
Abstract
A wide range of length and time scales are relevant to pharmacology, especially in drug development, drug design and drug delivery. Therefore, multiscale computational modeling and simulation methods and paradigms that advance the linkage of phenomena occurring at these multiple scales have become increasingly important. Multiscale approaches present in silico opportunities to advance laboratory research to bedside clinical applications in pharmaceuticals research. This is achievable through the capability of modeling to reveal phenomena occurring across multiple spatial and temporal scales, which are not otherwise readily accessible to experimentation. The resultant models, when validated, are capable of making testable predictions to guide drug design and delivery. In this review we describe the goals, methods, and opportunities of multiscale modeling in drug design and development. We demonstrate the impact of multiple scales of modeling in this field. We indicate the common mathematical and computational techniques employed for multiscale modeling approaches used in pharmacometric and systems pharmacology models in drug development and present several examples illustrating the current state-of-the-art models for (1) excitable systems and applications in cardiac disease; (2) stem cell driven complex biosystems; (3) nanoparticle delivery, with applications to angiogenesis and cancer therapy; (4) host-pathogen interactions and their use in metabolic disorders, inflammation and sepsis; and (5) computer-aided design of nanomedical systems. We conclude with a focus on barriers to successful clinical translation of drug development, drug design and drug delivery multiscale models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
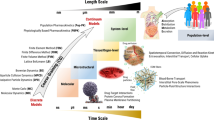
One of the biggest challenges in the current era of data abundance is revealing how the interactions between discrete biological system components result in integrated emergent effects on higher order systems. This is especially relevant in the setting of pharmacology and toxicology, where there have been no reasonable, efficient or cost-effective experimental or clinical strategies to facilitate prediction and development of therapeutic interventions. One of the reasons that projecting the effects of drugs on biological systems has been so difficult is because physiological processes occur over a wide range of length and time scales (Fig. 1). New approaches in multiscale modeling and simulation are now being developed to bridge these scales and allow for the first in silico predictions that can facilitate drug development and screening, predict drug mechanisms and responses, optimize drug delivery and therapeutic effect, and minimize toxicity. Mechanism-based multiscale models that include patient specific parameters are occurring in multiple domains ranging from excitable systems to cancer to metabolic disorders, inflammation and sepsis, and musculoskeletal systems. These modeling adjuncts to traditional clinical practice are an important complement to purely inferential (statistical) approaches to personalized medicine.

Multiscale computational models can span an enormous spatial range from populations downward and time scales from sub-second to decades.
Pharmacometric and Systems Pharmacology Models in Drug Development
Multiscale modeling is beginning to be applied more and more in the development of new drugs. The most advanced application is in the field commonly termed as “pharmacometrics”, i.e., “the branch of science concerned with mathematical models of biology, pharmacology, disease, and physiology used to describe and quantify interactions between xenobiotics and patients, including beneficial effects and side effects resultant from such interfaces”.119 This emerging discipline has been reviewed before121 and has influenced drug development, especially in the clinic, significantly.61,74 Together with the complementary discipline of systems pharmacology, which is perhaps best suited for the early discovery stages, pharmacometrics is helping drug discovery and development approach the aerospace industry in its reliance on computer modeling and design.127
This discussion will focus on drug development, as this is where multiscale models are most widely used, although their role in early stages is increasing rapidly. Models used in this context are multiscale models, but at the level of their statistical complexity, as opposed to structural behavior.
It has been recognized for some time that variability in drug dose-exposure–response relationships is hierarchical. While the time course of drug concentration (pharmacokinetics) and the time course of drug effect (pharmacodynamics) can most often be described, on average, by nonlinear differential equations with relatively few parameters, the parameters of these equations vary among individual patients (this is usually called BSV, between-subject variability). Moreover, real-world biological measurements are affected by error, which can also change with time (this is RUV, residual unknown variability). The statistical distribution of these parameters can become part of the model.50 The scientific context of these models is that of nonlinear mixed effects models, which are an extension of the linear mixed effects framework which has been so successful in experimental design in the statistical sciences.11
Briefly, nonlinear mixed effects models enable the estimation of means and variances of the statistical distributions of model parameters.78,79 An example would be the mean and variance in the population of the clearance of the drug being studied. This is particularly useful in the context of clinical trials, where a large number of subjects may have been studied, but no individual subject has a dense enough sampling schedule to reveal that patient’s individual parameters.
Latest developments in systems pharmacology, which has been described as the interface between Pharmacometrics and Systems Biology,119 aim at increasing the biological realism of the models underlying pharmacokinetics and pharmacodynamics. This usually requires more parameters and pathways to be added to the models, increasing their complexity. Systems pharmacology was the subject of a series of workshops at the National Institutes of Health, whose proceedings were summarized in a white paper.100 While systems pharmacology models are starting to be applied in the context of clinical data analysis and simulation, the application of pharmacometrics techniques has been occurring for longer. However, the therapeutic areas and applications where systems pharmacology results have been published are multiple: infectious diseases,115 cancer,66 cardiovascular disease23,76,77 and neurosciences43 among others. Future challenges122 include continuing to augment the biological realism of multiscale models used in drug development, as well as continue to improve integration between bioanalytical and laboratory sciences and model development experts. These salient issues are further demonstrated through specific illustrations of modeling technologies applied to various disease states in the sections that follow.
Models for Excitable Systems and Applications in Cardiac Disease
Multiscale modeling for drug prediction in excitable systems is critical because experimental approaches at individual system scales cannot solve the fundamental problem—that the effects of multifaceted drug interactions are emergent. Computational based methods under development to predict emergent effects of drugs on excitable rhythms may form an interactive technology driven process that can be used in industry for drug and disease screening, in academia for research and development and in the clinic for patient oriented medicine. There is potential for far-reaching implications because millions of people affected each year by arrhythmia would benefit from improved risk stratification for drug-based interventions. Effective pharmacological treatment of arrhythmia syndromes would reduce shocks from implantable defibrillators that reduce quality of life for so many individuals.
New computational methods are being developed for drug design and development that take advantage of high-performance computing technologies to reveal emergent mechanisms of disease and to facilitate prediction and development of therapeutic interventions.132 A primary goal in developing these computational approaches is to generate frameworks that can ultimately be scaled up and automated for prediction of drug design, development and drug effects that can be applied to industry, academia and in clinical settings.
In the context of the heart, there exists a long history of failure in predicting effective or harmful action of drugs. Antiarrhythmic drugs, which primary target cardiac ion channels,111,112 have been widely prescribed for arrhythmia syndromes arising from multiple underlying diseases that increase arrhythmia proclivity including coronary artery disease, cardiomyopathies, post-infarction injury and heart failure. For example, the CAST87 and SWORD123 clinical trials, showed that compared to placebo, common antiarrhythmic drugs increased mortality and risk of sudden cardiac death. Almost 30 years after the beginning of the first CAST trial, there is still no available approach to differentiate potentially useful and potentially harmful drugs for treating arrhythmia.
Much progress has been made in developing multiscale computational modeling and simulations approaches for prediction of the effects of cardiac ion channel blocking drugs (Fig. 2). Structural modeling of ion channel interactions with drugs is a critical approach for current and future drug discovery efforts. Modeling of drug receptor sites within an ion channel structure can be useful to identify key drug-channel interaction sites. Drug interactions with cardiac ion channels have been modeled at the atomic scale in simulated docking and molecular dynamics (MD) simulations, as well as at the level of the channel function to simulate drug effects on channel behavior.16,20,25,68,72,76,77,134–136 Modeling drug interactions at the molecular scale requires high-resolution structures of potassium and sodium channels that are used as templates for pairwise sequence alignments.69,97,124 Structural modeling of drug-channel interactions at the atomic scale may ultimately allow for design of novel high-affinity and subtype selective drugs for specific targeting of receptors for cardiac and neurological disorders.

A multiscale computational model for predicting cardiac pharmacology. The model may allow simulation and prediction from the small molecule scale of the drug, to protein structure, protein function, cell and tissue levels.
In order to accurately predict ion channel drug effects in higher dimensions, the intrinsic and explicit dynamical complexity of the drug kinetics is increasingly being considered in computational model representations. Early studies of drug effects on cardiac ion channels relied on pore-block models,12 which did not include the complex features of drug-channel kinetics that fundamentally emerge to alter cardiac rhythms in higher dimensions. Examples of emergent drug properties include well-known effects like use-dependent and frequency-dependent block, as well propensity to alternans and changes to action potential duration restitution and conduction velocity restitution in tissue.42,88,126,128
Emergent drug effects have been predicted at the cellular level by incorporating drug channel models into computational models of cardiac myocytes. Simulations have been used to test drug effects on cellular level parameters to search for antiarrhythmic or overt proarrhythmic potential.13,20,32,76,77,89,92 Although cellular level studies can plausibly suggest reduced or increased arrhythmia vulnerability, reentrant arrhythmias are fundamentally an emergent property of the cardiac system that can only be observed and studied in tissue. Thus, models have been developed to predict drug effects in higher dimensions that include spatial dimension and cellular coupling.
