
Abstract
Cell surface engineering was proven as the efficient strategy for enhanced production of target metabolites. In this study, we want to improve the yield of target protein by engineering cell surface in Bacillus licheniformis. First, our results confirmed that deletions of d-alanyl-lipoteichoic acid synthetase gene dltD, cardiolipin synthase gene clsA and CDP-diacylglycerol-serine O-phosphatidyltransferase gene pssA were not conducive to cell growth, and the biomass of gene deletion strains were, respectively, decreased by 10.54 ± 1.43%, 14.17 ± 1.51%, and 17.55 ± 1.28%, while the concentrations of total extracellular proteins were improved, due to the increases of cell surface net negative charge and cell membrane permeability. In addition, the activities of target proteins, nattokinase, and α-amylase were also improved significantly in gene deletion strains. Furthermore, the triplicate gene (dltD, clsA, and pssA) deletion strain was constructed, which further led to the 45.71 ± 2.43% increase of cell surface net negative charge and 26.45 ± 2.31% increase of cell membrane permeability, and the activities of nattokinase and α-amylase reached 37.15 ± 0.89 FU/mL and 305.3 ± 8.4 U/mL, increased by 46.09 ± 3.51% and 96.34 ± 7.24%, respectively. Taken together, our results confirmed that cell surface engineering via deleting dltD, clsA, and pssA is an efficient strategy for enhanced production of target proteins, and this research provided a promising host strain of B. licheniformis for efficient protein expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heterologous expression has been proven to be an effective approach for target protein production. Recently, many expression systems (Escherichia coli, Bacillus, Saccharomyces cerevisiae, insect cells, mammalian cells, etc.) have been developed for protein production [4], among these systems, Bacillus was regarded as the most characteristic host for large-scale production of target proteins (e.g., industrial enzymes, active peptides, vaccines, etc.), as it owns the features of non-toxicity, convenient for gene modification, high capability for protein secretion, and excellent fermentation characteristics [15, 35].
Previously, various strategies have been developed to improve heterologous protein production, and the promoters [40], 5′-UTR [17], signal peptides [14, 37], signal peptidases [28], secretory components [9] were engineered for high-level production of target proteins [20]. Currently, cell surface engineering has become an attractive and promising strategy for product production [32]. Overexpression of phospholipase C from Bacillus cereus enhanced the activities of β-galactosidase, CGTase, xylanase in E. coli [29]. Strengthening d,d-carboxypeptidase genes dacA, dacB expression enhanced both the outer and inner membrane permeability, which further benefited green fluorescent protein (GFP), recombinant amylase, and α-galactosidase production in E. coli [39]. Moreover, overexpressing phosphatidylserine synthase PssA improved the membrane integrity, surface potential, electrochemical potential, and hydrophobicity, which further improved the tolerance and octanoate yield in E. coli [31].
The net negative charge of cell surface served as the critical role in metabolite production, especially for protein expression [18]. d-Alanylation of lipoteichoic acid (LTA) mediated by dlt operon is a major process for LTA modification, which incorporates d-alanine into LTA, and resulting in the reduction of net negative charge on cell surface [23, 24]. Previous researches implied that deletion of dlt operon improved the cell surface net negative charge, and further improved the yields of nattokinase, α-amylase, β-mannanase, cyclodextrin glycosyltransferase rPA [11, 12, 33]. Bacillus has a very complex and variable membrane lipid composition, and it consists of 20–50% zwitterionic phospholipid phosphatidylethanolamine (PE), 15–45% phosphatidylglycerol (PG), 2–15% lysine acylphosphatidylglycerol (LysPG), 2–25% cardiolipin (CL), and 10–30% mono-, di-, and tri-glucosyl diacylglycerol (GL) [8, 22]. The phosphatidylserine synthetase (PssA) and cardiolipin synthase (ClsA) on the cell membrane are responsible for syntheses of two major phospholipid components PE and CL, and deletion of pssA and clsA greatly enhanced anionic membrane lipids, and further affected the properties and net negative charge of cell surface for promoting α-amylase secretion in B. subtilis [8]. In addition, previous results indicated that the improvement of negative charge was more conducive to the target protein with lower isoelectric point (PI) [8, 11].
B. licheniformis DW2 is an industrial strain for bacitracin production [43], which can also serve as the efficient host strain for protein expression [11]. In this study, the d-alanyl-lipoteichoic acid synthetase gene dltD of dlt operon, cardiolipin synthase gene clsA, and phosphatidylserine synthase gene pssA were blocked to improve target protein production in DW2, and our research indicated that cell surface engineering is an efficient strategy for enhanced production of target proteins.
