
Abstract
Rapamycin, as a macrocyclic polyketide with immunosuppressive, antifungal, and anti-tumor activity produced by Streptomyces hygroscopicus, is receiving considerable attention for its significant contribution in medical field. However, the production capacity of the wild strain is very low. Hereby, a computational guided engineering approach was proposed to improve the capability of rapamycin production. First, a genome-scale metabolic model of Streptomyces hygroscopicus ATCC 29253 was constructed based on its annotated genome and biochemical information. The model consists of 1003 reactions, 711 metabolites after manual refinement. Subsequently, several potential genetic targets that likely guaranteed an improved yield of rapamycin were identified by flux balance analysis and minimization of metabolic adjustment algorithm. Furthermore, according to the results of model prediction, target gene pfk (encoding 6-phosphofructokinase) was knocked out, and target genes dahP (encoding 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase) and rapK (encoding chorismatase) were overexpressed in the parent strain ATCC 29253. The yield of rapamycin increased by 30.8% by knocking out gene pfk and increased by 36.2 and 44.8% by overexpression of rapK and dahP, respectively, compared with parent strain. Finally, the combined effect of the genetic modifications was evaluated. The titer of rapamycin reached 250.8 mg/l by knockout of pfk and co-expression of genes dahP and rapK, corresponding to a 142.3% increase relative to that of the parent strain. The relationship between model prediction and experimental results demonstrates the validity and rationality of this approach for target identification and rapamycin production improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rapamycin, produced by Streptomyces hygroscopicus, is a 31-membered macrocyclic natural product with various biological and pharmacological activities, such as antifungal, immunosuppressive, and antitumor [8, 11, 36]. More importantly, as an immunosuppressant, rapamycin has a more efficient mode of action and lower biological toxicity compared with FK506 and cyclosporine A [32]. Recently, rapamycin has attracted much attention of many researchers and pharmaceutical companies owing to its pharmacological importance and broad applicability.
Currently, the low yield of rapamycin is still a bottleneck for further industrialization and commercialization, so much work has been done in strain modification and culture optimization. Four strategies have been mainly used in the rapamycin improvement: (a) traditional physical and chemical mutagenesis [44]; (b) optimization of fermentation process [24, 38]; (c) recombination by protoplast fusion [10]; and (d) genetic engineering methods [22, 27]. However, mutagenesis, fermentation, optimization, and recombination by protoplast fusion are laborious and tedious processes and sometimes require long and unpredictable durations. Due to the orientation and the short period, metabolic engineering technology has great advantages to improve productivity of natural products.
Indeed, metabolic engineering could efficiently regulate the key pathways of natural products, such as polyketides and nonribosomal peptides. For example, it was reported that overexpression of propionyl-CoA carboxylase promoted the accumulation of methylmalonyl-CoA, a key precursor of rapamycin and led to the marginally enhanced production of rapamycin in UV2-2 strain [22]. Irina et al. [7] improved production of actinorhodin and undecylprodigiosin by pfkA2 gene knockout in Streptomyces coelicolor. Although genetic manipulations may improve the precursor synthesis and production titer, validation of the target genes is usually uncertain and unpredictable. In addition, because microorganisms have complicated interconnected metabolic networks, manipulation of the specific gene may generate an effect on other metabolic pathways. Therefore, it is difficult to evaluate the cellular behaviors comprehensively and identify the accurate target genes for efficient strain improvement. Along with the maturity of the GSMM and the improvement of the model analysis method, systems biology plays an increasingly important role in strain metabolic engineering by changing metabolic flux distribution within a microorganism based on systematic strategies. The GSMM allows us to analyze the cell from a systems viewpoint to predict whole-cell effects of gene perturbations and to simulate known and hypothesized phenotypes. With the development of high-throughput sequencing technology and the reduction of sequencing costs, more and more microorganisms have been sequenced. The complete genome of Streptomyces rapamycinicus ATCC 29253 has been sequenced by Baranasic et al. [3], providing the foundation for constructing the genome-scale metabolic model and gaining comprehensive insight into Streptomyces hygroscopicus physiology. Stoichiometric models-based GSMM can be employed to interpret cellular metabolic response to genetic perturbation and unravel the underlying mechanism of undesired phenotypes. This approach not only saves time, labor, and research expenditure by decreasing the amounts of laboratory experiments but also can rationally guide the metabolic engineering. Currently, GSMMs have been used to identify metabolic engineering targets for many important industrial products, such as biofuels, amino acids, vitamins, and secondary metabolites [1, 28, 30, 45].