Computational studies have been carried out in tissue representations in one and two dimensions and even in high-resolution reconstructions of human virtual ventricles.12,35,76,82,132 Arrhythmia vulnerability parameters as described for one-dimensional tissue can be tracked in two dimensions.76,81,130,137 Two-dimensional simulations can be undertaken to predict if proarrhythmic phenomena observed in lower dimensions cause reentrant arrhythmias and/or spiral wave breakup. The change in voltage in space and time are tracked in the simulation.110 In two dimensions, reentry wavelength and period can also be tracked to investigate head–tail interactions. More recently, drug simulations in three-dimensional cardiac reconstructions from humans have been undertaken as computational resources are increasingly accessible.12,35,82,132
Because antiarrhythmic drugs exhibit complex kinetic interactions with ion channels that are modified by action potential properties including morphology, duration and frequency, strong bi-directional feedback exists because drugs alter the action potential waveform, which in turn affects the potency of drugs. In tissue, electronic coupling leads to unpredictable emergent responses to drug application. An example is the study by Moreno et al., which showed mild depression of single cell cellular excitability by flecainide, suggesting its therapeutic potential to suppress ectopic arrhythmia triggers.76 No overt proarrhythmic potential was ever observed in cells. In tissue level simulations, the outcome was dramatically different. Substantial use-dependent block with flecainide resulted in insufficient Na channel availability for successful conduction, a higher dimensional phenomenon that emerged as a result of increased electrotonic load in coupled tissue. Proarrhythmic conduction block sometimes led to development of tachycardia indicated by spiral wave reentry, verified experimentally in rabbit heart and in MRI-based human 3D ventricle models.76 These types of computational studies have begun to improve understanding of antiarrhythmic drug actions across multiple spatial scales of the cardiac system, from molecule, to channel, to cell, to tissue, to heart.
Models for Stem Cell Driven Complex Biosystems
While cardiovascular disease is the leading cause of death in the United States, cancer currently ranks second and is projected to become the top cause of death in the next few years.95 Multiscale modeling is crucial for simulating drug responses of stem cell driven biosystems and the corresponding applications in cancer therapeutics, regenerative medicines, and beyond. In such systems, stem cells play two roles: driving tissue growth through differentiation to functional cells, and maintaining regeneration potential by self-renewal. The micro-environment of stem cells (known as “stem cell niche”) provides physical, chemical, and biological cues with spatial and temporal102 patterns to regulate these two roles of stem cells, meanwhile the stem cell driven tissue growth will remodel such micro-environment and affect stem cell behaviors. Such complex biosystems demonstrate strong multiscale characters in both space and time. Spatially, at the molecular level, the biological or cellular cues in stem cell niches trigger intracellular signaling and modulate stem cell behaviors such as renewal, differentiation, apoptosis, and migration; at the cellular level, such stem cell behaviors, together with environmental cues, determine the numbers of stem cells, progenitor cells, and terminal cells as well as their behaviors; at the tissue level, the populations as well as the behaviors of these cells define the structural and thus the functional behavior of the tissue generated de novo. Temporally, at the second-to-minute scale, the external cues trigger signaling events; at the hour-to-day level, cells respond to such cues with various behaviors; at day-to-week scale, the downstream effects at tissue level as well as the remodeling of the cell microenvironments begin to show up. Such multiscale and highly dynamic biosystems require sophisticated drug delivery to generate desired spatial distribution and temporal patterns of drugs. Therefore, multiscale modeling on the spatial3,85,101,103,133 and temporal101,104 domains is the key strategy to predict the pharmaceutical inference of stem cell driven biosystems.
To illustrate the cutting-edge multiscale modeling approaches in simulating stem cell driven systems, two typical applications are presented in this section. The first one is the drug synergism analysis for cancer stem cell driven drug resistance.103 In this example, we demonstrate how to use agent-based models (ABMs) (Fig. 3) to analyze the complex spatial dynamics of myeloma initiating (stem) cells (MIC), bone marrow stromal cells (BMSC), progenitor cells (PC), and early multiple myeloma (MM) and terminal multiple myeloma (TMM) cancer cells, as well as the remodeling of the tissue stiffness in bone marrow during the development and drug treatment of multiple myeloma (Fig. 4). Each agent represented a tumor cell or a section of the elongated, network-like BMSCs, encapsulating intracellular signaling events within the agent, and, as a whole, responding to its microenvironment as cell behaviors such as proliferation, apoptosis, migration, contraction, and so on. Thus, an agent seamlessly incorporated the intracellular molecular scale events and the cellular scale behaviors, and provided the essential element to describe cell-to-cell interaction at the intercellular scale and finally the tumor development at the tissue scale. Knowledge and hypothesis were represented as “rules” of cell decision-making and realized using stochastic approaches such as Markov chain Monte Carlo (MCMC) methods. In this specific study, for a cell agent, the microenvironmental cues such as the concentrations of cytokines and drugs, the stiffness of the niche, and the types, number, and distance of neighbor cells, together with the current cell statuses (for example, during proliferation, during differentiation, etc.), were used as inputs by the pre-defined “rules” (equations) to calculate the probabilities of downstream cell behaviors. Such probabilities were then converted to cell decisions by random sampling. The MCMC approach thus readily connects the deterministic and continuous mathematical models (“rules”) to the discrete, stochastic cell decisions.

A sketch of the ABM model for the impact of the stiffness of the BMSC-formed MIC niches to the myeloma lineage expansion and drug responses. (ABM agent-based model, BMSC bone marrow stromal cell, MIC myeloma initiating cell, PC cancer progenitor cell, MM multiple myeloma cell, TMM terminal multiple myeloma cell, BZM Bortezomib, AMD AMD3100, CXCR4 C-X-C chemokine receptor type 4, SDF1 stromal cell-derived factor 1).

Simulation of myeloma development in three-dimensional bone marrow space. The tumor growth (a and c) associated with the stiffness profiles (b and d) and the activities of MICs at early (100 h, a and b) and later (500 h, c and d) stages. (e and f). Tumor and stiffness distributions after BZM treatment between time point 200 and 300 h. The tumor infiltrating frontiers were labeled by brown isosurfaces, condensed tumor region in yellow, and quiescent MICs in blue while proliferating MICs appear in red. The stiffness distributions were labeled with isosurfaces, blue denoted lower stiffness and red higher stiffness. (g) The distribution of stiffness in myeloma-associated bone marrow stiffness 400 h after initial myeloma seeding. (h) The distribution of myeloma cells and the activation of MBMSCs after 4 days 3D levitated culture. Blue: cell nuclei (stained with DAPI); red: F-actin fibers (stained with Alexa Fluor 594 conjugated phalloidin); white arrow: MBMSCs; green arrow: myeloma cells; yellow arrow: activated BMSCs marked by the formation of stress fiber.
The second example104 is to use a multiple temporal scale model to study the effects of sequential delivery of growth factors on a dual stem cell bone regeneration system (Fig. 5). The dynamic balance between the mesenchymal stem cell driven osteoblastic bone formation and the hematopoietic stem cell driven osteoclastic bone resorption were analyzed for the best timing of BMP2, Wnt, and TGFβ delivery (Fig. 6). Ordinary differential equations and Hill functions were used to describe the minutes-to-hours timescale intracellular signaling events and intercellular signaling, respectively; Hill functions were used to describe the days-scale stem cell differentiation; and finally stochastic differential equations were used to model the days-to-weeks scale dynamics of cell population and composition as well as the bone healing and remodeling. Key variables such as the section of cytokines and the population size of each cell type linked the three time scales to a consistent model.

Schematic illustration of intracellular and intercellular signaling and cellular dynamics in bone healing and bone remodeling. Bone regeneration or bone remodeling involves bone resorption by osteoclasts (OC) and the following bone formation by osteoblasts (OB) within basic multi-cellular units (BMU). Three cytokines were considered: TGFβ, Wnt and BMP2. Intracellular signaling pathway consists of Smad2/3, Smad1/5, β-Catenin, and Runx2 and Osx. Runx2 can promote the differentiation of mesenchymal stem cells (MSCs) into pre-osteoblasts (OBp) and can inhibit the differentiation of pre-osteoblasts into active osteoblasts (OBa). Osx also play a promoting role in the later stage of osteoblastic lineage which interacts with osteoclastic lineage through intercellular signaling pathway RANK-RANKL-OPG.

Synergy prediction on dual combinations of Wnt, BMP2, and TGFβ based on Bliss combination index. Wnt and BMP2 perform dose-dependent synergism. (a) BMP2 levels governed the synergism. When the BMP2 level was higher than 0, the two drugs were synergic, otherwise antagonistic. We also found that Wnt at high levels showed opposite effects in terms of synergism at different BMP2 levels. When BMP2 level was high, increasing Wnt level promoted the synergistic effects of the two drugs. In contrast, when BMP2 level was low, the more the Wnt was introduced, the stronger the antagonistic effect was. (b) Wnt/TGFβ and (c) BMP2/TGFβ combinations also showed dose-dependent synergism but much lower responses.
Taken together, systematic modeling of stem-cell-driven complex biosystems by incorporating multiple spatial and temporal scales casts new light onto basic and translational biomedical research in multiple aspects: it provides insight into the underlying mechanisms of diseases and cures, allows in silico predicting drug responses for drug screening and optimizing combination therapeutic designs, guides precise drug delivery, and helps to bridge the gap from bench-side knowledge to bed-side clinical practice.
Nanoparticle Delivery Models, with Applications to Angiogenesis and Cancer Therapy
Nanotechnology is another field advancing from bench-side knowledge to bed-side clinical practice. The use of targeted nanocarriers (NCs) for delivering therapeutic compounds to sites of pathology presents significant opportunities both in terms of personalizing medical care and in accessing multiscale modeling to refine the specifics of individual treatments. This is particularly applicable in cancer care, wherein the clinical successes of anticancer therapies are often limited by the marginal efficacy of the therapeutics and their various side effects. Prediction of the distribution, metabolism, absorption, excretion and toxicity of potential new drugs in early stages of development has attracted much attention.21,46 The therapeutic efficiency of nanomedicine is determined by the proper concentration of drug at the lesion site. Drug carriers need to be delivered directly to the desired tissues while minimizing deposition/uptake by other tissues. Targeted drug delivery using functionalized NCs coated with specific targeting ligands has been clinically identified as a promising approach in both therapeutic and diagnostic applications in cancer treatments.30 A typical process of targeted delivery to tumor site is illustrated in Fig. 7.