Materials and methods
Bacterial strains, plasmids, and cultivation conditions
The bacterial strains and plasmids used in this study are listed in Table 1. B. licheniformis DW2 was acted as the original strain for constructing recombinants. The plasmid T2(2)-Ori was used for gene knockout in B. licheniformis, and nattokinase and α-amylase expression vectors pP43SacCNK and pHY-SAT were attained in our previous research [5]. The primers used in this study are listed in Table S1 (seeing in the Supplementary Material). Lysogeny broth (LB) medium (10 g/L peptone, 5 g/L yeast extract, 10 g/L NaCl, pH 7.20) was used as the basic medium for bacterial growth, and the corresponding antibiotics (50 μg/mL ampicillin, 20 μg/mL tetracycline or 20 μg/mL kanamycin) were added when necessary. The medium E (ME medium) for cell growth assays contains 20 g/L glucose, 20 g/L sodium glutamate, 10 g/L sodium citrate, 7 g/L NH4Cl, 0.5 g/L K2HPO4·3H2O, 0.5 g/L MgSO4·7H2O, 0.04 g/L FeCl3·6H2O, 0.104 g/L MnSO4·H2O, 0.15 g/L CaCl2·2H2O, pH 7.2 [1]. The medium for nattokinase and α-amylase production was referred to our previous study [5]. The inoculum (3%) was added into 30 mL of fermentation medium in a 250 mL erlenmeyer flask, and incubated at 220 rpm and 37 °C for 48 h. All fermentation experiments were repeated at least three times.
Establishment of gene deletion strain
The method of gene knockout in B. licheniformis was referred to our previously reported research [2], and the procedure for constructing pssA-deficient strain was served as an example. Briefly, the upstream and downstream homology arms of gene pssA were amplified by corresponding primers (pssA-KF1/R1 and pssA-KF2/R2) (Table S1), and fused by splicing-overlap-extension PCR (SOE-PCR). The fused fragment was digested with restriction enzymes XbaI and SacI, and inserted into T2(2)-Ori. Diagnostic PCR and DNA sequence confirmed that the pssA deletion vector was constructed successfully, named as T2-pssA. Then T2-pssA was electrotransferred into B. licheniformis DW2, and verified by diagnostic PCR and plasmid extraction. The positive transformants were cultured in LB liquid medium containing 20 μg/mL kanamycin at 45 °C, and sub-cultured for three generations, and further transferred into kanamycin-free LB medium for another six generations at 37 °C. The cells were then cultivated on LB agar medium with or without kanamycin, and cultivated for 20 h. The pssA deletion strain was verified by diagnostic PCR and DNA sequence, designated as DW2ΔpssA. Similarly, the gene clsA deletion strain DW2ΔclsA and triplicate gene (dltD, clsA, pssA) deletion strain DW2ΔDCP were obtained by the same method.
Construction of gene overexpression strain
The method for constructing gene overexpression strain was referred to the previous study [3], and the construction procedure for PssA overexpression strain was served as an example. In brief, P43 promoter from B. subtilis 168, gene pssA and amyL terminator from B. licheniformis DW2 were amplified by corresponding primers (Table S1), and fused by SOE-PCR. The fused fragment was then inserted into pHY300 at the restriction sites EcoRI/XbaI, and diagnostic PCR and DNA sequence confirmed that the gene pssA overexpression vector pHY-pssA was constructed successfully. Then pHY-pssA was electrotransformed into DW2 to attain pssA overexpression strain, named as DW2/pHY-pssA. Similarly, the dltD and clsA overexpression strains, DW2/pHY-dltD and DW2/pHY-clsA, were obtained by the same method.
Determination of cell membrane permeability
The permeability of cell membrane was measured via determining the activity of extracellular β-d-galactosidase, according to the previous research [29]. 2-Nitrophenyl-β-d-galactopyranoside (ONPG) serves as the substrate for β-d-galactosidase, and it forms galactose and yellow o-nitrophenol (ONP) after hydrolyzation by β-d-galactosidase, and ONP has the ultraviolet absorption at 405–420 nm. Therefore, the amount of β-d-galactosidase that was exuded out of cells can be determined by measuring the absorbance at 405 nm. Briefly, the cells were diluted to OD600 = 0.5 by adding the mixture solution (containing 25 mg/L Tris–HCl and 0.5% NaCl, pH 7.2), and 100 μL bacterial cell solution was mixed with 40 μL 30 mmol/L ONPG and reacted at 37 °C for 2 h, and then the absorbance was measured at 405 nm wavelength.
Determination of cation binding ratio
The net negative charge of cell surface was measured by determining the cation binding ratio of cell surface, which was conducted by cationic dye alcian blue 8GX binding assay [11]. Briefly, bacteria cells at mid-logarithmic phase were washed twice with 20 mM morpholinepropanesulfonic acid (MOPS) buffer, and re-suspended to a final OD600 of 0.5, and then added to 65 mg/L alcian blue and rotated at the room temperature for 10 min at 3 rpm. Then it was centrifuged at 10,000g for 5 min, and the absorbance was detected under 530 nm wavelength. Acting as the negative control, MOPS buffer with 65 mg/L alcian blue was incubated under the same condition without bacteria. The cation binding ratio was calculated as A1 (the absorbance of sample containing bacteria)/A2 (the absorbance of negative control).