In this study, a GSMM of Streptomyces hygroscopicus ATCC 29253 was reconstructed to simulate the intracellular flux distribution. Then, several target genes were identified by FBA and MOMA prediction. These targets were then ranked and knockout target pfk, and overexpression targets dahP and rapK were selected to be engineered to improve production of rapamycin. Finally, the combined effects of pfk gene knockout and dahP and rapK overexpression on rapamycin biosynthesis were studied.
Materials and methods
Construction of genome-scale metabolic network
The genome-scale metabolic network for S. hygroscopicus ATCC 29253 was reconstructed according to Thiele et al. [39]. The first genome sequence draft of S. hygroscopicus ATCC 29253 has been reported recently. S. hygroscopicus ATCC 29253 genome length is 12.7 MB, coding 10,002 genes, with 70.5% GC content [3], which were stored in the NCBI, KEGG, and other databases. In this study, genes, enzyme complexes, and reactions were identified by pathway map of KEGG database, resulting in the initial network. Public databases, such as KEGG, BRENDA, and BiGG, were used to manually refine the draft metabolic network. Identifying network gaps and selecting candidate gap-filling reactions with strong supporting evidence are the biggest challenges for model refinement. GapFind and GapFill [26] algorithms were used to computationally identify and resolve gaps, thus minimizing the amount of manual curation needed. Candidate reactions suggested by GapFill were chosen from the BiGG database; this database contains genome-scale models that have undergone extensive refinement and validation, and thus is a resource of high-confidence reactions. Some reactions from the experimental data and published literature were also supplemented in the network. Besides, biosynthetic reactions for the biomass and rapamycin were included. Since there was no detailed information on the biomass composition of Streptomyces hygroscopicus ATCC 29253, DNA, RNA, protein, lipids, small molecules, and cell wall components (carbohydrate, peptidoglycan, and teichoic acid) were partly measured and partly referred from literature data. As for the rapamycin synthesis, the synthesis of specific precursors and overall reaction was added. At last, transport reactions and exchange reactions were added to the metabolic network to get a complete model. The refined model was shown in File 1S in Supplementary Materials.
Target gene prediction
The resulting model was analyzed using Constraint-Based Reconstruction and Analysis (COBRA). Gene prediction was performed using the COBRA Toolbox-2.0 in MATLAB, with GLPK and CPLEX as the optimization programming solvers [34]. For the overexpression target identification, the algorithm developed by Boghigian et al. [5] was employed. First, the model of xml format (File 2S in Supplementary Materials) was read on the MATLAB platform, and FBA algorithm was then employed to obtain an initial flux distribution with the maximization of the specific growth rate as the objective function. Subsequently, each non-zero reaction flux was amplified to some extent (for instance, twofold), and the quadratic programming problem was solved by MOMA algorithm [35], which was performed by searching for the minimal Euclidean distance between the parent metabolic model and model after gene perturbation. Finally, the overexpression targets were identified through comparing a fraction value, f PH (the ratio of weighted and dimensionless specific growth rate and specific rapamycin production rate). The knockout target identification was implemented by the same simulation method with some modifications. To be specific, each non-zero reaction flux was set to zero, and the quadratic programming problem was solved by MOMA algorithm. The knockout targets were identified through comparing f PH value. Target genes that had the higher f PH were the better candidates to be manipulated experimentally:
Strains, plasmids, and cultivation conditions
Strains and plasmids used in this study are listed in Table 1. E. coli DH5α was used to propagate all plasmids. E. coli ET12567/pUZ8002 was used as the non methylating plasmid donor strain for intergeneric conjugation with Streptomyces hygroscopicus. E. coli strains were cultured in Luria–Bertani (LB) medium at 37 °C. The integrative E. coli–Streptomyces vector pIB139 containing the ermE* promoter (PermE*) [41] was used for gene overexpression, and temperature-sensitive E. coli-Streptomyces shuttle vector pKC1139 was used for gene knockout in Streptomyces hygroscopicus.