Schematics demonstration of drug transport (a) through the circulation system, (b) blood vasculature, (c) across the vessel wall, (d) through interstitial space to the tumor site.
The biochemical and physiological properties of a tumor’s microenvironment, including the vasculature and the interstitial extracellular matrix (ECM), are the key regulators of anti-cancer drug distribution and efficacy.54,56 Blood vessels provide the primary passage for drugs to be delivered to the tumor and other tissues. However, heterogeneous microvascular function within tumors can compromise delivery and undermine the effects of therapeutic agents.54 Enhanced permeability and retention (EPR) in leaky vessels has facilitated the targeting of macromolecular therapies.26,36,114 Yet, elevated interstitial fluid pressure (IFP), induced by vessel abnormalities, fibrosis and contraction of the interstitial matrix, also hinders the drug delivery to the tumor.18,49 In fact, IFP is known as the main barrier for drug delivery to tumor sites. Moreover, targeting of NCs to endothelium remains an important design challenge in pharmacological and biomedical sciences since functionalized NCs offers a wide range of tunable design parameters such as size, shape, type, and functional coating.106
Mechanistic mathematical and computational modeling at multiple scales, from gene to protein to tissue and organ, and eventually the whole body is becoming an effective if not a required tool for examining the impact of various biophysical features of the tumor tissue and biochemical properties of drug compounds on drug delivery efficacy.21,39,40,46,109,125 Numerical simulations are well-suited and cheaper, compared to laboratory experiments, for testing combinations of multiple parameters that can be varied simultaneously in a controlled manner and over a wide range of values. These in silico screenings can be helpful to optimize the drug design so that it is efficient in interstitial transport, or make decisions regarding the most effective drug combinations and scheduling protocols.
The NC targeted delivery in vascular system involves interplay of transport, hydrodynamic force, and multivalent interactions with targeted biosurfaces. Thus, drug delivery to tumor sites is a complex and challenging process over various spatial scales, including organ, tissue, cell, and intracellular levels. After systemic administration, drugs have to go through a few processes before arriving at the targeted tumor sites: (1) transport in the circulation system, (2) extravasation across the vessel wall, and (3) transport through interstitial space to the tumor site. Therefore, the modeling targeted drug delivery process spans physics across continuum vascular flow, particle Brownian adhesion dynamics, to molecular level ligand-receptor binding and cellular uptake.
We focus our discussion of multiscale drug targeting on three interconnected processes happening in various biological spatial scales: drug carrier transport in circulation system, drug transport through interstitial space, binding dynamics and cellular uptake. The targeted drug techniques such as MD, Brownian motion, and stochastic approaches such as Monte Carlo simulation can be used to simulate nano, micro, and macroscale interactions between carrier and target site.
Using nanoparticles in biomedical applications involves physical translocation processes of nanoparticles and the cellular uptake of particles. Models at this scale characterize the different interactions such as drug-carrier, carrier-medium (biological) and drug-medium. These molecular-scale models deal with length scales in the order of nm ~ μm and time scales of ns ~ μs. For instance, Yang and Ma et al. 129 used computer simulation to investigate nanoparticle penetration through cell membranes where the translocation processes of nanospheres, nano-ellipsoids, nanorods, nanodiscs and pushpin-like NCs across a lipid bilayer were studied by dissipative particle dynamics (DPD). It was reported that the shape anisotropy and initial orientation of the particle are crucial to the interactions between the particle and lipid bilayer.
Before reaching the targeted tumor region, drug carriers have to transport through interstitial space. Generally, conservation of mass is adopted as the mathematical equation to govern the drug delivery in tissue or tumor scale. In the most general description, changes in the amount of drug present in the tissue depend on three values: the amount of drug entering the tissue (drug production), how the drug moves within the tissue (drug transport), and the amount leaving the tissue (drug elimination),57 as shown in Eq. (1):

Drugs can be carried through the tissue with the interstitial fluid flow (advective transport) or move randomly due to the Brownian motion of drug molecules (diffusive transport). Drug elimination from the tissue can take place due to its natural half-life (decay), binding to the ECM (degradation or deactivation), or cellular uptake. Pozrikidis presented a theoretical framework describing blood flow through an irregular vasculature of a solid tumor where capillary leakage due to the transmural pressure is considered.86 NC binding to the vessel wall lumenal surface incorporating ligand receptor interactions were modeled in Ref. 64, 106.
NCs loaded with drugs have been widely used to target tumor cells due to manufacturing and synthesis capabilities to control size, shape, and surface chemistry.26,44 This necessitates a multiparameter optimization for achieving efficient vascular targeting.9,17 Studying adhesion dynamics, Liu et al. estimated NP binding affinity to endothelial cells.64 King et al. studied multiparticle adhesion dynamics and applied it to leukocyte rolling.58,59 Fogelson et al. coupled ligand-receptor binding with platelet aggregation.41 Most theoretical studies of NC deposition are limited to simple spherical particles under ideal shear flows,27–29 or combined Brownian motion with hydrodynamics in cylindrical tube flow.117
At the macroscale, continuum convection-diffusion-reaction and particulate98 models have been widely used in modeling drug delivery process. A significant aspect of modeling nanoparticle motion in vasculature is the accurate evaluation of the associated momentum forces from which different translational and rotational motions arise. These macroscopic models deal with length scales in the order of μm ~ mm. In the continuum assumption, blood flow is characterized through the Navier-Stokes (NS) equations, while the drug is described as a variable denoted as concentration. The NS equations are coupled with convection diffusion reaction equations so that the distribution of NCs along the vascular network can be predicted.51 Particulate models also can be used to analyze drug deposition in complex vascular geometries.98 Shipley and Chapman94 and Modok et al. 75 modeled delivery of spherical NCs in tumor. Tan et al. 108 used a coupled continuum and particulate model to study NC transport and binding dynamics. Mahmoudi et al. 67 and Li et al. 62 performed computational fluid dynamics studies of magnetic NCs in vascular flow.
Blood is not a simple Newtonian fluid but is comprised of different cells, proteins, and nutrients. Explicit blood components have to be considered particularly if we are interested in the physical interaction between cells and NCs in microcirculation. Cell models have been considered in recent studies using DPD,80 Lattice Boltzmann,53 and Immersed Boundary107 methods. These studies showed that the margination and adhesion probability depends on the cell concentration, particle size, shape, and shear rates. Haun and Hammer48 also have investigated the kinetic rate constants of attachment and detachment of nanocarriers as a function of receptor density, ligand density on surface, and flow shear rate. They also showed the time dependence of the detachment rate due to multivalent binding.
It is difficult to fully comprehend and integrate the complex, nonlinear, and often unintuitive processes involved in the cellular and physiological disposition of drug carriers, without the use of a multiscale, mechanism-based mathematical model. For instance, Shah et al. 93 built a multiscale pharmacokinetic–pharmacodynamic model of antibody drug conjugates for its preclinical to clinical translation efficacy. They not only characterized the biodisposition of antibody drug conjugates and payload at the cellular and physiological level, but also provided translation of preclinical efficacy data to the clinic. Moreover, Liu et al. 63 have used the Metropolis Monte Carlo (MC) strategy in conjunction with the weighted histogram analysis method (WHAM) to compute the free energy landscape associated with the multivalent antigen–antibody interactions.
Overall, multiscale modeling to optimize NC delivery for cancer therapy and to alter angiogenesis requires that a wide range of length and time scales be accessed to describe the physics of hydrodynamic and microscopic molecular interactions mediating NC motion in blood flow, binding, uptake and offloading of the deliverable. As with other clinically relevant simulations discussed herein, optimization for clinical applications must include relevant anatomical, physiological and pharmacological features into computational models bridging the relevant multiple scales. Simulations can limit the need for large scale in vivo and in vitro experimentation in designing effective NP- or NC-based treatment.
Models for Host-Pathogen Interactions and Their Use in Metabolic Disorders, Inflammation and Sepsis
Simulation can also advance our knowledge and clinical capabilities in treating infectious and metabolic diseases. The ability of organisms to respond to and recover from damage is a fundamental biological function. The ubiquity of the inflammatory process, and its role as a pathway to healing, across the entire range of tissues and in response to a plethora of external and internal threats, is a testament to this fact. The pervasive role of inflammation is also becoming increasingly clear in virtually all the significant disease processes that challenge us today; from the hyper-acute disruptions seen in Ebola and sepsis, to the punctuated equilibrium dynamics of cancer, to the chronic indolent nature of obesity, auto-immune diseases, Alzheimer’s and cardiovascular disease. The Janus-faced visage of inflammation resides in every tissue, but with different controls and set-points, and modulating inflammation perhaps represents the prototypical dilemma of delivering the appropriate control at the right time, to the right place, with a minimum of collateral damage.
Evolution has dictated that a constant source of danger for multi-cellular organisms comes from the microbial world. However, while long being considered the primary threat to human health (not unfounded or incorrect), the recent understanding of the role of our resident microbiomes as partners in maintaining human health has added a new dimension to the relationship between host and microbe (see Fig. 8). In fact, the inevitable evolutionary response of the microbial world to our attempts to eradicate them, manifest as the growing challenge of antibiotic resistance, suggests that it has become necessary for us to develop more nuanced means of engaging in trans-kingdom relations in the interest of human health, strategies that owe more to ecological understanding than eradicative strategies.

Overview of the multiple scales and classes of processes involved in host-microbial interactions. Modeling efforts would necessarily integrate metabolic processes, microbial community dynamics, host responses involving inflammation as they manifest in different tissues. Figure reproduced from19 under the Creative Commons Attribution License.