Analytical methods
Biomass was measured by measuring the optical density at 600 nm. The volume of 2 mL of fermentation broth was centrifuged at 10,000g for 5 min, and re-suspended in an appropriate volume of physiological saline, and OD600 was measured using a spectrophotometer (Bio-Rad, USA). The activities of nattokinase and α-amylase were measured based on the previously reported methods [5]. The concentrations of total extracellular proteins were measured by the means of bovine serum albumin (BSA), based on our previous research [11].
Statistical analysis
All samples were analyzed in triplicate, and the data were presented as the mean ± standard deviation for each sample point. Data analysis was done using one-way analysis of variance (ANOVA) and Duncan’s multiple range using SPSS® 19.0 at p ≤ 0.05 [41].
Results
Deletions of dltD, clsA, and pssA improved the concentrations of total extracellular proteins
Lipoteichoic acid (LTA) is a critical component of cell wall in Gram-positive bacteria, and its d-alanylation mediated by dlt operon is a major process for LTA modification. The phospholipid components CL and PE are important components for cell membrane, which syntheses are controlled by cardiolipin synthase gene clsA and CDP-diacylglycerol–serine O-phosphatidyltransferase gene pssA, respectively [8]. In this study, the genes clsA, pssA were, respectively, deleted in B. licheniformis DW2; diagnostic PCR and DNA sequence confirmed that the gene deletion strains were constructed successfully, named as DW2ΔclsA and DW2ΔpssA, respectively. In addition, the dltD deficient strain DW2ΔdltD was attained in our previous research [11], and the genes dltD, clsA, and pssA overexpression strains DW2/pHY-dltD, DW2/pHY-clsA and DW2/pHY-pssA were constructed simultaneously.
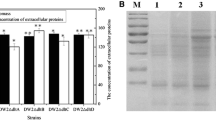
Then the cell biomass and concentrations of total extracellular proteins of these recombinant strains were determined in ME medium, as well as the controls DW2 and DW2/pHY300. Based on our results of Fig. 1a, deletions of these genes were not conducive to cell growth, and the maximum biomass (OD600) of DW2ΔpssA (9.753 ± 0.233), DW2ΔclsA (9.356 ± 0.215), and DW2ΔdltD (8.983 ± 0.282) were, respectively, 10.54 ± 1.43%, 14.17 ± 1.51%, and 17.55 ± 1.28% lower than that of DW2 (10.90 ± 0.25), indicating the critical role of these genes for cell growth. Furthermore, the concentration of total extracellular proteins was measured at 24 h, and our results showed that the concentrations of gene deletion strains DW2ΔdltD, DW2ΔclsA, and DW2ΔpssA were 248.4 ± 9.5 mg/L, 259.4 ± 8.7 mg/L, 286.7 ± 9.4 mg/L, increased by 15.91 ± 3.21%, 21.04 ± 3.03%, 33.76 ± 4.25% compared to DW2 (214.3 ± 7.5 mg/L), respectively (Fig. 1a). In addition, our results implied that the cell biomass of DW2/pHY300 was lower than that of DW2, which might be due to the increase in cell maintain metabolism in the strain harboring free plasmid [42], and this phenomenon was positively correlated with our previous results [7]. Moreover, our results implied that the concentrations of total extracellular proteins were, respectively, decreased by 12.62 ± 1.15%, 17.81 ± 1.34%, and 20.52 ± 1.64% in the genes dltD, clsA, and pssA overexpression strains, although the biomass were all improved compared to DW2/pHY300 (Fig. 1b).

Effects of dltD, clsA, and pssA deletion/overexpression on cell growth and concentrations of total extracellular proteins. a Biomass and concentrations of total extracellular proteins in gene deletion strains; b biomass and concentrations of total extracellular proteins in gene overexpression strains
Deletions of dltD, clsA, and pssA improved the net negative charges and permeabilities of cell surface
Then these gene deletion and overexpression strains were cultivated in LB medium to determine the net negative charges and permeabilities of cell surface. The net negative charge of cell surface was measured by determining the cation binding ratio via cationic dye alcian blue 8GX binding assay. Based on our results of Fig. 2a, the cation binding ratio of DW2 was 0.3518 ± 0.0065, and which of dltD, pssA and clsA deletion strains were, respectively, 17.54 ± 0.89%, 26.15 ± 1.85%, and 31.98 ± 2.47% higher than that of DW2, reached 0.4135 ± 0.0083, 0.4438 ± 0.0068, and 0.4643 ± 0.0078 (Fig. 2a), indicating that the cell surface net negative charge was significantly increased in gene deletion strains. In addition, over expression of DltD, ClsA, and PssA decreased the cation binding ratio, which reduced by 9.664 ± 0.582%, 14.08 ± 0.85%, and 25.02 ± 2.16%, compared to DW2/pHY300, respectively (Fig. 2b).