Wild-type S. hygroscopicus was cultivated on the agar slant medium for about 14–18 days to harvest spores. Fresh slants were washed into 50 mL un-baffled shake flask containing glass beads with 5 mL sterile water, and it was shaken and filtered to get spore suspension. An aliquot of 1 mL of the spore suspension (~108/mL) was inoculated into a 250 mL un-baffled shake flask containing 30 mL seed medium, and incubated at 28 °C and 220 rpm for 60 h. Then, 3 mL seed culture was transferred into a 250 mL un-baffled shake flask containing 30 mL fermentation medium, and cultured at 28 °C and 220 rpm for 120 h.
Slant and plate medium contained 4.0 g/L mannitol, 20 g/L oat flour, 2.5 g/L yeast extract, and 20 g/L agar and pH 7.0. The seed medium was composed of 20 g/L glucose, 20 g/L soluble starch, 10 g/L corn flour, 6 g/L yeast powder, 6 g/L peptone, 1.5 g/L casein hydrolysate, 0.5 g/L MgSO4, and 1.0 g/L K2HPO4 and pH 7.0. The fermentation medium contained 45 g/L glucose, 15 g/L soluble starch, 40 g/L soybean meal, and 1 g/L (NH4)2SO4 and pH 7.0. In the process of medium preparation, soybean meal was first dissolved quantitatively, autoclaved, and then centrifuged at 8000×g for 10 min at 4 °C to remove the insoluble ingredients. After that, other medium compositions were quantitatively added into the supernatant of soybean meal and autoclaved again.
Gene cloning, plasmid construction, and transformation
General DNA manipulation was performed according to the standard protocols [23]. All primers used in this work were listed in Table 2. To delete gene pfk, deletion plasmid was constructed by amplifying the upstream and downstream flanking regions from genomic DNA of Streptomyces hygroscopicus. The upstream and downstream flanking regions of pfk were amplified using the primers pfk-LF/pfk-LR and pfk-RF/pfk-RR, respectively. The above two fragments were excised with XbaI–BamHI and KpnI–EcoRI, respectively, sequentially ligated into the pUC119-KanR which possessed the kanamycin resistance cassette. After digestion with the restriction enzymes XbaI–EcoRI, the fragments were ligated into pKC1139 that had been digested with the same restriction enzymes, resulting in the deletion plasmid pΔpfk. This plasmid was then transferred into S. hygroscopicus ATCC 29253 through conjugal transfer [23]. The double crossover mutant was selected as described previously [25], and verified by PCR amplification and DNA sequencing.
When target genes were overexpressed, rapK and dahP were amplified from genomic DNA of S. hygroscopicus ATCC 29253 using primer pairs of srapK-F/srapK-R and dahP-F/dahP-R, respectively. The PCR products were digested by restriction enzymes NdeI and XbaI. Then, it was cloned into pIB139 which was digested with the same restriction enzymes to get pIB139R and pIB139D. When co-expressing the target genes rapK and dahP, the ribosome binding site was added in the upstream sequence of rapK gene, and the redesigned primer pairs of rapK gene were listed in Table 2. To delete the BamHI restriction site, pUC18 was excised with BamHI, linearized, blunt-ended and ligated, generating pUC18M. For the construction of PIB139-DR carrying double genes, PCR product of dahP was excised with NdeI–XbaI and transferred to the same sites of pUC18M, generating pUC18M-D. Then, PCR product of rapK (primer pairs of crapK-F/crapK-R containing a ribosome bind site) was digested with BglII–XbaI and transferred to the BamHI–XbaI sites of pUC18M-D, generating pUC18M-DR. The NdeI–XbaI fragment of the dahP and rapK genes was excised from the pUC18M-DP and ligated into pIB139 to yield pIB139DR. Each constructed plasmid was transferred into E. coli ET12567/pUZ8002, which was subsequently introduced into S. hygroscopicus ATCC 29253 through conjugal transfer [23]. The positive exconjugants were verified by PCR amplification and DNA sequencing with primer pair pIB-F/pIB-R. The construction process of the overexpression plasmids and the location of pIB-F/pIB-R in pIB139 are shown in File 3S in Supplementary Materials.