Taken together, the goals of dealing with the microbial world and our host-side inflammatory mechanisms reacting to damage and threat, have potential implications across a wide range of pathophysiological processes that occur at multiple levels of biological organization. Therefore, system-level, multiscale perspectives are invaluable in our attempts to engineer safe and effective controls for these systems, and require the concurrent use of multiple complementary modeling methods. The comprehensive integration of a multiscale workflow is still far in the future, but below we present some selected examples that address some identified focus areas in the attempt to move towards that greater goal.
Taking a modeling approach to integrate systems biology and cheminformatics can provide new leverage for understanding and manipulating host-microbe interactions. The use of small-molecule therapeutics to shift how a host-microbe ecosystem behaves is a long established clinical approach to advantaging host immune response (e.g., antibiotics that target core conserved metabolic process for energy acquisition or biomass production) or dampening overactive immune response. In developing antibiotics such as isoniazid, which targets fatty acid biosynthetic pathways in Mycobacterium tuberculosis, the primary consideration has been within a single chemical scale, namely development of therapeutics that interfere with the chemical structures of the small molecules or enzymes that underpin critical microbial persistence or virulence pathways. With the proliferation of omics technology enabling a systems scale view of host-pathogen interactions, and high-throughput chemotherapeutic modulation of those interactions, the need to consider and incorporate the multiscale effects of small-molecules on microbial persistence has led to the increasing integration of the fields of cheminformatics and systems biology.14,37,38,84 Here we focus on modeling platforms that consider the biochemical consequence of introduction of the chemotherapeutic agent into the organism, as opposed to models used to solely identify potential drug targets. As an example, systems chemical biology (SCB) and specifically computational SCB integrates chemical biology and computational systems biology to investigate the consequential outcome of small molecule disruption of metabolic pathways of pathogenic organisms (see Fig. 9).71,83,84 Given a target metabolic pathway, the computational SCB platform provides a tool for multiscale targeted drug design beginning with an automated cheminformatics virtual screening pipeline consisting of WOMBAT and SciFinder tools for generation of candidate small molecules, OMEGA and the Protein Data Bank (PDB) to generate 3D ligand and protein structures, and GRID and FRED to analyze structures and perform virtual docking studies. Molecular scale protein–ligand interaction data is integrated into metabolic scale models to capture the dynamic impact of small molecule inhibition on vital biochemical processes. Use of SCB modeling methods has been applied to explore the interplay of environmental and chemotherapeutic stress on M. tuberculosis growth and persistence,70 and given the ability to use multiscale models to capture dynamic adaptation of the pathogen system, further extensions to studying the role of small molecule therapeutics on pathogen persistence and resistance is feasible. The continued integration of chemical informatics and system scale modeling will serve as a critical step in the development of personalized candidate interventions. Multiscale modeling of host-pathogen interactions in the presence of therapeutic agents can play an important role in discovering desirable and undesirable mechanisms of action for existing antibiotics and antivirals, in determining differences between successful vs. unsuccessful treatments, and in understanding the dynamic interactions involved in the emergence of multidrug resistant pathogenic strains.

The computational systems chemical biology (SCB) workflow integrates cheminformatics platforms for model-based identification of small molecule therapeutics with dynamic simulation of the system scale outcome of targeted inhibition. The SCB approach has been used to theoretically investigate the metabolic consequence of multiple chemotherapeutic agents on persistent and non-persistent M. tuberculosis.
Recognizing that microbial communities act as endogenous bioreactors establishes a pathway for applying new modeling paradigms. As noted above, there seems to be no end to the systemic effects of our endogenous microbiomes, particularly with respect to the metabolic impact of our gut microbiota. The gut microbiome can be thought of as the first-pass metabolic organ affecting both our nutrient intake and our resistance to toxicants, and is likely to be both a signifier and player in the health differences arising from different diets.
A key issue for understanding the dynamics of the gut microbiome is that many of the microbial players are unknown and, even for well-characterized microbes, knowledge of their metabolic capacity is incomplete. While the quickening pace of sequencing technology has been extremely valuable, a major challenge is that genomes are all annotated based on similarity to known genes and function. We understand the function of metabolism in a handful (of related) microbes well, but for many others we have knowledge of only their central metabolism, and even those processes are not characterized sufficiently. Moreover, we have limited knowledge of regulation of metabolism, let alone how a microbial community is regulated as a whole.
In part due to this lack of knowledge and in part due to lack of physical theories that operate on the scale of biological processes, social theory (e.g., game theory) has been used to try and understand the dynamics of microbial communities. However, social theory falls short in that the molecular mechanisms that are responsible for emergent behavior of the community are not directly modeled (or understood), making this approach less useful for drug design.
Intermediate between social theory and complete physical models of microbial metabolism is constraint-based flux modeling,99 in which the external fluxes into and out of a system are used as constraints and internal fluxes are then optimized with respect to those constraints. If additional internal constraints are known, through isotope labeling experiments for example, then these fluxes can also be used as constraints. While these approaches have been enormously valuable in understanding microbial metabolism, they provide no information on metabolite concentrations and have large solution spaces even when additional physical constraints are added.
Physics based modeling has taken two opposing approaches to modeling microbial dynamics. The naive, but theoretically well-grounded, choice is to attempt to use mass action dynamics of metabolism to model enzymatic reactions. However, this approach is not feasible beyond small reaction networks due to the requirement for knowledge of the hundreds to tens of thousands of rate constants that are involved. An alternative choice is to model functional guilds, which can be thought of as microbial species grouped together based on shared/common metabolic capabilities and treated as a single modeling entity, an approach that was developed before genome sequencing was available and has continued to evolve.99 In modeling functional guilds, only a few summary reactions are used to represent the metabolic capability of each organism. Clearly, there is a tremendous need for development of more rigorous113 and complete models of microbial metabolism.
There are existing models of the systemic inflammatory response on which to build. Even with microbial détente the host must deal with persistent incursions from resident microbiota, to say nothing about the constant wear and tear on its own constituent tissues. The inflammatory response, at baseline, retains a persistent counterpoise between effective responsiveness (pro-inflammation) and sufficient attenuation (anti-inflammation), with recovery (healing) intimately tied to the anti-inflammatory response. Inflammation is therefore a classic robust adaptive control structure that has been evolutionarily optimized.24,33,34 Paradoxically, enhancements of human health have shifted that fitness landscape such that pre-technological adaptations for evolutionary fitness have been superseded so that inflammation enhances, if not directly contributes to, a newer class of diseases. Dysregulation of inflammation plays a role in a host of indolent diseases that most likely result from an inappropriate resetting of homeostatic set points. However, at an even more fundamental level, there is a challenge of parsing the dynamic range of a particular individual’s response, and determining where and how within their pathway control structure different trajectories are determined. To this end, we turn to perhaps the most dramatic case of inflammatory dysregulation, sepsis, and the role of multiscale modeling for dynamic knowledge representation and conceptual model verification.
Despite being the focus of extensive basic research and the target of over 20 clinical trials of anti-mediator therapies, there currently exists no approved pharmacological treatment specific for sepsis. The failure of the initial set of anti-mediator clinical trials in the 1990s may be one of the first explicit examples of the Translational Dilemma, i.e., the inability to effectively and efficiently translate basic mechanistic knowledge, developed based on in vitro data, into clinically successful therapeutics,6,7 and spurred the initial steps in the computational multiscale modeling of acute systemic inflammation.4,5,22,60 One of the primary insights gained from that experience is the need for multiscale computational modeling as a means of testing whether the mechanistic hypotheses concatenated from pre-clinical experimental results actually behave in the manner expected. This approach was demonstrated in an early example of cell-level agent-based modeling used to generate an in silico trial population.5 An ABM of systemic inflammation was developed based on the prevailing conceptual model of how systemic inflammation worked, and, importantly, represented the conceptual basis for the design of anti-mediator therapies for sepsis that ultimately failed. The purpose of the ABM was to determine whether, if these interventions behaved exactly as they were supposed to have behaved, would they have impacted survival in a simulated clinical population? In addition to the existing clinical trials, potential multi-modal/combination therapies were simulated in an attempt to address the concern that pathway redundancies limited the efficacy of an intervention. The result that none of the simulated interventions demonstrated a survival benefit is both not surprising (since it was known that the trials had failed) and enlightening (since none of the combination therapies worked either). The conclusion drawn was that there was a fundamental conceptual flaw in the design of those interventions, a flaw that would have been evident had such a means of dynamic knowledge representation been utilized as part of the standard drug development pipeline. This approach has been expanded to other disease processes8,52,73,118,138 and shows promise as an additional means of assessing the impact of pathway complexity/cell population heterogeneity on putative control strategies/therapies.
Multi-tissue signaling must also be incorporated into clinically relevant modeling. Our persistent theme in this section has been the concept that both microbial and inflammatory systems produce both beneficial and detrimental effects. To add to the complexity of these interactions is the fact that the different tissues and body compartments are in constant communication. Engineering effective therapeutics requires us to acknowledge this reality and develop rational strategies for predicting and capitalizing on that communication. One such strategy involves understanding the systemic transport of locally produced signals. Systemic effects arise when local signals are transported to other parts of the body, primarily via the systemic circulation.
Understanding, and being able to model and hence predict, this systemic communication is key to understanding systemic multiscale and “systems biology” effects. The movement of chemicals through the body is generally represented using Physiologically-Based Pharmacokinetic (PBPK) modeling techniques, which were developed primarily to model the movement of drugs through the body.91 The same techniques can be applied to modeling the movement of locally generated signals that give rise to systemic responses (hence coupling local behaviors to other bodily sites) and in modeling the time evolution of clinical markers used to guide clinical interventions.90 There is a critical need to develop robust “hardened” models that, in addition to xenobiotics, can model the transfer of systemic signals. The conversion of “pharmacokinetic” to “metabolokinetic” modeling is needed to understand systemic signaling and clinical markers, to capture the systemic communication between host and microbiome, and ultimately to predict local concentrations of agents (both exogenous and endogenous) at the site of inflammation.