Effects of dltD, clsA, and pssA deletion/overexpression on the net negative charges and permeabilities of cell surface. a The net negative charges and β-d-galactosidase activities in gene deletion strains; b the net negative charges and β-d-galactosidase activities in gene overexpression strains
Meanwhile the cell membrane permeability was analyzed by measuring the activity of extracellular β-d-galactosidase via determining the absorbance at 405 nm [29]. Based on our results, the β-d-galactosidase activities of genes dltD, pssA, and clsA deletion strains reached 63.54 ± 1.37 U/L, 63.76 ± 1.28 U/L, and 69.95 ± 2.01 U/L, which, respectively, increased by 9.653 ± 1.523%, 17.10 ± 1.14%, and 20.71 ± 1.28% compared to DW2 (63.54 ± 1.54 U/L), indicating the increase in cell membrane permeabilities in gene deletion strains (Fig. 2a). In addition, overexpression of dltD, pssA, and clsA decreased β-d-galactosidase activities and further cell membrane permeability (Fig. 2b). Taken together, all these above results confirmed that deletions of dltD, pssA, and clsA improved cell surface net negative charges and cell membrane permeabilities, which were beneficial for protein secretion; thus, the concentrations of total extracellular proteins were enhanced in gene deletion strains.
The activities of nattokinase and α-amylase were improved in dltD, clsA, and pssA deletion strains
Furthermore, nattokinase (PI: 8.60) and α-amylase (PI: 6.26) were served as the target proteins to evaluate the effects of individual gene deletion on target protein production. Nattokinase and α-amylase expression vectors, pP43SacCNK and pHY-SAT, were attained in our previous research [5], and then, respectively, electrotransferred into these gene deletion strains. Our results showed that deletion of these genes benefited target protein production. The nattokinase produced by DW2ΔpssA/pP43SacCNK, DW2ΔclsA/pP43SacCNK, and DW2ΔdltD/pP43SacCNK were 29.65 ± 0.74 FU/mL, 32.54 ± 0.84 FU/mL, and 34.42 ± 0.89 FU/mL, increased by 16.59 ± 2.25%, 27.96 ± 2.62%, and 35.35 ± 3.63% compared with that of DW2/pP43SacCNK (25.43 ± 0.68 FU/mL), respectively. The nattokinase activity of per gram cell (1 OD600 = 0.39 g/L) was 23.76 ± 2.54%, 43.35 ± 3.27%, and 54.21 ± 4.74% increases in these gene deletion strains, compared with that of DW2/pP43SacCNK, respectively. Additionally, the α-amylase produced by these gene mutant strains was also significantly enhanced, and the α-amylase produced by per gram cell in dltD, clsA, and pssA deficient strains reached 66.91 ± 2.54 U/g DCW, 75.05 ± 3.87 U/g DCW, and 94.63 ± 5.37 U/g DCW, increased by 36.73 ± 2.57%, 53.36 ± 4.86%, 93.39 ± 6.74%, respectively. Additionally, the results of SDS-PAGE in Fig. 3b and d were positively correlated with enzyme activities.

Nattokinase and α-amylase produced by DW2, and dltD, clsA and pssA deletion strains. a Nattokinase activities and biomass; b SDS-PAGE analysis, M: protein marker (170, 130, 100, 70, 55, 40, 35, 25, and 15 kDa), line 1: DW2/pP43SacCNK, line 2: DW2ΔdltD/pP43SacCNK, line 3: DW2ΔclsA/pP43SacCNK, line 4: DW2ΔpssA/pP43SacCNK; the molecular weight of nattokinase is 27.78 KDa. c α-Amylase activities and biomass; d SDS-PAGE analysis, M: protein markers (170, 130, 100, 70, 55, 40, 35, 25, and 15 kDa), lane 1: DW2/pHY-SAT, lane 2: DW2ΔdltD/pHY-SAT, lane 3: DW2ΔpssA/pHY-SAT, lane 4: DW2ΔclsA/pHY-SAT. The molecular weight of α-amylase is 55.61 KDa
Construction of the triplicate gene deletion strain and their effects on net negative charge and permeability
Furthermore, the triplicate gene superposition knockout approach was performed to obtain the genes dltD, clsA, and pssA deletion strain, attained DW2ΔdltDΔclsAΔpssA (abbreviation DW2ΔDCP). Our results in Fig. 4 show that the cation binding ratio in DW2ΔDCP is increased to 0.5138 ± 0.0135 by 45.60 ± 4.37%, compared with that of DW2 (0.3518 ± 0.0065); moreover, the cell membrane permeability of DW2ΔDCP is increased by 26.41 ± 1.79%. Meanwhile, the cation binding ratio and cell membrane permeability of DW2ΔDCP were also higher than those of individual gene deletion strains, indicating that superposition knockout of these three genes has a positive effect on the increase of net negative charge and permeability of cell surface.