Analytical methods
Dry cell weight (DCW) was measured by sampling 10 mL fermentation broth. The fermentation broth was centrifuged at 8000×g for 10 min and washed twice with distilled water, then dried to constant weight at 80 °C. The total residual sugar content was measured by phenol–sulfuric acid method with glucose as the standard [12]. The concentration of rapamycin was measured by high-performance liquid chromatography (HPLC) (1200; Agilent Technologies, USA) equipped with a Zorbax SB-C18 analytical column (250 mm × 4.6 mm; Agilent Technologies) [47]. Briefly, 2 mL of culture samples was mixed with 2 mL methanol, and then was shaken intermittently in a water bath at 50 °C for 120 min. After that, the mixed liquor was centrifuged at 6000×g for 10 min and the supernatant was subjected to HPLC analysis. The mobile phase was composed of methanol, acetonitrile, and water (70:15:15, v/v/v) with a flow rate of 1.0 mL/min, using UV detector at 277 nm, and the column temperature was 40 °C. The concentration of rapamycin in the samples was measured using a calibration curve generated by authentic rapamycin standards (MP Biomedicals, USA).
Analysis of in vitro enzyme activities
The culture samples were harvested at the exponential phase (60 h) and the stationary phase (120 h) by centrifugation at 8000×g for 10 min, washed twice with 100 mM Tris–HCl (pH 7) containing 20 mM KCl, 5 mM MnSO4, 2 mM DTT, and 0.1 mM EDTA, and then resuspended in the same buffer. Cells were broken by ultrasonication (UH-250A ULTRASONIC PROCESSOR) under the power output of 250 W on ice bath for 10 min. Cell debris was then removed by centrifugation at 10,000×g for 10 min at 4 °C. The supernatant was further centrifuged at 10,000×g for 20 min at 4 °C, and the resulting supernatant (crude enzyme extract) was used for assay of enzyme activity and the protein concentration. Total protein concentration was quantified by Bradford assay with a reagent solution (Quick Start Bradford Dye, BioRad) [42].
The standard assay for DAHP (3-deoxy-d-arabino-heptulosonate-7-phosphate) synthase activity was spectrophotometrically (549 nm) monitored by following the oxidation with NaIO4 and reaction with thiobarbituric acid at 100 °C. [29]. Chorismatase activity was spectrophotometrically determined by following the decrease in NADH concentration with lactate dehydrogenase as auxiliary enzyme, and the decrease of NADH concentration was followed for 5 min at 340 nm and 25 °C [21]. As described before [37], the standard assay for 6-phosphofructokinase activity was spectrophotometrically determined by following the decrease in NADH concentration with aldolase, triosephosphate isomerase, and glycerophosphate dehydrogenase as auxiliary enzymes, and the decrease of NADH concentration was followed at 340 nm.
Results
GSMM construction for S. hygroscopicus ATCC 29253
In this study, S. hygroscopicus ATCC 29253 GSMM was constructed by integrating the genome annotation results and literature data. A model, including 1003 reactions and 711 metabolites, was obtained after being refined. Here, the biomass composition (File 1S in Supplementary Materials) was obtained by integrating the corresponding results related to other Streptomyces from experiments and references. The energetic coefficients for both cell growth and maintenance were also evaluated based on the previous report [6]. Subsequently, the model constraint conditions were set to ensure accurate calculation. According to the fermentation experiments of S. hygroscopicus ATCC 29253, the specific glucose uptake rate was set to 0.8 mmol/g DCW/h and the specific rapamycin synthetic rate to 5 × 10−4 mmol/g DCW/h as the lower bound. The maximum specific growth rate calculated by FBA was 0.0535 h−1, very close to the experimental results 0.0512 h−1 with an error less than 4.5%, manifesting the high accuracy of our model.