Computer-Aided Design of Nanomedical Systems
Concepts from multiscale modeling for NP and NC targeted delivery as well as host-pathogen interactions described above can also be intertwined in developing nanomedical systems, including nanoparticle vaccines, nanoreactors, and nanocapsules for the targeted delivery of small therapeutic molecules, genes, or imaging agents. To achieve the clinical objective of these nanosystems, a computer-aided design (CAD) approach is needed to meet the many, often conflicting, requirements; for example, nanomedical systems must (1) avoid complexing with nontarget proteins and cells, (2) strongly interact with target objects, and (3) have longtime thermal-chemical stability. Since purely laboratory or clinically based approaches are time and resource demanding, a CAD approach is of great interest.
A CAD approach generally involves a computational model and ways to translate the computer simulation results into clinical or laboratory relevant information (Fig. 10).45 In this section, the special multiscale challenge and simulation for nanomedical system are illustrated with the nanoparticle-based vaccines CAD, although the discussion readily generalizes to other nanomedical systems.

Schematic computer-aided vaccine design workflow. Candidate nanoparticles are simulated and the most promising ones are synthesized experimentally and immunologically tested.
Nanoparticles free of genetic information have been in clinical use to provide protection against human papillomavirus (HPV),15,31,65 and others are in development or clinical trials. The general objective behind nanoparticle-based vaccine research is to design a nanoparticle that elicits a neutralizing antibody response from the immune system. However, this effort suffers from two challenges. First, a nanomedical system with surrounding microenvironment contains millions of atoms and thus using conventional computer simulation methods is a burden on computational resources, especially in light of the many longtime simulations in the course of a CAD study. The second challenge is to use the simulation results to provide a preclinical assay that predicts vaccine efficacy. In the following, these challenges will be addressed via multiscale MD simulations and an integrated physics-based bioinformatics methodology, respectively.1,45
Multiscale MD approaches are employed for performing nanoparticle (NP) simulation.1 Successful vaccines of the NP type are based on a NP which, along with its microenvironment, constitutes a supramillion atom system (Fig. 11). Conventional atom-resolved molecular dynamics provides a possibility of a calibration-free approach, i.e., only a well-tested interatomic force field should be required and there is a wealth of experience in the development of interatomic force fields.10,105,120 However, the large number of atoms involved, the longtime simulation, and the many simulations needed in a typical CAD study make conventional MD an impractical basis of vaccine CAD.

Snapshots of the initial and final configurations of (a) human papillomavirus (HPV) T = 1 virus like particle (VLP) undergoing thermal fluctuations. After 29 ns, the system has slightly changed signifying the VLP is stable and has reached equilibrium, and (b) P22 T = 1 VLP significantly evolving from its initial symmetrical icosahedral form, which implies the VLP is unstable.
Multiscale methods1,2 are making all-atom MD increasingly efficient, reducing one or more orders of magnitude in simulation time without (1) significant loss of accuracy, (2) the need for calibration with each new system considered, and (3) uncertainties in hypothesized coarse-grained (CG) governing equation. Also, the availability of atom-resolved imaging technologies now enables rigorous testing of multiscale algorithms. Notions such as quasiequivalence131 are making rigorous comparison of multiscale software feasible (notably since even two conventional MD simulations may defer due to the orbital instability of classical trajectories). Thus an accurate, atom-resolved model free of CG phenomenology and the need for experiment laboratory testing is essential for arriving at a practical nanomedical system CAD.1
A promising solution to the nanomedical system computational challenges is the multiscale factorization (MF) algorithm.2 MF is based on the notion that CG variables guide the ensemble of microstates, while the latter provide the Newtonian mechanics and interatomic force fields that make the simulation well-grounded in physical principles. This scheme also involves the use of special CG variables which depend on the microstate (i.e., the positions and momenta of all atoms). These CG variables are designed to evolve with minimal microstate-generated stochastics fluctuations.96 Minimization of the noise effects enables the CG state to be advanced with large timesteps. Lie-Trotter factorization47,116 is used to rigorously coevolve CG and microscopic states in MF.1,2
Multiscale bioinformatics methods have utility for assessing immunogenicity.45,55 To achieve vaccine CAD, an approach is needed that enables predication of the clinically relevant neutralizing antibody response from microscale information on a NP. This connection is via a complex network that is not yet completely qualitatively understood, nor are completely quantitatively model available that connect cell membrane-localized receptor cite processes to the neutralizing antibody response. To address this gap, an essential step in vaccine CAD, a bioinformatics method is being developed as follows.45 Epitopes are peptide sequences read, e.g., by B-cells, which elicits neutralizing antibody responses. The idea of the immunoinformatics approach is to establish a correlation between epitope molecular-scale properties and the immune response. Such a correlation has been proposed, the epitope fluctuation-immunogenicity correlation (Fig. 12).45 The fluctuations intensity can only be reliably used when a molecular metric is computed for a nanoparticle that has been well-equilibrated. This suggests the role of multiscale MD to achieve the atom-resolved, longtime, and equilibration simulations.1

Experimentally determined immunogenicity is inversely correlated with calculated mean-square fluctuations, a measure of flexibility. The experimental immunogenicity is coarse-grained from 0–1 to provide a semiquantitative link between experimental and computational measurements.
Summary
A variety of multiscale modeling and simulation techniques from the atomic scale to the protein, cell, tissue, organ and even the whole body are now being developed to allow for in silico drug design and development. A variety of modeling approaches are being employed, including molecular dynamics, bioinformatics, stochastic approaches, ordinary and partial differential equation approaches and dynamical systems and statistical analyses, among others. The goals of these efforts is to develop computational multiscale models and processes that will ultimately allow for improved efficiency, specificity, sensitivity, accuracy and cost-effectiveness in drug development, screening and delivery. The multiscale modeling and simulation approaches that are currently in development may ultimately be applied to various contexts of use for multiple physiological and pathophysiological systems.
There are still multiple barriers that must be overcome prior to successful clinical translation of drug development, drug design and drug delivery multiscale models. These include intrinsic limitations of the different in silico techniques on which the models are based. For example, MD simulations of ion channel interactions for drug development and evaluation can be constrained by the limitations of current force-fields. The quantum mechanics/molecular mechanics (QM/MM) approach further enables study of chemical processes involving proteins and solutions and can be employed to address these limitations. Likewise, the inherent loss of atomistic information that occurs in CG simulations could potentially be addressed by conversion of CG to atomistic simulations (for which tools exist), followed by performing minimization to enhance system accuracy. Other limitations for modeling in general include the enormous computing power and memory required to perform certain computational tasks, and the problems associated with “big data” in how to glean insight from massive data sets generated from these techniques. There is also a pressing need for community engagement to specifically define the context of use and application for computational multiscale models. Models need to be extensively tested and thoroughly validated within the defined context of use. Some of the modeling approaches that are being developed are extendable to include specific patient data to personalize the models to improve predictive value. Indeed, mechanism-based multiscale models that include patient specific parameters are an important complement to purely inferential (statistical) approaches to personalized medicine. Collection of patient data might include genotype, hormone/metabolic/endocrine status, co-morbidities and associated disease states and this collection may constitute an additional barrier to multiscale model implementation. Once these, and other identified barriers, are overcome, the computational processes are expected to have broad impact in the regulatory process prior to drug approval, in academia for research, in industry for drug and disease screening, and for patient oriented medicine in the clinic.
Abbreviations
- ABM:
-
Agent-based model
- AMD:
-
AMD3100
- BMSC:
-
Bone marrow stromal cell
- BSV:
-
Between-subject variability
- BZM:
-
Bortezomib
- CAD:
-
Computer-aided design
- CG:
-
Coarse-grained
- CXCR4:
-
C-X-C chemokine receptor type 4
- DPD:
-
Dissipative particle dynamics
- ECM:
-
Extracellular matrix
- EPR:
-
Enhanced permeability and retention
- HPV:
-
Human papillomavirus
- IFP:
-
Interstitial fluid pressure
- MC:
-
Monte Carlo
- MCMC:
-
Markov chain Monte Carlo
- MD:
-
Molecular dynamics
- MF:
-
Multiscale factorization
- MIC:
-
Myeloma initiating cell
- MM:
-
Multiple myeloma
- NC:
-
Nanocarrier
- NP:
-
Nanoparticle
- NS:
-
Navier–Stokes
- PBPK:
-
Physiologically-based pharmacokinetic
- PC:
-
Cancer progenitor cell
- PDB:
-
Protein Data Bank
- QM/MM:
-
Quantum mechanics/molecular mechanics
- RUV:
-
Residual unknown variability
- SCB:
-
Systems chemical biology
- SDF1:
-
Stromal cell-derived factor 1
- TMM:
-
Terminal multiple myeloma cell
- WHAM:
-
Weighted histogram analysis method
References
Abi Mansour, A. A., Sereda, Y. V., Yang, J. & Ortoleva, P. J. Prospective on multiscale simulation of virus-like particles: Application to computer-aided vaccine design. Vaccine, 2015. doi:10.1016/j.vaccine.2015.05.099.
Abi Mansour, A., and P. J. Ortoleva. Multiscale factorization method for simulating mesoscopic systems with atomic precision. J. Chem. Theory Comput. 10:518–523, 2014. doi:10.1021/ct400615a.
Adra, S., T. Sun, S. MacNeil, M. Holcombe, and R. Smallwood. Development of a three dimensional multiscale computational model of the human epidermis. PLoS ONE 5:e8511, 2010. doi:10.1371/journal.pone.0008511.