Effects of triple gene deletion on the net negative charge and permeability of cell surface
Effects of triplicate gene deletion on nattokinase and α-amylase production
Furthermore, the nattokinase and α-amylase expression vectors pP43SacCNK and pHY-SAT were electrotransformed into B. licheniformis DW2ΔDCP. Our results showed that DW2ΔDCP/pP43SacCNK produced 37.15 ± 0.89 FU/mL nattokinase, 46.09 ± 3.51% higher compared to DW2/pP43SacCNK (25.43 ± 0.76 FU/mL) (Fig. 5a). The α-amylase produced by DW2ΔDCP/pHY-SAT reached 305.3 ± 8.4 U/mL, increased by 96.34 ± 7.24% compared with that of DW2/pHY-SAT (155.5 ± 5.6 U/mL) (Fig. 5b). Additionally, the nattokinase and α-amylase activities of DW2ΔDCP were higher than those of the individual gene deletion strains.

Effects of triple gene deletion on nattokinase and α-amylase production. a Nattokinase activities and biomass; b SDS-PAGE analysis, M: protein marker (170, 130, 100, 70, 55, 40, 35, 25, and 15 kDa), line 1: DW2/pP43SacCNK, line 2: DW2ΔDCP/pP43SacCNK; c α-amylase activities and biomass; d SDS-PAGE analysis, M: protein markers (170, 130, 100, 70, 55, 40, 35, 25, and 15 kDa), lane 1: DW2/pHY-SAT, lane 2: DW2ΔDCP/pHY-SAT
The enzyme activities and biomass of DW2ΔDCP/pP43SacCNK and DW2ΔDCP/pHY-SAT were determined during the fermentation processes, as well as control strains DW2/pP43SacCNK and DW2/pHY-SAT. Based on the results of Fig. 6, the biomass of DW2ΔDCP/pP43SacCNK was lower than those of DW2/pP43SacCNK throughout the fermentation process, and the maximum biomass was reduced by 18.80 ± 1.37%, compared to DW2/pP43SacCNK. While, the nattokinase activities were enhanced in DW2ΔDCP/pP43SacCNK, and nattokinase produced by per gram cell reached 7.632 ± 0.534 FU/g DCW, increased by 79.92 ± 5.64% compared with that of DW2/pP43SacCNK (4.242 ± 0.279 FU/g DCW) (Fig. 6a). In addition, the maximum α-amylase activity was enhanced by 96.34 ± 7.24% in DW2ΔDCP/pHY-SAT, and the α-amylase produced by per gram cell reached 117.8 ± 8.3 U/g DCW, increased by 1.413 ± 0.131-fold (Fig. 6b).

Fermentation process curves of recombinant strains and controls during nattokinase and α-amylase production. a Nattokinase; b α-amylase
Discussion
Bacillus acted as the efficient host strain for protein production, as it owns the features of non-toxicity, convenience for gene modification, short fermentation cycle on cheap nutrition, superior capacity of protein secretion, and robustness in industrial fermentation [4, 13]. Modification of host strain was regarded as the effective tactic to improve target production, due to its universality and efficiency [4]. Previously, the signal peptidases SipV, SipT and signal peptide peptidase SppA were overexpressed to improve nattokinase, β-mannanase, and α-amylase production [5, 6, 28], and overexpression of the elements in Sec pathway and chaperone PrsA benefited α-amylase, parvulin-type peptidyl-prolyl cis/trans isomerases (PPIase), and lipase production [9, 26, 34]. In this study, it was confirmed that the concentrations of total extracellular proteins, nattokinase, and α-amylase activities were all enhanced in dltD, pssA, and clsA deletion strains, in which yields were further improved in the triplicate-gene deletion strain DW2ΔDCP. Taken together, this study provided a promising host strain for target protein production.
Along with the development of modern biotechnology, various strategies have been developed for product production, such as in cofactor engineering, structural biotechnology, synthetic RNA switch, etc. [10]. Cell surface engineering is a promising approach of strain improvement for metabolite production [32], which is conducted by changing the components of cell membrane or cell wall. Previously, the cutinase from Thermobifida fusca was expressed without an appended signal peptide, which strengthened the hydrolysis of phospholipid component, and further led to the increase of cell membrane permeability in E. coli BL21 [30]. Also, overexpression of CDP-diacylglycerol–glycerol-3-phosphate 3-phosphatidyltransferase gene pgsA and clsA benefited CL content in cell membrane, and ultimately led to the significant increase in hyaluronic acid titer with high molecular weight [38]. Overexpression of intramembrane protease RasP led to a 3-fold increases yield of serine protease from Bacillus clausii, and 2.5- to 10-fold increased production of α-amylase from Paenibacillus curdlanolyticus [27]. In addition, overexpression of pssA decreased the synthesis capability of non-native trans-unsaturated fatty acids (TUFA) and membrane fluidity, which led to the increase in the strain tolerance to industrially relevant inhibitors, such as furfural, acetate, toluene, and ethanol, and further enhanced the fatty acid tolerance and yield [31]. Altering the relative distribution of saturated and unsaturated fatty acids tails was also an effective tactic in alleviating membrane leakage during fatty acid production, although fatty acids yield was not increased [25]. In addition, the recent research of our group implied that overexpression of dlt operon benefited poly γ-glutamic acid production in B. licheniformis [16]. In this research, the net negative charges and permeabilities of cell surface were enhanced in dltD, clsA, and pssA deletion strains, which both benefited protein secretion; therefore, our results indicated that cell surface engineering is an efficient approach for enhanced production of target protein. Meanwhile, the increase rates of nattokinase (PI: 8.60) were significantly lower than those of α-amylase (PI: 6.26) which indicated that improving cation binding ratio of host strain was more conducive to the target protein with lower PIs, and our result was correlated positively with the previous researches [8, 11].