Target gene identification based on in silico simulations
Preliminary simulation results were obtained using FBA and MOMA algorithm. Target genes for improving rapamycin production were identified with the aid of f PH [5]. Reactions that produced an f PH value greater than 1 were chosen as the potential targets. Figure 1 shows f PH of several important reactions closely related to the rapamycin overproduction. The prediction targets include 4 knockout targets and 13 overexpression targets. As shown in Fig. 1, pfk target has the highest ratio (f PH = 3.2) among knockout targets. Therefore, the gene pfk was selected as the experimental knockout target for rapamycin improvement. Peng et al. [43] increased flux of malonyl-CoA by deletion of gene fum; malonyl-CoA is a key precursor to the synthesis of rapamycin, which indirectly confirms the rationality of the knockout target fum. Overexpression targets were divided into primary and secondary metabolic targets. Among these eight primary metabolic targets, ppC and accA were responsible for synthesis of precursors of rapamycin methylmalonyl-CoA and malonyl-CoA, respectively. Jung et al. [22] improved production of rapamycin by overexpression gene ppc, which indicates that overexpression target ppc is correct. It was reported that malonyl-CoA was identified as a key metabolite of rapamycin synthesis by comparative metabolic profiling analysis [47], which indirectly confirmed that the target accA was rational. Targets asd and lysC promoted the synthesis of lysine, which can be transformed into pipecolic acid, a precursor for the synthesis of rapamycin. Target gene dahP, which encodes the first enzyme in the shikimate synthesis pathway, had the highest ratio (f PH = 14.7). Therefore, the gene dahP was selected as the target of primary metabolism for rapamycin improvement. As shown in Fig. 1, f PH of all the secondary metabolic targets are the same. Target genes rapA/B/C were responsible for the polymerization of methylmalonyl-CoA and malonyl-CoA to the polyketide chain, according to [27] rapamycin synthesis was promoted by overexpression of rapH gene, and rapH gene plays a positive role in gene overexpression of rapA/B, indicated that rapamycin synthesis could be promoted by overexpression of rapA/B. Lysine cyclodeaminase encoded by rapL provided pipecolic acid for rapamycin synthesis. Huang et al. [19] promoted the synthesis of FK506 by overexpression of lysine cyclodeaminase in Streptomyces tsukubaensis, which indirectly confirmed that the target rapL was rational because of the close biosynthetic and structural relationships between FK506 and rapamycin. Gene rapK encodes chorismatase, a key enzyme responsible for converting chorismate to 4,5-dihydroxycyclohexa-1,5-dienecarboxylic acid (DCDC), which is followed by the conversion to (4R, 5R)-4,5-dihydroxycyclohex-1-enecarboxylic acid (DHCHC:rapamycin starting unit). As an experimental study, rapK was selected as the secondary metabolic target. Detailed information about genes pfk, dahP, and rapK was shown in Table 3.