An, G. Agent-based computer simulation and SIRS: Building a bridge between basic science and clinical trials. Shock 16:266–273, 2001. doi:10.1097/00024382-200116040-00006.
An, G. In silico experiments of existing and hypothetical cytokine-directed clinical trials using agent-based modeling. Crit. Care Med. 32:2050–2060, 2004.
An, G., J. Bartels, and Y. Vodovotz. In silico augmentation of the drug development pipeline: examples from the study of acute inflammation. Drug Dev. Res. 72:187–200, 2011. doi:10.1002/ddr.20415.
An, G., and S. Christley. Addressing the translational dilemma: dynamic knowledge representation of inflammation using agent-based modeling. Crit. Rev. Biomed. Eng. 40:323–340, 2012.
An, G. Introduction of a framework for dynamic knowledge representation of the control structure of transplant immunology: employing the power of abstraction with a Solid Organ Transplant Agent-based Model (SOTABM). Front. Immunol. 6, 2015. doi:10.3389/fimmu.2015.00561.
Arap, W., R. Pasqualini, and E. Ruoslahti. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279:377–380, 1998.
Baker, C. M., V. M. Anisimov, and A. D. MacKerell. Development of CHARMM polarizable force field for nucleic acid bases based on the classical drude oscillator model. J. Phys. Chem. B 115:580–596, 2010. doi:10.1021/jp1092338.
Beal, S. L., and L. B. Sheiner. Estimating population-kinetics. Crc. Crit. Rev. Biomed. Eng. 8:195–222, 1982.
Brennan, T., M. Fink, and B. Rodriguez. Multiscale modelling of drug-induced effects on cardiac electrophysiological activity. Eur. J. Pharm. Sci. 36:62–77, 2009. doi:10.1016/j.ejps.2008.09.013.
Britton, O. J., et al. Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc. Natl. Acad. Sci. USA 110:E2098–E2105, 2013. doi:10.1073/pnas.1304382110.
Brown, J. B., and Y. Okuno. Systems biology and systems chemistry: new directions for drug discovery. Chem. Biol. 19:23–28, 2012. doi:10.1016/j.chembiol.2011.12.012.
Brown, D. R., et al. The humoral response to Gardasil® over four years as defined by Total IgG and competitive Luminex immunoassay. Hum. Vaccines 7:230–238, 2011. doi:10.4161/hv.7.2.13948.
Carpenter, T. S., E. Y. Lau, and F. C. Lightstone. Identification of a possible secondary picrotoxin-binding site on the GABAA receptor. Chem. Res. Toxicol. 26:1444–1454, 2013. doi:10.1021/tx400167b.
Champion, J. A., and S. Mitragotri. Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. USA 103:4930–4934, 2006.
Chauhan, V. P., et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 7:383–388, 2012. doi:10.1038/nnano.2012.45.
Christley, S., C. Cockrell, and G. An. Computational studies of the intestinal host-microbiota interactome. Computation 3:2079–3197, 2015. doi:10.3390/computation3010002.
Clancy, C. E., Z. I. Zhu, and Y. Rudy. Pharmacogenetics and anti-arrhythmic drug therapy: a theoretical investigation. Am. J. Physiol. Heart Circ. Physiol. 292:H66–H75, 2007. doi:10.1152/ajpheart.00312.2006.
Clegg, L. E., and F. Mac Gabhann. Molecular mechanism matters: Benefits of mechanistic computational models for drug development. Pharmacol. Res. 99:149–154, 2015. doi:10.1016/j.phrs.2015.06.002.
Clermont, G., et al. In silico design of clinical trials: a method coming of age. Crit. Care Med. 32:2061–2070, 2004.
Collins, T. A., et al. Modeling and simulation approaches for cardiovascular function and their role in safety assessment. CPT Pharmacomet. Syst. Pharmacol. 4:e00018, 2015. doi:10.1002/psp4.18.
Csete, M., and J. Doyle. Bow ties, metabolism and disease. Trends Biotechnol. 22:446–450, 2004. doi:10.1016/j.tibtech.2004.07.007.
Davis, I. W., and D. Baker. RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 385:381–392, 2009. doi:10.1016/j.jmb.2008.11.010.
Davis, M. E., Z. Chen, and D. M. Shin. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat. Rev. Drug. Discov. 7:771–782, 2008.
Decuzzi, P., and M. Ferrari. The adhesive strength of non-spherical particles mediated by specific interactions. Biomaterials 27:5307–5314, 2006. doi:10.1016/j.biomaterials.2006.05.024.
Decuzzi, P., S. Lee, B. Bhushan, and M. Ferrari. A theoretical model for the margination of particles within blood vessels. Ann. Biomed. Eng. 33:179–190, 2005. doi:10.1007/s10439-005-8976-5.
Decuzzi, P., S. Lee, M. Decuzzi, and M. Ferrari. Adhesion of microfabricated particles on vascular endothelium: a parametric analysis. Ann. Biomed. Eng. 32:793–802, 2004.
Deisboeck, T. S., Wang, Z., Macklin, P. & Cristini, V. Multiscale cancer modeling. Annu. Rev. Biomed. Eng. 13, 2011.
Deschuyteneer, M., et al. Molecular and structural characterization of the L1 virus-like particles that are used as vaccine antigens in Cervarix™, the AS04-adjuvanted HPV-16 and -18 cervical cancer vaccine. Hum. Vaccines 6:407–419, 2010.
Di Veroli, G. Y., M. R. Davies, H. Zhang, N. Abi-Gerges, and M. R. Boyett. High-throughput screening of drug-binding dynamics to HERG improves early drug safety assessment. Am. J. Physiol. Heart Circ. Physiol. 304:H104–H117, 2013. doi:10.1152/ajpheart.00511.2012.
Doyle, J., and M. Csete. Motifs, control, and stability. PLoS Biol. 3:e392, 2005. doi:10.1371/journal.pbio.0030392.
Doyle, J. C., and M. Csete. Architecture, constraints, and behavior. Proc. Natl. Acad. Sci. USA 108(Suppl 3):15624–15630, 2011. doi:10.1073/pnas.1103557108.
Dux-Santoy, L., R. Sebastian, J. Felix-Rodriguez, J. M. Ferrero, and J. Saiz. Interaction of specialized cardiac conduction system with antiarrhythmic drugs: a simulation study. IEEE Trans. Bio-med. Eng. 58:3475–3478, 2011. doi:10.1109/TBME.2011.2165213.
Fang, J., H. Nakamura, and H. Maeda. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 63:136–151, 2011.
Fang, X., A. Wallqvist, and J. Reifman. A systems biology framework for modeling metabolic enzyme inhibition of Mycobacterium tuberculosis. BMC Syst. Biol. 3:92, 2009. doi:10.1186/1752-0509-3-92.
Fang, X., A. Wallqvist, and J. Reifman. Modeling synergistic drug inhibition of Mycobacterium tuberculosis growth in murine macrophages. Mol. BioSyst. 7:2622–2636, 2011. doi:10.1039/c1mb05106g.
Finley, S. D., P. Angelikopoulos, P. Koumoutsakos, and A. S. Popel. Pharmacokinetics of anti-VEGF agent aflibercept in cancer predicted by data driven, molecular-detailed model. CPT Pharmacomet. Syst. Pharmacol. 4(11):641–649, 2015.
Finley, S. D., L. H. Chu, and A. S. Popel. Computational systems biology approaches to anti-angiogenic cancer therapeutics. Drug Discov. Today 20:187–197, 2015. doi:10.1016/j.drudis.2014.09.026.
Fogelson, A. L., and R. D. Guy. Immersed-boundary-type models of intravascular platelet aggregation. Comput. Method Appl. M 197:2087–2104, 2008. doi:10.1016/j.cma.2007.06.030.
Garfinkel, A., et al. Preventing ventricular fibrillation by flattening cardiac restitution. Proc. Natl. Acad. Sci. USA 97:6061–6066, 2000. doi:10.1073/pnas.090492697.
Geerts, H., A. Spiros, P. Roberts, and R. Carr. Has the time come for predictive computer modeling in CNS drug discovery and development? CPT Pharmacomet. Syst. Pharmacol. 1:e16, 2012. doi:10.1038/psp.2012.17.
Geng, Y., et al. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2:249–255, 2007.
Grosch, J., J. Yang, A. Shen, Y. V. Sereda, and P. Ortoleva. Broad spectrum assessment of the epitope fluctuation—immunogenicity hypothesis. Vaccine 2015. doi:10.1016/j.vaccine.2015.06.111.
Haddish-Berhane, N., J. L. Rickus, and K. Haghighi. The role of multiscale computational approaches for rational design of conventional and nanoparticle oral drug delivery systems. Int. J. Nanomed. 2:315, 2007.
Hall, B. Lie Groups, Lie Algebras, and Representations: An Elementary Introduction. Berlin: Springer, 2004.
Haun, J. B., and D. A. Hammer. Quantifying nanoparticle adhesion mediated by specific molecular interactions. Langmuir 24:8821–8832, 2008.
Heldin, C.-H., K. Rubin, K. Pietras, and A. Ostman. High interstitial fluid pressure [mdash] an obstacle in cancer therapy. Nat. Rev. Cancer 4:806–813, 2004.
Holford, N. H. G., H. C. Kimko, J. P. R. Monteleone, and C. C. Peck. Simulation of clinical trials. Annu. Rev. Pharmacol. 40:209–234, 2000. doi:10.1146/Annurev.Pharmtox.40.1.209.
Hossain, S. S., et al. In silico vascular modeling for personalized nanoparticle delivery. Nanomedicine 8:343–357, 2013.