The cell wall of Bacillus is a multilayer structure that consisted of a copolymer of peptidoglycan and an anionic polymer, which contains lipoteichoic acid and protein [21]. The charge density of bacterial cell wall and cross-linking index of peptidoglycan layer determine the secretion efficiency of target protein [29]. Previously, it has been proven that deletion of dlt operon enhanced the expression of PrsA, and thereby enhancing the folding and stability of secreted proteins [19]. In addition, Bacillus has a very complex and variable membrane lipid composition, and inactivation of pssA and clsA affected the distribution of phospholipids on the cell membrane surface, which in turn changed the composition and cell integrity of cell membrane, and further increased cell membrane permeability and target protein production [8]. Therefore, deletions of dltD, clsA, and pssA enhanced the net negative charges and permeabilities of cell surface for protein secretion. Since deletion of dlt operon improved the cell autolysis, the biomass of dlt deletion strain was decreased by 10.54 ± 1.43% compared with that of DW2 [36]. Also, deletions of clsA and pssA destroyed the cell membrane integrity, and further reduced cell biomass (14.17 ± 1.51% and 17.55 ± 1.28%, respectively). Unfortunately, the composition of phospholipid, phospholipid phosphatidylethanolamines (PE), and cardiolipin (CL) has not been detected in this research, based on our current instrument. Previously, Cao et al. have confirmed the function roles of pssA and clsA in phospholipid compositions [8]; here, we provided a strategy as well as the host strain for efficient protein production. Despite this, we will continue to measure these substances in our future research. Compared to DW2ΔdltD and DW2ΔclsA, DW2ΔpssA owned the lowest biomass, since gene pssA is responsible for the synthesis of PE, the largest proportion of the membrane phospholipids, which might be the reason for the lowest biomass of DW2ΔpssA. In addition, the net negative charge and permeability of cell surface in DW2ΔpssA were the highest among the individual gene deletion strains, indicating the best performance of DW2ΔpssA on protein secretion, and our results were positively correlated with the previous researches [8, 31].
Conclusion
In this study, the dlt operon gene dltD, phosphatidylserine synthase gene pssA, and cardiolipin synthase gene clsA associated with cell wall/membrane compositions were deleted to improve the target protein production. According to our results, the net negative charges and permeabilities of cell surface were all enhanced in the genes dltD, clsA, and pssA deletion strains, as well as the yields of nattokinase and α-amylase. Moreover, a triplicate gene deletion strain DW2ΔDCP was constructed, which further led to the 46.09 ± 3.51% increase of nattokinase activity and 96.34 ± 7.24% increase of α-amylase activity. Taken together, our results indicated that cell surface engineering is an efficient strategy for enhanced production of heterologous protein, and this study provided a promising host strain for target protein production.
References
Birrer GA, Cromwick AM, Gross RA (1994) Gamma-poly(glutamic acid) formation by Bacillus licheniformis 9945a: physiological and biochemical studies. Inter J Biol Macromol 16:265–275
Cai D, Chen Y, He P, Wang S, Mo F, Li X, Wang Q, Nomura CT, Wen Z, Ma X, Chen S (2018) Enhanced production of poly-gamma-glutamic acid by improving ATP supply in metabolically engineered Bacillus licheniformis. Biotechnol Bioeng 115:2541–2553
Cai D, He P, Lu X, Zhu C, Zhu J, Zhan Y, Wang Q, Wen Z, Chen S (2017) A novel approach to improve poly-γ-glutamic acid production by NADPH Regeneration in Bacillus licheniformis WX-02. Sci Rep 7:43404
Cai D, Rao Y, Zhan Y, Wang Q, Chen S (2019) Engineering Bacillus for efficient production of heterologous protein: current progress, challenge and prospect. J Appl Microbiol 126:1632–1642
Cai D, Wang H, He P, Zhu C, Wang Q, Wei X, Nomura CT, Chen S (2017) A novel strategy to improve protein secretion via overexpression of the SppA signal peptide peptidase in Bacillus licheniformis. Microb Cell Fact 16:70
Cai D, Wei X, Qiu Y, Chen Y, Chen J, Wen Z, Chen S (2016) High-level expression of nattokinase in Bacillus licheniformis by manipulating signal peptide and signal peptidase. J Appl Microbiol 121:704–712