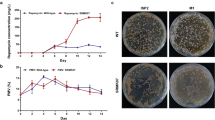
Prediction effect of single gene perturbation on the specific rapamycin production and f PH. The enzymes encoded by these genes are as follows: mdh: malate dehydrogenase; fum: fumarate hydratase gad: glutamate decarboxylase pfk: 6-phosphofructokinase ppC: propionyl-CoA carboxylase; ilvB: acetolactate synthase asd: aspartate-semialdehyde dehydrogenase; ligK: 4-hydroxy-4-methyl-2-oxoglutarate aldolase; metK: S-adenosylmethionine synthetase; lysC: aspartate kinase; accA: acetyl-CoA carboxylase; dahP: 3-deoxy-d-arabinoheptulosonate-7-phosphate synthase; rapK: chorismatase; rapA\B\C: polyketide synthase; rapL: lysine cyclodeaminase (color figure online)
Improving rapamycin production by the perturbation of target genes
To rationally increase the rapamycin production, the target gene pfk was inactivated by the kanamycin resistance cassette without affecting the expression of the downstream genes. The two targets rapK and dahP were overexpressed under the promoter ermE* in Streptomyces hygroscopicus ATCC 29253 using conjugation from Escherichia coli ET12567/pUZ8002. To avoid any unexpected effect caused by the existence of multiple copies of the plasmid itself, the parent strain harboring pIB139 (named S. hygroscopicus-P) was used as the control strain. There were no differences in rapamycin production, cell growth, and morphology between S. hygroscopicus-P and wild-type strain when cultured in fermentation medium (not shown).
There was no change in morphological phenotype between the pfk deletion mutant S. hygroscopicus-Δk and the parent strain ATCC 29253. However, the growth of S. hygroscopicus-Δk strain was slightly affected, with a lower biomass concentration (8.1 ± 0.6 g/L at 120 h) as compared with the parent strain (8.8 ± 0.3 g/L at 120 h) (Fig. 2). In addition, the 6-phosphofructokinase activities of S. hygroscopicus-Δk strain at 60 and 120 h were assayed to 0.15 ± 0.03 and 0.11 ± 0.02 U/mg, respectively, while these were 0.23 ± 0.03 and 0.17 ± 0.02 U/mg of parent strain (Table 4), indicating that the target gene pfk was replaced by the resistance cassette. 6-phosphofructokinase was not completely inactivated by deletion of pfk gene due to the presence of isogenes in S. hygroscopicus. Rapamycin production increased by 30.8%, up to 135.4 ± 6.2 mg/L at 120 h, compared with the parent strain (103.5 ± 4.2 mg/L). The result suggested that altered glucose metabolism by knockout of pfk gene could improve the production of rapamycin.

Experimental effect of genetic perturbation on rapamycin production and cell growth. The data are the average values of at least three series of parallel tests, and error bars represent standard deviations (color figure online)
In the dahP-overexpressed strain S. hygroscopicus-D, the dahP synthase activity was 0.21 ± 0.03 and 0.16 ± 0.02 U/mg at 60 and 120 h, respectively, approximately 1.5 times higher than those of wild type (0.15 ± 0.02 and 0.11 ± 0.02 U/mg at 60 and 120 h, respectively) (Table 4). It indicated that overexpressed gene dahP functioned well in S. hygroscopicus-D. As shown in Fig. 2, the cell growth was changed little for the engineered strain (S. hygroscopicus-D: 8.4 ± 0.3 g/L vs. parent strain ATCC 29253: 8.8 ± 0.3 g/L at 120 h). Importantly, rapamycin production increased by approximately 45%, up to 149.8 ± 6.2 mg/L at 120 h, compared with the parent strain. Accordingly, the specific rapamycin production rate (0.163 ± 0.01 μmol/g DCW/h) for S. hygroscopicus-D was higher than that for the corresponding parent strain ATCC 29253 (0.107 ± 0.005 μmol/g DCW/h). The result suggested that dahP gene overexpression played a positive role in rapamycin production and shikimate pathway may be one of the rate-limited steps in the synthesis of rapamycin.
As shown in Fig. 2, the S. hygroscopicus-R strain showed an increase in rapamycin titer (141.1 ± 6.5 mg/L), which was approximately 36.2% higher than that in the wild-type strain after 120 h cultivation. The chorismatase activity of the resulting strain S. hygroscopicus-R was 0.23 ± 0.02 and 0.19 ± 0.02 U/mg at 60 and 120 h, respectively, approximately 2.1 times those of wild type (0.11 ± 0.02 and 0.09 ± 0.01 U/mg at 60 and 120 h, respectively) (Table 4). Enzyme activity analysis confirmed that the activity of chorismatase in S. hygroscopicus-R was significantly improved compared with the control strain ATCC 29253. The first step of rapamycin biosynthesis is the conversion of chorismate to DHCHC. These findings clearly show that the polyketide starter unit plays a key role in rapamycin biosynthesis.
