interconnected networks that undergo constant remodeling through fission and fusion. These processes are governed by distinct sets of proteins whose mechanism and regulation we are only beginning to fully understand. Early studies on mitochondrial dynamics were performed in yeast and simple mammalian cell culture models that allowed easy visualization of these intricate networks. Equipped with this core understanding, the field is now expanding into more complex systems. Cardiac cells are a particularly interesting example because they have unique energetic and spatial demands that make the study of their mitochondria both challenging and potentially very fruitful. This review will provide an overview of mitochondrial fission and fusion as well as recent developments in the understanding of these processes in the heart.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondria have been observed in cells since early light microscopic studies could visualize subcellular structures. From their earliest identification, they were known to occur in different shapes and sizes, ranging from long thread-like structures to smaller grain-like structures [1–3]. Shortly after these initial observations, recordings in living cells showed that mitochondria are in constant motion and are capable of changes in both shape and size. Throughout the years, efforts have been made to characterize this dynamic network in different cell types and diseases, but it was not until recently that the machinery responsible for these processes has come to light, ushering in a resurgence of interest in this incredible system.
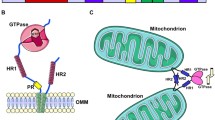
The mitochondrial network is governed by the processes of fission and fusion, which are mediated by two distinct groups of proteins (which will be discussed in depth below; Fig. 1). These processes are known to be essential: knocking out the protein components of the fission and fusion machinery causes a lethal phenotype [4, 5]. But the underlying question of why these organelles are so dynamic continues to be of great interest. It was discovered early on that this dynamic network serves as a means of distribution of mitochondrial DNA (mtDNA) to the progeny of both yeast and mammalian cells [6, 7]. This function of the fission/fusion processes is highlighted by experiments demonstrating that a loss or dysfunction of these proteins can result in a decrease or total loss of mtDNA from cells [8–11]. The dynamic network may also act as a distribution system for other molecules and act in the process of mitochondrial quality control [12, 13]. As the centers of oxygen consumption in the cell via oxidative phosphorylation, mitochondria sustain a tremendous amount of oxidative damage to the molecules of mtDNA, lipids, and proteins over the course of their lifetimes. It is thought that by circulating mitochondria through a dynamic network, the damaged mitochondria will be able to repair and replenish their DNA, lipids, and proteins from the healthy pool [12, 13]. This hypothesis is substantiated by the fact that blocking fission in cells results in the accumulation of oxidized proteins [14] and that in the absence of OPA1 in flies there is a large increase in ROS production [15, 16]. In wild-type cells, overly damaged mitochondria are excluded from the network by the requirement of membrane potential and pH gradient for normal mitochondrial fusion [17, 18] and can be degraded by autophagy [14]. This method of maintaining a healthy mitochondrial population through fission and fusion of the network allows for tight regulation and modulation of the system and helps explain why the proteins that govern these processes are so important to cellular survival.

Schematic of the fusion and fission processes and major proteins involved in mammalian cells
The protein machinery
Since the initial work in the field of mitochondrial dynamics was performed in yeast, flies, worms, and mammals, there are several differing names for the fission and fusion proteins (Table 1). The primary conserved fusion proteins are called mitofusins (Mfn1 and Mfn2) and Opa1 in mammals and Fzo1 and Mgm1 in yeast. The major fission proteins are Drp1, Mff, and hFis1 in mammals and Dnm1, Mdv1, and Fis1 in yeast. Mfn1/2, Opa1, and Drp1 are all large GTPases that are known to have the properties of self-assembly and assembly-stimulated GTP hydrolysis. Their homo- and heteromeric assembly causes formation of the structures that are responsible for the mechanics of membrane fusion and fission.
Mitochondrial fusion
Lipid bilayer fusion events are complicated molecular processes that are difficult to characterize, and this is confounded by the double membrane structure of the mitochondria. Fzo was the first described essential fusion protein when it was characterized in Drosophila in 1997 as a regulator of mitochondrial fusion in sperm development [19]. This was followed by the discovery of yeast (Fzo1) and mammalian (Mfn1/2) orthologs in 1998 and 2001, respectively [20, 21]. With the identification of these proteins across a large range of species, it became apparent that a common fusion mechanism had been conserved throughout eukaryotic evolution.