Cai D, Zhu J, Zhu S, Lu Y, Zhang B, Lu K, Li J, Ma X, Chen S (2019) Metabolic engineering of main transcription factors in carbon, nitrogen, and phosphorus metabolisms for enhanced production of bacitracin in Bacillus licheniformis. ACS Synth Biol 8:866–875
Cao H, van Heel AJ, Ahmed H, Mols M, Kuipers OP (2017) Cell surface engineering of Bacillus subtilis improves production yields of heterologously expressed alpha-amylases. Microb Cell Fact 16:56
Chen J, Fu G, Gai Y, Zheng P, Zhang D, Wen J (2015) Combinatorial Sec pathway analysis for improved heterologous protein secretion in Bacillus subtilis: identification of bottlenecks by systematic gene overexpression. Microb Cell Fact 14:92
Chen X, Gao C, Guo L, Hu G, Luo Q, Liu J, Nielsen J, Chen J, Liu L (2018) DCEO biotechnology: tools to design, construct, evaluate, and optimize the metabolic pathway for biosynthesis of chemicals. Chem Rev 118:4–72
Chen Y, Cai D, He P, Mo F, Zhang Q, Ma X, Chen S (2018) Enhanced production of heterologous proteins by Bacillus licheniformis with defective d-alanylation of lipoteichoic acid. World J Microbiol Biotechnol 34:135
Craynest M, Jorgensen S, Sarvas M, Kontinen VP (2003) Enhanced secretion of heterologous cyclodextrin glycosyltransferase by a mutant of Bacillus licheniformis defective in the d-alanylation of teichoic acids. Lett Appl Microbiol 37:75–80
Cui W, Han L, Suo F, Liu Z, Zhou L, Zhou Z (2018) Exploitation of Bacillus subtilis as a robust workhorse for production of heterologous proteins and beyond. World J Microbiol Biotechnol 34:145
Degering C, Eggert T, Puls M, Bongaerts J, Evers S, Maurer KH, Jaeger KE (2010) Optimization of protease secretion in Bacillus subtilis and Bacillus licheniformis by screening of homologous and heterologous signal peptides. Appl Environ Microbiol 76:6370–6376
Harwood CR, Cranenburgh R (2008) Bacillus protein secretion: an unfolding story. Trends Microbiol 16:73–79
He P, Wan N, Cai D, Hu S, Chen Y, Li S, Chen S (2019) (13)C-metabolic flux analysis reveals the metabolic flux redistribution for enhanced production of poly-gamma-glutamic acid in dlt over-expressed Bacillus licheniformis. Front Microbiol 10:105
Hoshida H, Kondo M, Kobayashi T, Yarimizu T, Akada R (2017) 5-UTR introns enhance protein expression in the yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol 101:241–251
Hyyrylainen HL, Pietiainen M, Lunden T, Ekman A, Gardemeister M, Murtomaki-Repo S, Antelmann H, Hecker M, Valmu L, Sarvas M, Kontinen VP (2007) The density of negative charge in the cell wall influences two-component signal transduction in Bacillus subtilis. Microbiology 153:2126–2136
Hyyrylainen HL, Vitikainen M, Thwaite J, Wu H, Sarvas M, Harwood CR, Kontinen VP, Stephenson K (2000) d-Alanine substitution of teichoic acids as a modulator of protein folding and stability at the cytoplasmic membrane/cell wall interface of Bacillus subtilis. J Biol Chem 275:26696–26703
Kang Z, Yang S, Du G, Chen J (2014) Molecular engineering of secretory machinery components for high-level secretion of proteins in Bacillus species. J Ind Microbiol Biotechnol 41:1599–1607
Kasahara J, Kiriyama Y, Miyashita M, Kondo T, Yamada T, Yazawa K, Yoshikawa R, Yamamoto H (2016) Teichoic acid polymers affect expression and localization of dl-endopeptidase LytE required for lateral cell wall hydrolysis in Bacillus subtilis. J Bacteriol 198:1585–1594
Kawai F, Shoda M, Harashima R, Sadaie Y, Hara H, Matsumoto K (2004) Cardiolipin domains in Bacillus subtilis marburg membranes. J Bacteriol 186:1475–1483
Kiriukhin MY, Neuhaus FC (2001) d-alanylation of lipoteichoic acid: role of the d-alanyl carrier protein in acylation. J Bacteriol 183:2051–2058
Kovacs M, Halfmann A, Fedtke I, Heintz M, Peschel A, Vollmer W, Hakenbeck R, Bruckner R (2006) A functional dlt operon, encoding proteins required for incorporation of d-alanine in teichoic acids in gram-positive bacteria, confers resistance to cationic antimicrobial peptides in Streptococcus pneumoniae. J Bacteriol 188:5797–5805
Lennen RM, Pfleger BF (2013) Modulating membrane composition alters free fatty acid tolerance in Escherichia coli. PLoS One 8:e54031
Ma RJ, Wang YH, Liu L, Bai LL, Ban R (2018) Production enhancement of the extracellular lipase LipA in Bacillus subtilis: effects of expression system and Sec pathway components. Protein Expr Purif 142:81–87