Effect of combined genetic modifications on rapamycin synthesis
The impact of individual genetic modification on cellular phenotype can be accumulative due to the interactions among the interconnected pathways [20]. S. hygroscopicus-DR with co-expression of genes dahP and rapK, S. hygroscopicus-Δk-DR with gene pfk knockout, and co-expression of genes dahP and rapK were constructed to study their combined effects on the rapamycin synthesis. Furthermore, fermentation kinetics between the S. hygroscopicus ATCC 29253 and strain S. hygroscopicus-Δk-DR were studied.
As shown in Fig. 2, DCW reached 8.1 g/L in engineering strain S. hygroscopicus-DR and 8.8 g/L in the wild-type strain at 120 h. However, rapamycin production reached 210.8 mg/L in S. hygroscopicus-DR at 120 h. Production of rapamycin was significantly improved by co-expression of dahP and rapK. Interestingly, compared with wild-type strain, increment of rapamycin production in S. hygroscopicus-DR exceeded the sum of increments in S. hygroscopicus-D and S. hygroscopicus-R (Fig. 2). This result indicated that co-expression of genes dahP and rapK had a positively synergistic effect on rapamycin production. Enzyme activity of S. hygroscopicus-DR was analyzed to explore the potential mechanism of the synergistic effect. The DAHP synthase activity of S. hygroscopicus-DR was 0.28 ± 0.03 and 0.19 ± 0.02 U/mg at 60 and 120 h, greater than that in S. hygroscopicus-D (0.21 ± 0.03 and 0.16 ± 0.02 U/mg at 60 and 120 h, respectively) (Table 4). However, activity of chorismatase in S. hygroscopicus-DR was about the same as that in S. hygroscopicus-R. We concluded that synergistic effect may be due to the further increase of the DAHP synthase activity in S. hygroscopicus-DR.
As shown in Fig. 2, rapamycin production reached 250.8 mg/L in S. hygroscopicus-Δk-DR at 120 h. However, DCW reached 7.4 g/L in engineering strain S. hygroscopicus-Δk-DR and 8.8 g/L in the wild-type strain at 120 h. Furthermore, fermentation characteristic curve of S. hygroscopicus-Δk-DR was studied. As shown in Fig. 3, the growth of S. hygroscopicus ATCC 29253 and S. hygroscopicus-Δk-DR is basically synchronous but DCW decreased obviously with S. hygroscopicus-Δk-DR. The results suggested that intracellular fluxes in S. hygroscopicus-Δk-DR might be altered through the biomass precursors towards rapamycin biosynthesis, thus resulting in increased rapamycin biosynthesis at the expense of decreased biomass as the cell growth and the synthesis of rapamycin share some common precursors.

Fermentation characteristics between the wild-type S. hygroscopicus ATCC 29253 and strain S. hygroscopicus-Δk-DR under the same culture condition. WT represents wild-type strain S. hygroscopicus ATCC 29253, and com represents S. hygroscopicus-Δk-DR. The data are the average values of at least three series of three parallel tests, and error bars represent standard deviations (color figure online)
Discussion
GSMM provides an approach for strain optimization—system metabolic engineering. This approach can be used to identify target genes in synthesis of target product thus improve product yield on the basis of metabolic engineering. In fact, this strategy has been successfully used to identify target genes for the improvement of biochemical products, such as lysine, flavanone, and putrescine [4, 14, 31]. In the present work, the GSMM of S. hygroscopicus ATCC 29253 was constructed and used to identify gene targets for improved rapamycin production.