Outer membrane fusion
Mitofusins reside in the outer mitochondrial membrane (OMM) and are the proteins that are primarily responsible for OMM fusion. They are large GTPases with two membrane-spanning domains that orient both the N- and C-termini in the cytosol [22]. Both the cytosolic C-terminal coiled-coil and GTPase domains are required for fusion [4, 17, 23]. Mfns interact to tether mitochondria for fusion and act in both cis (Mfn molecules on the same mitochondria) and trans (Mfn molecules on different mitochondria) manners to form oligomeric structures [17, 24, 25]. The need for trans-interactions was demonstrated by elegant in vitro fusion assays that demonstrated mitochondria from wild-type and Fzo1 mutant yeast were not capable of fusing together [17]. In mammals, it has been recorded that the mitochondrial tethering capabilities of Mfn1 and Mfn2 differ [4], and the ratio of these two proteins in different cell types may help explain the wide variety of mitochondrial network phenotypes cells [26].
Mutations in Mfn2 protein are the primary cause of the disease Charcot-Marie-Tooth type 2A (CMT2A), an inherited neuropathy [27, 28]. Disease-causing mutations in Mfn2 cause an impairment of the fusion capabilities of Mfn2 and result in cells with fragmented and often aggregated mitochondria [29]. These mutations cause the formation of dysfunctional Mfn2 homo-oligomers, but can form functional hetero-oligomers with Mfn1 [29]. However, these Mfn2 mutations cause disease despite the background of wild-type Mfn1 in CMT2A patients. It is thought that affected cells in CMT2A have a particular dependence on Mfn2, likely due to the low expression of Mfn1. This idea is supported through work in the cerebellum showing that Purkinje cell development is dependent on Mfn2 levels in the presence of low endogenous levels of Mfn1 [30].
The expression of Mfn2 is regulated by both transcriptional and posttranslational methods. One of the regulators of mitochondrial biogenesis, peroxisome proliferator-activated receptor γ coactivator-1β, is known to control the expression of Mfn2 [31], as is the related peroxisome proliferator-activated receptor δ [32]. This regulation is also controlled by exercise to aid in the maintenance of mitochondria in muscles during this stress [33, 34]. Posttranslational modification to Mfns can also control protein abundance. Both Fzo1 and the Mfns have been shown to undergo proteolytic degradation by both proteasome-dependent and proteasome-independent processes. The Mfns in Drosophila [35, 36] and mammals (Tanaka and Youle, unpublished) appear to be substrates of Parkin, an E3 ubiquitin ligase implicated in mitochondria quality control. Fzo1 in yeast is also controlled by ubiquitination and degradation in a pathway including Mdm30 as a regulator of ubiquitination [37, 38]. Further characterization of when and how these mitochondrial OMM fusion proteins are regulated and how they in turn modulate the structure of mitochondria within the cell is essential to our basic understanding of these organelles and their role in disease.
Inner membrane fusion
Fusion of the mitochondrial inner membrane (IMM) is controlled by a different large GTPase: Mgm1 in yeast and Opa1 in mammals [39, 40]. Both proteins have multiple isoforms that are present as either IMM or intermembrane space (IMS) proteins and are products of complex proteolytic processing [8, 41]. In yeast, a fraction of Mgm1 is constitutively cleaved by PCP1 [8], while the mammalian counterpart Opa1 is cleaved by Yme1 [42–44]. Both full-length and cleaved forms of Mgm1 are required for efficient fusion, and the ratio between these forms is critical [8]. For a thorough review of this proteolytic maintenance and alternate processing upon decreases in membrane potential, see [45]. In an analogous manner to Fzo1, in vitro fusion assays have also shown that Mgm1 functions in both cis and trans manners [46]. In addition, these assays revealed the distinct nature of OMM and IMM fusion events. IMM fusion defects can result in single OMM structures enclosing multiple IMM-bound matrices [17], implying that in order to efficiently fuse mitochondria, distinct membrane fusion events must occur at the interfaces of both membranes (Fig. 1). Though the OMM and IMM fusion events are separable, it is likely that some proteins confer their synchronous regulation. In yeast, Ugo1 is a likely candidate for this role. It has been shown to be essential for fusion [47] and to bind both Mgm1 and Fzo1 [48]. Unlike Mgm1 and Fzo1, Ugo1 is not required on both fusing membranes and is thus not involved in tethering the mitochondria together [49], but is required as a partner for the regulation of other fusion components. There is no identifiable ortholog of Ugo1 in mammalian cells, but it is predicted that some protein fills a similar role.