Hunt, C. A., R. C. Kennedy, S. H. Kim, and G. E. Ropella. Agent-based modeling: a systematic assessment of use cases and requirements for enhancing pharmaceutical research and development productivity. Wiley Interdiscip Rev Syst Biol Med 5:461–480, 2013. doi:10.1002/wsbm.1222.
Hyakutake, T., and S. Nagai. Numerical simulation of red blood cell distributions in three-dimensional microvascular bifurcations. Microvasc. Res. 97:115–123, 2015. doi:10.1016/j.mvr.2014.10.001.
Jain, R. K. Barriers to drug delivery in solid tumors. Sci. Am. 58–65:e29528, 1994.
Joshi, H., S. Cheluvaraja, E. Somogyi, D. R. Brown, and P. J. Ortoleva. A molecular dynamics study of loop fluctuation in human papillomavirus type 16 virus-like particles: a possible indicator of immunogenicity. Vaccine 29:9423–9430, 2011. doi:10.1016/j.vaccine.2011.10.039.
Kakde, D., D. Jain, V. Shrivastava, R. Kakde, and A. Patil. Cancer therapeutics-opportunities, challenges and advances in drug delivery. J. Appl. Pharm. Sci. 1(9):1–10, 2011.
Kim, M., R. J. Gillies, and K. A. Rejniak. Current advances in mathematical modeling of anti-cancer drug penetration into tumor tissues. Front. Oncol. 3:120, 2013.
King, M. R., and D. A. Hammer. Multiparticle adhesive dynamics: hydrodynamic recruitment of rolling leukocytes. Proc. Natl. Acad. Sci. USA 98:14919–14924, 2001. doi:10.1073/pnas.261272498.
King, M. R., S. D. Rodgers, and D. A. Hammer. Hydrodynamic collisions suppress fluctuations in the rolling velocity of adhesive blood cells. Langmuir 17:4139–4143, 2001. doi:10.1021/La010234b.
Kumar, R., G. Clermont, Y. Vodovotz, and C. C. Chow. The dynamics of acute inflammation. J. Theor. Biol. 230:145–155, 2004. doi:10.1016/j.jtbi.2004.04.044.
Lalonde, R. L., et al. Model-based drug development. Clin. Pharmacol. Ther. 82:21–32, 2007. doi:10.1038/sj.clpt.6100235.
Li, X. L., K. L. Yao, and Z. L. Liu. CFD study on the magnetic fluid delivering in the vessel in high-gradient magnetic field. J. Magn. Magn. Mater. 320:1753–1758, 2008. doi:10.1016/j.jmmm.2008.01.041.
Liu, J., R. Bradley, D. M. Eckmann, P. S. Ayyaswamy, and R. Radhakrishnan. Multiscale modeling of functionalized nanocarriers in targeted drug delivery. Curr. Nanosci. 7:727, 2011.
Liu, J., et al. Computational model for nanocarrier binding to endothelium validated using in vivo, in vitro, and atomic force microscopy experiments. Proc. Natl. Acad. Sci. 107:16530–16535, 2010. doi:10.1073/pnas.1006611107.
Lowy, D. R., and J. T. Schiller. Prophylactic human papillomavirus vaccines. J. Clin. Invest. 116:1167–1173, 2006. doi:10.1172/JCI28607.
Lu, D., et al. Model-based meta-analysis for quantifying Paclitaxel dose response in cancer patients. CPT Pharmacomet. Syst. Pharmacol. 3:e115, 2014. doi:10.1038/psp.2014.14.
Mahmoudi, M., et al. Multiphysics flow modeling and in vitro toxicity of iron oxide nanoparticles coated with poly(vinyl alcohol). J. Phys. Chem. C 113:2322–2331, 2009. doi:10.1021/jp809453v.
Malisi, C., et al. Binding pocket optimization by computational protein design. PLoS ONE 7:e52505, 2012. doi:10.1371/journal.pone.0052505.
Mandell, D. J., E. A. Coutsias, and T. Kortemme. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 6:551–552, 2009.
May, E. E., A. Leitao, A. Tropsha, and T. I. Oprea. A systems chemical biology study of malate synthase and isocitrate lyase inhibition in Mycobacterium tuberculosis during active and NRP growth. Comput. Biol. Chem. 47:167–180, 2013. doi:10.1016/j.compbiolchem.2013.07.002.
May, E., et al. Understanding virulence mechanisms in M. tuberculosis infection via a circuit-based simulation framework. Conf. Proc. IEEE Eng. Med. Biol. Soc. 4953–4955:2008, 2008. doi:10.1109/IEMBS.2008.4650325.
Meiler, J., and D. Baker. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins 65:538–548, 2006. doi:10.1002/prot.21086.
Mi, Q., B. Riviere, G. Clermont, D. L. Steed, and Y. Vodovotz. Agent-based model of inflammation and wound healing: insights into diabetic foot ulcer pathology and the role of transforming growth factor-beta1. Wound Repair Regen. 15:671–682, 2007. doi:10.1111/j.1524-475X.2007.00271.x.
Milligan, P. A., et al. Model-based drug development: a rational approach to efficiently accelerate drug development. Clin. Pharmacol. Ther. 93:502–514, 2013. doi:10.1038/clpt.2013.54.
Modok, S., et al. Transport kinetics of four-coordinate and six-coordinate platinum compounds in the multicell layer tumour model. Br. J. Cancer 97:194–200, 2007. doi:10.1038/sj.bjc.6603854.
Moreno, J. D., et al. A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci. Trans. Med. 3:98ra83, 2011. doi:10.1126/scitranslmed.3002588.
Moreno, J. D., et al. Ranolazine for congenital and acquired late INa-linked arrhythmias: in silico pharmacological screening. Circ. Res. 113:e50–e61, 2013. doi:10.1161/CIRCRESAHA.113.301971.
Mould, D. R. Model-based meta-analysis: an important tool for making quantitative decisions during drug development. Clin. Pharmacol. Ther. 92:283–286, 2012. doi:10.1038/clpt.2012.122.
Mould, D. R. Models for disease progression: new approaches and uses. Clin. Pharmacol. Ther. 92:125–131, 2012. doi:10.1038/clpt.2012.53.
Muller, K., Fedosov, D. A. & Gompper, G. Margination of micro- and nano-particles in blood flow and its effect on drug delivery. Sci. Rep. 4: doi:10.1038/srep04871. http://www.nature.com/srep/2014/140502/srep04871/abs/srep04871.html—supplementary-information, 2014.
Nakamura, H., et al. progesterone regulates cardiac repolarization through a nongenomic pathway: an in vitro patch-clamp and computational modeling study. Circulation 116:2913–2922, 2007. doi:10.1161/circulationaha.107.702407.
Obiol-Pardo, C., J. Gomis-Tena, F. Sanz, J. Saiz, and M. Pastor. A multiscale simulation system for the prediction of drug-induced cardiotoxicity. J. Chem. Inf. Model. 51:483–492, 2011. doi:10.1021/ci100423z.
Oprea, T. I., E. E. May, A. Leitao, and A. Tropsha. Computational systems chemical biology. Methods Mol. Biol. 672:459–488, 2011. doi:10.1007/978-1-60761-839-3_18.
Oprea, T. I., A. Tropsha, J. L. Faulon, and M. D. Rintoul. Systems chemical biology. Nat. Chem. Biol. 3:447–450, 2007. doi:10.1038/nchembio0807-447.
Osborne, J. M., et al. A hybrid approach to multi-scale modelling of cancer. Philos. Trans. A Math. Phys. Eng. Sci. 368:5013–5028, 2010.
Pozrikidis, C. Numerical simulation of blood and interstitial flow through a solid tumor. J. Math. Biol. 60:75–94, 2010.
Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N. Engl. J. Med. 321:406–412, 1989.
Qu, Z., J. N. Weiss, and A. Garfinkel. Cardiac electrical restitution properties and stability of reentrant spiral waves: a simulation study. Am. J. Physiol. 276:H269–H283, 1999.
Recanatini, M., A. Cavalli, and M. Masetti. Modeling HERG and its interactions with drugs: recent advances in light of current potassium channel simulations. ChemMedChem 3:523–535, 2008. doi:10.1002/cmdc.200700264.
Remien, C. H., F. R. Adler, L. Waddoups, T. D. Box, and N. L. Sussman. Mathematical modeling of liver injury and dysfunction after acetaminophen overdose: Early discrimination between survival and death. Hepatology 56:727–734, 2012. doi:10.1002/hep.25656.
Sager, J. E., J. Yu, I. Ragueneau-Majlessi, and N. Isoherranen. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications and model verification. Drug Metab. Dispos. 2015. doi:10.1124/dmd.115.065920.
Sarkar, A. X., and E. A. Sobie. Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: a computational analysis. Heart Rhythm. 8:1749–1755, 2011. doi:10.1016/j.hrthm.2011.05.023.
Shah, D. K., N. Haddish-Berhane, and A. Betts. Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab-vedotin. J. Pharmacokinet Pharmacodyn. 39:643–659, 2012.
Shipley, R. J., and S. J. Chapman. Multiscale modelling of fluid and drug transport in vascular tumours. Bull. Math. Biol. 72:1464–1491, 2010. doi:10.1007/s11538-010-9504-9.
Siegel, R. L., K. D. Miller, and A. Jemal. Cancer statistics, 2015. CA Cancer J. Clin. 65:5–29, 2015. doi:10.3322/caac.21254.
Singharoy, A., H. Joshi, Y. Miao, and P. J. Ortoleva. Space warping order parameters and symmetry: application to multiscale simulation of macromolecular assemblies. J. Phys. Chem. B 116:8423–8434, 2012. doi:10.1021/jp2119247.
Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics (Oxford, England) 21:951–960, 2005.