Neef J, Bongiorni C, Goosens VJ, Schmidt B, van Dijl JM (2017) Intramembrane protease RasP boosts protein production in Bacillus. Microb Cell Fact 16:57
Song Y, Fu G, Dong H, Li J, Du Y, Zhang D (2017) High-efficiency secretion of beta-mannanase in Bacillus subtilis through protein synthesis and secretion optimization. J Agric Food Chem 65:2540–2548
Su L, Jiang Q, Yu L, Wu J (2017) Enhanced extracellular production of recombinant proteins in Escherichia coli by co-expression with Bacillus cereus phospholipase C. Microb Cell Fact 16:24
Su L, Woodard RW, Chen J, Wu J (2013) Extracellular location of Thermobifida fusca cutinase expressed in Escherichia coli BL21(DE3) without mediation of a signal peptide. Appl Environ Microbiol 79:4192–4198
Tan Z, Khakbaz P, Chen Y, Lombardo J, Yoon JM, Shanks JV, Klauda JB, Jarboe LR (2017) Engineering Escherichia coli membrane phospholipid head distribution improves tolerance and production of biorenewables. Metab Eng 44:1–12
Ueda M (2016) Establishment of cell surface engineering and its development. Biosci Biotechnol Biochem 80:1243–1253
Vitikainen M, Hyyrylainen HL, Kivimaki A, Kontinen VP, Sarvas M (2005) Secretion of heterologous proteins in Bacillus subtilis can be improved by engineering cell components affecting post-translocational protein folding and degradation. J Appl Microbiol 99:363–375
Vitikainen M, Lappalainen I, Seppala R, Antelmann H, Boer H, Taira S, Savilahti H, Hecker M, Vihinen M, Sarvas M, Kontinen VP (2004) Structure-function analysis of PrsA reveals roles for the parvulin-like and flanking N- and C-terminal domains in protein folding and secretion in Bacillus subtilis. J Biol Chem 279:19302–19314
Wang H, Wang Y, Yang R (2017) Recent progress in Bacillus subtilis spore-surface display: concept, progress, and future. Appl Microbiol Biotechnol 101:933–949
Wecke J, Perego M, Fischer W (1996) d-alanine deprivation of Bacillus subtilis teichoic acids is without effect on cell growth and morphology but affects the autolytic activity. Microb Drug Resist 2:123–129
Wei X, Zhou Y, Chen J, Cai D, Wang D, Qi G, Chen S (2015) Efficient expression of nattokinase in Bacillus licheniformis: host strain construction and signal peptide optimization. J Ind Microbiol Biotechnol 42:287–295
Westbrook AW, Ren X, Moo-Young M, Chou CP (2018) Engineering of cell membrane to enhance heterologous production of hyaluronic acid in Bacillus subtilis. Biotechnol Bioeng 115:216–231
Yang H, Lu X, Hu J, Chen Y, Shen W, Liu L (2018) Boosting secretion of extracellular protein by Escherichia coli via cell wall perturbation. Appl Environ Microbiol 84:e01382
Yang S, Du G, Chen J, Kang Z (2017) Characterization and application of endogenous phase-dependent promoters in Bacillus subtilis. Appl Microbiol Biotechnol 101:4151–4161
Zhan Y, Sheng B, Wang H, Shi J, Cai D, Yi L, Yang S, Wen Z, Ma X, Chen S (2018) Rewiring glycerol metabolism for enhanced production of poly-gamma-glutamic acid in Bacillus licheniformis. Biotechnol Biofuels 11:306
Zhou S, Ding R, Chen J, Du G, Li H, Zhou J (2017) Obtaining a panel of cascade promoter-5′-UTR complexes in Escherichia coli. ACS Synth Biol 6:1065–1075
Zhu S, Cai D, Liu Z, Zhang B, Li J, Chen S, Ma X (2019) Enhancement of bacitracin production by NADPH generation via overexpressing glucose-6-phosphate dehydrogenase Zwf in Bacillus licheniformis. Appl Biochem Biotechnol 187:1502–1514
Acknowledgements
This study was funded by the National Key Research and Development Program of China (2018YFA0900303, 2015CB150505), the Technical Innovation Special Fund of Hubei Province (No. 2018ACA149), China Postdoctoral Science Foundation (2018M642802), and the Science and Technology Project of Hubei Tobacco Company (027Y2019-018).
Author information
Authors and Affiliations
Contributions
XM and SC designed and supervised the study. FM, DC, PH, FY, and YC performed the experiments. FM, DC, PH, XM, and SC analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mo, F., Cai, D., He, P. et al. Enhanced production of heterologous proteins via engineering the cell surface of Bacillus licheniformis. J Ind Microbiol Biotechnol 46, 1745–1755 (2019). https://doi.org/10.1007/s10295-019-02229-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-019-02229-8