As with the fusion machinery of the OMM, Opa1 mutation causes human disease, optic atrophy type 1 (Opa1). This disease was characterized before the underlying genetic mutation was discovered to be the gene encoding a large GTPase that was a mitochondrial protein involved in fusion (Opa1) [40, 50]. It is now understood that disease-causing mutations in this protein result in mitochondrial fission and several other functional defects [15, 16, 51]. The disease is caused by haploinsufficiency, explaining how a mild defect does not cause more widespread deficits. However, why retinal ganglia cells are so dependent on mitochondrial fusion remains to be understood. It has also been observed that Opa1+/− mice have an increased number of autophagasomes in their retinal ganglion cells, implying a defect in not only fusion, but mitochondrial quality control may contribute to this disease [52].
Mitochondrial fission
Another large GTPase is primarily responsible for mitochondrial fission, Drp1 in mammals and Dnm1 in yeast. Both proteins are known to self-assemble and form large oligomeric structures. In yeast, Dnm1 localizes in puncta caused by this self-assembly in the cytosol and on the OMM and is enriched on the mitochondria in areas of constriction [53, 54]. In mammalian cells, Drp1 is more diffusely localized in the cytosol, but is also enriched at constriction sites of mitochondrial division [55]. Dnm1 forms structures similar to dynamin: curved filaments in the GDP-bound state and spirals in the GTP-bound state [56]. These spirals of GTP-bound Dnm1 form at the constriction sites of mitochondria, and the formation of these spirals on liposomes in vitro is sufficient to cause the constriction of liposomes [57]. Drp1 self-assembles as well, but into smaller diameter circular/spiral structures [55, 58]. Knocking out either Dnm1 or Drp1 results in an elongated mitochondrial phenotype, confirming their roles in mitochondrial fission [5, 54].
Another protein that appears to be required for mitochondrial fission is Fis1 [59–61]. In both yeast and mammals, this mitochondrial OMM protein has a short C-terminus in the IMS and an N-terminus exposed to the cytosol [59, 62]. At least in yeast, this protein appears to be the mitochondrial receptor for Dnm1 as it is required for the proper association of Dnm1 with the mitochondrial membrane [59]. Overexpression of Fis1 results in small fragmented mitochondria [61, 62], but these results could be a consequence of overexpression. The structure of this protein is helix-rich and contains two tetratrico-peptide repeat motif folds [63, 64]. These structures suggest several interfaces with which Fis1 may bind to other fission proteins and act as a scaffold for assembly of the fission machinery. Aside from Dnm1, Fis1 has been shown to bind to Mdv1 and Caf4 in yeast [65]. Mdv1 and Caf4 are also required for mitochondrial fission [66, 67], but there are no identified correlates to these proteins in mammalian cells, raising the question of whether Fis1 is playing the same scaffolding role for Drp1 in mammalian cells as in yeast. However, the OMM protein Mff has recently been shown to be required for fission in mammals where it could be playing a similar scaffolding role [68].
Unlike mitochondrial fusion, there are no recognized inner membrane proteins in yeast or mammals involved in fission. Interestingly, primitive algae and Dictyostelium retain a bacterial division homologue, FtsZ, in the matrix of mitochondria that is required for fission [69–71]. Although the current model is that the constrictions of Drp1 apply to both OMM and IMM and result in the concurrent fission of both membranes, it is possible that some IMM proteins are needed to facilitate the formation of constriction sites or mediate the location of these sites relative to mtDNA nucleoids.