Sohrabi, S., J. Zheng, E. A. Finol, and Y. Liu. Numerical simulation of particle transport and deposition in the pulmonary vasculature. J. Biomech. Eng. 136:121010, 2014.
Song, H.-S., W. Cannon, A. Beliaev, and A. Konopka. Mathematical modeling of microbial community dynamics: a methodological review. Processes 2:711, 2014.
Sorger, P. K. et al. Quantitative and systems pharmacology in the post-genomic era: new approaches to discovering drugs and understanding therapeutic mechanisms. An NIH White Paper by the QSP Workshop Group. (2011). https://www.nigms.nih.gov/training/documents/systemspharmawpsorger2011.pdf.
Stamatakos, G. S., E. A. Kolokotroni, D. D. Dionysiou, E. Georgiadi, and C. Desmedt. An advanced discrete state-discrete event multiscale simulation model of the response of a solid tumor to chemotherapy: mimicking a clinical study. J. Theor. Biol. 266:124–139, 2010. doi:10.1016/j.jtbi.2010.05.019.
Su, J. & Henson, M. A. Circadian gating of the mammalian cell cycle restriction point: a mathematical analysis. Life Sci. Lett. IEEE:1–1, 2015, doi:10.1109/LLS.2015.2449511.
Su, J., et al. Targeting the biophysical properties of the myeloma initiating cell niches: a pharmaceutical synergism analysis using multi-scale agent-based modeling. PLoS ONE 9:e85059, 2014.
Sun, X., et al. Cytokine combination therapy prediction for bone remodeling in tissue engineering based on the intracellular signaling pathway. Biomaterials 33:8265–8276, 2012.
Szklarczyk, O. M., S. J. Bachmann, and W. F. van Gunsteren. A polarizable empirical force field for molecular dynamics simulation of liquid hydrocarbons. J. Comput. Chem. 35:789–801, 2014. doi:10.1002/jcc.23551.
Tan, J., S. Shah, A. Thomas, H. D. Ou-Yang, and Y. Liu. The influence of size, shape and vessel geometry on nanoparticle distribution. Microfluidics Nanofluidics 14:77–87, 2013.
Tan, J., A. Thomas, and Y. Liu. Influence of red blood cells on nanoparticle targeted delivery in microcirculation. Soft. Matter. 8(6):1934–1946, 2012.
Tan, J., S. Wang, J. Yang, and Y. Liu. Coupled particulate and continuum model for nanoparticle targeted delivery. Comput. Struct. 122:128–134, 2013.
Tang, L., et al. Computational modeling of 3D tumor growth and angiogenesis for chemotherapy evaluation. PLoS ONE 9:e83962, 2014. doi:10.1371/journal.pone.0083962.
ten Tusscher, K. H. W. J., and A. V. Panfilov. Alternans and spiral breakup in a human ventricular tissue model. Am. J. Physiol. Heart Circ. Physiol. 291:H1088–H1100, 2006.
The Sicilian gambit. A new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. Task Force of the Working Group on Arrhythmias of the European Society of Cardiology. Circulation 84:1831–1851, 1991.
The ‘Sicilian Gambit’. A new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanisms. The Task Force of the Working Group on Arrhythmias of the European Society of Cardiology. Eur. Heart J. 12:1112–1131, 1991.
Thomas, D. G., S. Jaramillo-Riveri, D. J. Baxter, and W. R. Cannon. Comparison of optimal thermodynamic models of the tricarboxylic acid cycle from heterotrophs, cyanobacteria, and green sulfur bacteria. J. Phys. Chem. B 2014. doi:10.1021/jp5075913.
Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 63:131–135, 2011.
Trivedi, A., R. E. Lee, and B. Meibohm. Applications of pharmacometrics in the clinical development and pharmacotherapy of anti-infectives. Expert Rev. Clin. Phar. 6:159–170, 2013. doi:10.1586/ECP.13.6.
Trotter, H. F. On the product of semi-groups of operators. Proc. Am. Math. Soc. 10:545–551, 1959. doi:10.1090/S0002-9939-1959-0108732-6.
Uma, B., T. N. Swaminathan, R. Radhakrishnan, D. M. Eckmann, and P. S. Ayyaswamy. Nanoparticle Brownian motion and hydrodynamic interactions in the presence of flow fields. Phys Fluids 23:073602, 2011. doi:10.1063/1.3611026.
Uppal, A., S. C. Wightman, S. Ganai, R. R. Weichselbaum, and G. An. Investigation of the essential role of platelet-tumor cell interactions in metastasis progression using an agent-based model. Theor. Biol. Med. Model 11:17, 2014. doi:10.1186/1742-4682-11-17.
van der Graaf, P. H. CPT: pharmacometrics and systems pharmacology. CPT Pharmacomet. Syst. Pharmacol 1:e8, 2012. doi:10.1038/psp.2012.8.
Vanommeslaeghe, K., et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31:671–690, 2009.
Vicini, P., and B. P. Smith. Whither pharmacometrics?: present state and future choices. Clin. Pharmacol. Ther. 95:567–571, 2014. doi:10.1038/clpt.2014.72.
Vicini, P., and P. H. van der Graaf. Systems pharmacology for drug discovery and development: paradigm shift or flash in the pan? Clin. Pharmacol. Ther. 93:379–381, 2013. doi:10.1038/clpt.2013.40.
Waldo, A. L., et al. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol. Lancet 348:7–12, 1996.
Wang, C., P. Bradley, and D. Baker. Protein-protein docking with backbone flexibility. J. Mol. Biol. 373:503–519, 2007. doi:10.1016/j.jmb.2007.07.050.
Wang, Z., and T. S. Deisboeck. Mathematical modeling in cancer drug discovery. Drug Discov. Today 19:145–150, 2014. doi:10.1016/j.drudis.2013.06.015.
Weiss, J. N., et al. Electrical restitution and cardiac fibrillation. J. Cardiovasc. Electrophysiol. 13:292–295, 2002.
Woltosz, W. S. If we designed airplanes like we design drugsaEuro broken vertical bar. J. Comput. Aid. Mol. Des. 26:159–163, 2012. doi:10.1007/s10822-011-9490-5.
Xie, Y., L. T. Izu, D. M. Bers, and D. Sato. Arrhythmogenic transient dynamics in cardiac myocytes. Biophys. J . 106:1391–1397, 2014. doi:10.1016/j.bpj.2013.12.050.
Yang, K. & Ma, Y.-Q. Computer simulation of the translocation of nanoparticles with different shapes across a lipid bilayer. Nat. Nano 5:579–583. http://www.nature.com/nnano/journal/v5/n8/abs/nnano.2010.141.html—supplementary-information, 2010.
Yang, P. C., J. Kurokawa, T. Furukawa, and C. E. Clancy. Acute effects of sex steroid hormones on susceptibility to cardiac arrhythmias: a simulation study. PLoS Comput. Biol. 6:e1000658, 2010. doi:10.1371/journal.pcbi.1000658.
Yang, J., A. Singharoy, Y. V. Sereda, and P. J. Ortoleva. Quasiequivalence of multiscale coevolution and ensemble MD simulations: a demonstration with lactoferrin. Chem. Phys. Lett. 616–617:154–160, 2014. doi:10.1016/j.cplett.2014.10.020.
Zemzemi, N., et al. Computational assessment of drug-induced effects on the electrocardiogram: from ion channel to body surface potentials. Br. J. Pharmacol. 168:718–733, 2013. doi:10.1111/j.1476-5381.2012.02200.x.
Zeng, X., and S. Li. Multiscale modeling and simulation of soft adhesion and contact of stem cells. J. Mech. Behav. Biomed. Mater. 4:180–189, 2011. doi:10.1016/j.jmbbm.2010.06.002.
Zhang, J. Z., et al. Structure-function map of the receptor site for beta-scorpion toxins in domain II of voltage-gated sodium channels. J. Biol. Chem. 286:33641–33651, 2011. doi:10.1074/jbc.M111.282509.
Zhang, J. Z., et al. Mapping the interaction site for a beta-scorpion toxin in the pore module of domain III of voltage-gated Na(+) channels. J. Biol. Chem. 287:30719–30728, 2012. doi:10.1074/jbc.M112.370742.
Zhou, Q., G. C. Bett, and R. L. Rasmusson. Markov models of use-dependence and reverse use-dependence during the mouse cardiac action potential. PLoS ONE 7:e42295, 2012. doi:10.1371/journal.pone.0042295.
Zhu, Z. I., and C. E. Clancy. L-type Ca2+channel mutations and T-wave alternans: a model study. Am. J. Physiol. Heart Circ. Physiol. 293:H3480–H3489, 2007. doi:10.1152/ajpheart.00476.2007.
Ziraldo, C., et al. A computational, tissue-realistic model of pressure ulcer formation in individuals with spinal cord injury. PLoS Comput. Biol. 11:e1004309, 2015. doi:10.1371/journal.pcbi.1004309.
Acknowledgments
This work was supported in part by NIH grants R01CA138264 (ASP), U01HL126273 (CEC), U01EB016027 (DME), R01EB006818 (DME), R01-GM-115839 and P30-DK-42086 (GA), R01GM077138 (JPS) and R15EB015105 (YL) as well as EPA grant R835001 (JPS). WRC was funded under the Laboratory Directed Research Program at the Pacific Northwest National Laboratory. PNNL is operated by Battelle for the U.S. Department of Energy under Contract DE-AC06-76RLO.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Associate Editor Thomas Yankeelov oversaw the review of this article.
Rights and permissions
About this article

Cite this article
Clancy, C.E., An, G., Cannon, W.R. et al. Multiscale Modeling in the Clinic: Drug Design and Development. Ann Biomed Eng 44, 2591–2610 (2016). https://doi.org/10.1007/s10439-016-1563-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10439-016-1563-0