Mitochondrial fission and fusion in the heart
Unlike yeast or most of the mammalian cell lines used to study mitochondrial morphology, cardiac cells have unique spatial constraints in the myofilament-dominated cytosol. The mitochondria in heart cells are packed tightly either between the myofibrils (interfibrillar, Fig. 2a) or between the myofibrils and the cell membrane (subsarcolemmal, Fig. 2b) [72]. This fact, coupled with the difficulties in culture and transfection of myocytes, has made the study of fission and fusion very difficult in adult heart cells. Mitochondria in adult cardiac cells do not form the large mitochondrial networks that are present in some other cell types, but rather form distinct electrical units. While these cells express all the necessary proteins for mitochondrial dynamics, catching mitochondria in the act of fission or fusion has proven an elusive challenge. Since heart mitochondria have such a heavy load of oxidative phosphorylation, and thus oxidative damage, it stands to reason that maintenance of a healthy mitochondrial network by fission and fusion is essential in these cells. While recent efforts have not allowed the direct observation of fission or fusion in adult heart cells, current research suggests that there may be roles for these processes in both healthy and diseased hearts.

Electron microscopy of adult canine heart. a Subsarcolemmal mitochondria can be observed pressed between the myofilament proteins and the cell membrane. b Interfibrillar mitochondria are packed densely between bundles of the myofilament contractile apparatus. Given these spatial constraints, large mitochondrial networks and fission/fusion events have been difficult to observe in live cardiac cells. White scale bars represent 0.5 μm. EM images courtesy of Geoffrey Hesketh
Early studies of the mitochondria in cardiac cells were focused mainly on their size and morphology, and how disease or dysregulation could affect these properties. It was observed that with various treatments or disease, heart cells accumulated either abnormally large or abnormally small mitochondrial populations [73–76]. This implies that the mitochondria in adult cardiomyocytes are not entirely static and, at least in the case of extreme stresses, can undergo some forms of fission/fusion. Several groups have more recently shown the existence of dynamic mitochondria in both neonatal cardiac myocytes and cultured cells lines of cardiac lineage [77–80]. These are both cells that express cardiac-specific proteins, but are easier to culture and observe than adult cardiomyocytes. However, these cell types lack the well-organized sarcomeres that are present in adult cells and therefore do not have a similar dense packing and structural arrangement of their mitochondria. These distinctions between HL-1, neonatal cardiomyocytes, and adult cardiomyocytes make the findings of fission and fusion in these cells less directly applicable to the adult cell phenotype. However, skeletal muscle cells do have a similar dense packing to heart cells, and recent work by Chen et al. [9] revealed an important role for Mfns in maintaining mtDNA mass and fidelity in these cells. They also showed that loss of Mfn1/2 results in muscle atrophy, respiratory deficiency, and an attempt at compensation through mitochondrial proliferation. This study indicates that in densely packed skeletal muscle cells, Mfns play important roles in mitochondrial maintenance.
To study whether mitochondrial fission and fusion play a role in heart cells, Beraud et al. [78] recently studied the dynamics of mitochondria in both adult and non-beating HL-1 cells using confocal microscopy techniques. Although the mitochondria in adult cells exhibit some movements, these mitochondria remain electrically distinct from one another and did not appear to form a network. Using the dye MitoTracker GreenTM to mark all of the mitochondria (independently of membrane potential), they observed the fluorescent centers of these mitochondria constantly making small movements, but not fusing together.
Though Beraud et al. were unable to observe fusion or fission of mitochondria in healthy adult heart cells, there is emerging evidence that these processes exist and play some important role in disease. Several different groups have recently found that there are changes in the proteins involved in fission and fusion in various forms of heart disease. Chen et al. [81] found that a high-coronary ligation model in adult rats caused a decrease in levels of the fusion protein Opa1 and a fragmentation of mitochondria in heart cells (as observed by EM). They went on to use H9c2 cells (another cardiac myogenic cell line) to show that ischemia can cause a loss of Opa1 protein which also corresponds to a fragmented mitochondrial phenotype. If Opa1 is overexpressed in these cells prior to ischemia induction, the mitochondrial network is partially preserved. This group also observed that in human patients with ischemic cardiomyopathy (ICM), there is a loss of Opa1 protein and a fragmented mitochondrial phenotype in heart cells. These results imply that ischemia can affect the stability or production of Opa1 protein and suggest that Opa1 could play a role in the pathology of ICM. Another group recently showed that the mutations in the fission protein Drp1 result in elongated mitochondria, reduced levels of oxidative phosphorylation proteins, decreased cardiac ATP, and cause a form of dilated cardiomyopathy in mice [82].
Similar to the Opa1 finding above, Ong et al. [80] also recently observed that ischemia will cause fragmentation of mitochondria in a cardiac-derived cell line (HL-1). Following 2 h of ischemia, they observed almost exclusively cells with fragmented mitochondria. However, if they overexpressed a dominant negative form of the fission protein Drp1 (Drp1K38A), this ischemia-induced fission almost entirely disappeared. They showed that overexpression of the fusion-inducing proteins Mfn1, Mfn2, or Drp1K38A protected these cells from death caused by ischemia and increased the time until mitochondrial permeability transition pore (MPTP) opening, whereas overexpression of the fission protein hFis1 resulted in a higher percentage of cell death during ischemia. These experiments imply that in cardiac-derived cells, modulation of mitochondrial phenotype can change the ability of these cells to resist the stress of ischemia. They went on to show that a Drp1 inhibitor, mdivi-1 [83], can protect against mitochondrial fragmentation, MPTP opening, and ischemia-induced cell death in both HL-1 cells and cultured adult rat cardiomyocytes. More importantly, this fission inhibitor was shown to act in vivo by reducing the myocardial infarct size in mouse hearts following 30 min of regional ischemia (followed by 120 min of reperfusion). In other cell types, several groups have shown that tipping the balance of fission/fusion toward fusion not only produces elongated mitochondria but also induces a protection against apoptosis [84]. It would appear that mdivi-1 is performing a similar function in the heart. The authors also show that mdivi-1 causes elongation of the mitochondria in vivo by using electron microscopy to determine the length of mitochondria as compared to the length of the adjacent sarcomeres. This length measurement may not be entirely accurate as it is made based only on the two-dimensional area of the mitochondria provided by the tissue sectioning for microscopy. However, it does display that there is a stark difference in shape between the control and mdivi-1-treated hearts, with the mdivi-1 treatment resulting in generally larger mitochondria. Overall, Ong et al. show that mdivi-1 can act as a cardioprotective or preconditioning agent: a drug that, when given prior to ischemia, can protect the heart (or other tissue) from damage. There has been great interest in preconditioning and its mechanisms over the past few years as researchers attempt to gain an understanding of how to protect the human heart in the context of surgery and disease [85, 86]. Several of the known pharmaceutical preconditioning agents are known to have effects on the mitochondria (for a review, see [87]), but to date, mdivi-1 seems to be the first to have direct effects on mitochondrial morphology. Other agents are known to preserve mitochondrial function or structural integrity during ischemia. For example, nitric oxide (NO) has been studied for years for its roles in cardioprotection; as a preconditioning agent, downstream signal of ischemic preconditioning and posttranslational modification of protein thiol groups (S-nitrosylation, S-NO) [88]. NO and S-NO have also been implicated in mitochondrial morphology control via inhibitory S-NO of Drp1 [89, 90], though this finding and its effects remain controversial [91]. This NO-mediated regulation of Drp1 has also been observed in myoblasts and is critical for myogenic differentiation [92]. It is tempting to speculate that the results of the study by Ong et al. on mdivi-1 and the known actions of NO imply an overarching theme: that the preservation of an elongated mitochondrial phenotype is the basis for myocardial protection. Although, too much elongation is clearly detrimental to myocytes as the recent paper from Ashrafian et al. [82] indicates that mutations in Drp1 and permanent elongation of cardiac mitochondria result in heart failure. The balance of fission and fusion that maintains normal mitochondrial morphology is essential, and in the cardiac cell, perturbations of this balance can result in either cardioprotection or failure.
Conclusions
Mitochondria exist in most cell types not as the small ovals that appear in textbooks but as a rich and dynamic network. Through the actions of multiple proteins, mitochondria fuse and divide to maintain the health of this network. In the adult heart, there is some controversy over the size and interconnectedness of the mitochondria, but studies are starting to show that there is at least some evidence for fission and fusion. Future studies are required to prove that these processes do occur in adult myocytes, and how and why these processes are dysfunctional in disease.
References
Goodpasture EW (1918) Observations on mitochondria of tumors. J Med Res 38:213–224, 211
Lewis MR, Lewis WH (1914) Mitochondria in tissue culture. Science 39:330–333
Lewis MR, Lewis WH (1915) Mitochondria (and other cytoplasmic structures) in tissue cultures. Am J Anat 17(3):245–401, 339–401
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11:958–966
Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, Walter P (1997) Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell 8:1233–1242
Margineantu DH, Gregory Cox W, Sundell L, Sherwood SW, Beechem JM, Capaldi RA (2002) Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion 1:425–435
Herlan M, Vogel F, Bornhovd C, Neupert W, Reichert AS (2003) Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem 278:27781–27788
Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141:280–289
Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM (1998) Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol 143:359–373
Jones BA, Fangman WL (1992) Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes Dev 6:380–389
Detmer SA, Chan DC (2007) Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8:870–879
Tatsuta T, Langer T (2008) Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 27:306–314
Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446
Yarosh W, Monserrate J, Tong JJ, Tse S, Le PK, Nguyen K, Brachmann CB, Wallace DC, Huang T (2008) The molecular mechanisms of OPA1-mediated optic atrophy in Drosophila model and prospects for antioxidant treatment. PLoS Genet 4:e6
Tang S, Le PK, Tse S, Wallace DC, Huang T (2009) Heterozygous mutation of Opa1 in Drosophila shortens lifespan mediated through increased reactive oxygen species production. PLoS ONE 4:e4492
Meeusen S, McCaffery JM, Nunnari J (2004) Mitochondrial fusion intermediates revealed in vitro. Science 305:1747–1752
Legros F, Lombes A, Frachon P, Rojo M (2002) Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 13:4343–4354
Hales KG, Fuller MT (1997) Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell 90:121–129
Rapaport D, Brunner M, Neupert W, Westermann B (1998) Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J Biol Chem 273:20150–20155
Santel A, Fuller MT (2001) Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114:867–874
Rojo M, Legros F, Chateau D, Lombes A (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115:1663–1674
Griffin EE, Chan DC (2006) Domain interactions within Fzo1 oligomers are essential for mitochondrial fusion. J Biol Chem 281:16599–16606
Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305:858–862
Ishihara N, Eura Y, Mihara K (2004) Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci 117:6535–6546
Kuznetsov AV, Hermann M, Saks V, Hengster P, Margreiter R (2009) The cell-type specificity of mitochondrial dynamics. Int J Biochem Cell Biol 41:1928–1939
Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A. Nat Genet 36:449–451
Cartoni R, Martinou JC (2009) Role of mitofusin 2 mutations in the physiopathology of Charcot–Marie–Tooth disease type 2A. Exp Neurol 218:268–273
Detmer SA, Chan DC (2007) Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol 176:405–414
Chen H, McCaffery JM, Chan DC (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130:548–562
Liesa M, Borda-d’Agua B, Medina-Gomez G, Lelliott CJ, Paz JC, Rojo M, Palacin M, Vidal-Puig A, Zorzano A (2008) Mitochondrial fusion is increased by the nuclear coactivator PGC-1beta. PLoS ONE 3:e3613
Li Y, Yin R, Liu J, Wang P, Wu S, Luo J, Zhelyabovska O, Yang Q (2009) Peroxisome proliferator-activated receptor delta regulates mitofusin 2 expression in the heart. J Mol Cell Cardiol 46:876–882
Cartoni R, Leger B, Hock MB, Praz M, Crettenand A, Pich S, Ziltener JL, Luthi F, Deriaz O, Zorzano A, Gobelet C, Kralli A, Russell AP (2005) Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J Physiol 567:349–358
Ding H, Jiang N, Liu H, Liu X, Liu D, Zhao F, Wen L, Liu S, Ji LL, Zhang Y (2010) Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta 1800:250–256
Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 5:e10054
Ziviani E, Tao RN, Whitworth AJ (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci USA 107:5018–5023
Neutzner A, Youle RJ (2005) Instability of the mitofusin Fzo1 regulates mitochondrial morphology during the mating response of the yeast Saccharomyces cerevisiae. J Biol Chem 280:18598–18603
Cohen MM, Leboucher GP, Livnat-Levanon N, Glickman MH, Weissman AM (2008) Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol Biol Cell 19:2457–2464
Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J (2000) The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol 151:341–352
Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26:207–210
Ishihara N, Fujita Y, Oka T, Mihara K (2006) Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J 25:2966–2977
Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM (2009) Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 187:959–966
Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI, Langer T (2009) Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol 187:1023–1036
Song Z, Chen H, Fiket M, Alexander C, Chan DC (2007) OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol 178:749–755
McBride H, Soubannier V (2010) Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health. Curr Biol 20:R274–R276
Meeusen S, DeVay R, Block J, Cassidy-Stone A, Wayson S, McCaffery JM, Nunnari J (2006) Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 127:383–395
Sesaki H, Jensen RE (2001) UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol 152:1123–1134
Sesaki H, Jensen RE (2004) Ugo1p links the Fzo1p and Mgm1p GTPases for mitochondrial fusion. J Biol Chem 279:28298–28303
Hoppins S, Horner J, Song C, McCaffery JM, Nunnari J (2009) Mitochondrial outer and inner membrane fusion requires a modified carrier protein. J Cell Biol 184:569–581
Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26:211–215
Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, Wissinger B, Pinti M, Cossarizza A, Vidoni S, Valentino ML, Rugolo M, Carelli V (2008) OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 131:352–367
White KE, Davies VJ, Hogan VE, Piechota MJ, Nichols PP, Turnbull DM, Votruba M (2009) OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Invest Ophthalmol Vis Sci 50:2567–2571
Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM (1999) The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol 1:298–304
Otsuga D, Keegan BR, Brisch E, Thatcher JW, Hermann GJ, Bleazard W, Shaw JM (1998) The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J Cell Biol 143:333–349
Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143:351–358
Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J (2005) Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol 170:1021–1027
Lackner LL, Horner JS, Nunnari J (2009) Mechanistic analysis of a dynamin effector. Science 325:874–877
Yoon Y, Pitts KR, McNiven MA (2001) Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell 12:2894–2905
Mozdy AD, McCaffery JM, Shaw JM (2000) Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol 151:367–380
Jakobs S, Martini N, Schauss AC, Egner A, Westermann B, Hell SW (2003) Spatial and temporal dynamics of budding yeast mitochondria lacking the division component Fis1p. J Cell Sci 116:2005–2014
James DI, Parone PA, Mattenberger Y, Martinou JC (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278:36373–36379
Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23:5409–5420
Suzuki M, Neutzner A, Tjandra N, Youle RJ (2005) Novel structure of the N terminus in yeast Fis1 correlates with a specialized function in mitochondrial fission. J Biol Chem 280:21444–21452
Suzuki M, Jeong SY, Karbowski M, Youle RJ, Tjandra N (2003) The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J Mol Biol 334:445–458
Tieu Q, Okreglak V, Naylor K, Nunnari J (2002) The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J Cell Biol 158:445–452
Griffin EE, Graumann J, Chan DC (2005) The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol 170:237–248
Tieu Q, Nunnari J (2000) Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J Cell Biol 151:353–366
Gandre-Babbe S, van der Bliek AM (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19:2402–2412
Beech PL, Nheu T, Schultz T, Herbert S, Lithgow T, Gilson PR, McFadden GI (2000) Mitochondrial FtsZ in a chromophyte alga. Science 287:1276–1279
Gilson PR, Yu XC, Hereld D, Barth C, Savage A, Kiefel BR, Lay S, Fisher PR, Margolin W, Beech PL (2003) Two Dictyostelium orthologs of the prokaryotic cell division protein FtsZ localize to mitochondria and are required for the maintenance of normal mitochondrial morphology. Eukaryot Cell 2:1315–1326
Nishida K, Takahara M, Miyagishima SY, Kuroiwa H, Matsuzaki M, Kuroiwa T (2003) Dynamic recruitment of dynamin for final mitochondrial severance in a primitive red alga. Proc Natl Acad Sci USA 100:2146–2151
Hoppel CL, Tandler B, Fujioka H, Riva A (2009) Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol 41:1949–1956
Kraus B, Cain H (1980) Giant mitochondria in the human myocardium—morphogenesis and fate. Virchows Arch B Cell Pathol Incl Mol Pathol 33:77–89
Coleman R, Silbermann M, Gershon D, Reznick AZ (1987) Giant mitochondria in the myocardium of aging and endurance-trained mice. Gerontology 33:34–39
Kanzaki Y, Terasaki F, Okabe M, Otsuka K, Katashima T, Fujita S, Ito T, Kitaura Y (2010) Giant mitochondria in the myocardium of a patient with mitochondrial cardiomyopathy: transmission and 3-dimensional scanning electron microscopy. Circulation 121:831–832
Bhimji S, Godin DV, McNeill JH (1986) Isoproterenol-induced myocardial alterations in alloxan-diabetic rabbits. Can J Cardiol 2:313–319
Yu T, Sheu SS, Robotham JL, Yoon Y (2008) Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res 79:341–351
Beraud N, Pelloux S, Usson Y, Kuznetsov AV, Ronot X, Tourneur Y, Saks V (2009) Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J Bioenerg Biomembr 41:195–214
Hom J, Yu T, Yoon Y, Porter G, Sheu SS (2010) Regulation of mitochondrial fission by intracellular Ca(2+) in rat ventricular myocytes. Biochim Biophys Acta 1797:913–921
Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–2022
Chen L, Gong Q, Stice JP, Knowlton AA (2009) Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res 84:91–99
Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, Lygate CA, Hough T, Townsend S, Williams D, Wells S, Norris D, Glyn-Jones S, Land J, Barbaric I, Lalanne Z, Denny P, Szumska D, Bhattacharya S, Griffin JL, Hargreaves I, Fernandez-Fuentes N, Cheeseman M, Watkins H, Dear TN (2010) A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet 6:e1001000
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14:193–204
Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22:1577–1590
Hausenloy DJ, Yellon DM (2009) Preconditioning and postconditioning: underlying mechanisms and clinical application. Atherosclerosis 204:334–341
Huffmyer J, Raphael J (2009) Physiology and pharmacology of myocardial preconditioning and postconditioning. Semin Cardiothorac Vasc Anesth 13:5–18
Murphy E, Steenbergen C (2007) Preconditioning: the mitochondrial connection. Annu Rev Physiol 69:51–67
Burwell LS, Brookes PS (2008) Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia–reperfusion injury. Antioxid Redox Signal 10:579–599
Nakamura T, Cieplak P, Cho DH, Godzik A, Lipton SA (2010) S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 10:573–578
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324:102–105
Bossy B, Petrilli A, Klinglmayr E, Chen J, Lutz-Meindl U, Knott AB, Masliah E, Schwarzenbacher R, Bossy-Wetzel E (2010) S-nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer’s disease. J Alzheimers Dis 20(Suppl 2):S513–S526
De Palma C, Falcone S, Pisoni S, Cipolat S, Panzeri C, Pambianco S, Pisconti A, Allevi R, Bassi MT, Cossu G, Pozzan T, Moncada S, Scorrano L, Brunelli S, Clementi E (2010) Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ. 2010 May 14. doi:10.1172/JC140373
Kanazawa T, Zappaterra MD, Hasegawa A, Wright AP, Newman-Smith ED, Buttle KF, McDonald K, Mannella CA, van der Bliek AM (2008) The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet 4:e1000022
McQuibban GA, Lee JR, Zheng L, Juusola M, Freeman M (2006) Normal mitochondrial dynamics requires rhomboid-7 and affects Drosophila lifespan and neuronal function. Curr Biol 16:982–989
Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM (1999) C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4:815–826
Acknowledgments
The authors’ would like to thank Geoffrey Hesketh for the images used in Fig. 2. Work in the authors’ laboratory is funded by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health.
Disclosure of potential conflict of interests
The authors declare no conflict of interests related to this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kane, L.A., Youle, R.J. Mitochondrial fission and fusion and their roles in the heart. J Mol Med 88, 971–979 (2010). https://doi.org/10.1007/s00109-010-0674-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-010-0674-6