Among the initial predicted targets, 4 knockout targets and 13 overexpression targets were identified using FBA and MOMA algorithm raised by [5]. Rapamycin production increased by 30.8% with pfk gene knockout. Irina et al. [7] improved production of actinorhodin and undecylprodigiosin by pfkA2 gene knockout in Streptomyces coelicolor. 6-Phosphofructokinase encoded by pfk played an important role in the flux distribution of EMP and PPP glycolytic pathways. We suggested that PPP glycolytic pathway was enhanced by deletion of pfk resulting in formation of more NADPH, as a redox precursor which promoted the production of rapamycin. A role for NADPH in enhancement of antibiotic production has also been suggested before [15].
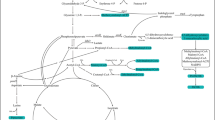
Target gene dahP encodes DAHP synthase, the first enzyme in the shikimate pathway. As shown in Fig. 4, the reaction catalyzed by DAHP synthase may play an important role in carbon flux distribution by connecting the pentose phosphate pathway and shikimate pathway. It was reported that aroG (encoding DAHP synthase) overexpression could significantly increase the flux of shikimate pathway in Escherichia coli [9]. In addition, exogenously feeding shikimate can promote rapamycin production in Streptomyces hygroscopicus [13, 47]. In fact, the primary shikimate pathway played a key role in the supply of precursor chorismate, which was used as the donor in DHCHC biosynthesis [2]. In addition, dahP overexpression has been previously applied to improve FK506 yields by enhancing shikimate pathway [18]. However, the yield increment was not so much as predicted by the algorithm. The bottleneck may be that metabolic regulation on the model was not taken into account in the process of simulation. In addition, DAHP synthase may be strictly controlled by feedback inhibition. Three DAHP synthase isozymes encoded by aroF, aroG, and aroH are sensitive to tyrosine, phenylalanine, and tryptophan, respectively, in E. coli. [40]. Increase of DAHP synthase activity was limited due to feedback inhibition in S. hygroscopicus-D which resulted in the difference between experimental and simulation results. It is necessary to apply specific metabolic adaptations to resist feedback inhibition [17]. Fortunately, it has been revealed that some antibiotic biosynthetic clusters contain another isoenzyme gene, which is not inhibited by aromatic amino acids [16, 33, 46]. Therefore, it provides an opportunity to further enhance rapamycin production. The other target gene rapK encodes chorismatase, which catalyzes hydrolysis of chorismate into DCDC that can be converted to DHCHC. DHCHC, as the polyketide starter unit, was considered to be one of the important precursors to promote the synthesis of FK506 [19]. As shown in Fig. 4, chorismate not only can be transformed into aromatic amino acids for microbial growth, but also can be converted to DHCHC for the synthesis of rapamycin. As a common precursor of microbial growth and rapamycin synthesis, the high efficient transformation from chorismate to DHCHC may be an important factor to promote the production of rapamycin. Overexpression of gene rapK resulted in a 36.2% increase in rapamycin yield in S. hygroscopicus-R. The result indicated that intracellular formation of DHCHC seemed to be one of the rate-limiting factors. Crystal Structure and amino acid site-directed mutagenesis of chorismatase had been studied by Juneja et al. [21]. This gives us some insight into improvement of rapamycin by enhancing the specific activity of chorismatase.

Schematic overview of rapamycin biosynthetic pathway in Streptomyces hygroscopicus ATCC 29253. The experimental overexpression targets for improved rapamycin production are shown in green. The experimental knockout target for improved rapamycin production is shown in red. The shaded boxes represent precursors of rapamycin biosynthesis (color figure online)
With the help of GSMM, production of rapamycin was promoted by genetic modification on target genes. However, the observed improvements in the specific rapamycin production rate were different from the predicted improvements. These differences might be due to the fact that effects of metabolic regulation on the model were not taken into account in the process of simulation. Therefore, there existed some limitations in our constraint-based metabolic model. Even so, the observed improvement in the rapamycin production proved the validity of the metabolic engineering target. In addition, rapamycin, as a secondary metabolite, has a complex bio-synthetic pathway, and there may be a number of rate-limiting steps in the synthetic pathway. The production of rapamycin may not be promoted significantly if we only pay attention to a certain gene modification alone. Recently, strategy of multiple gene perturbation was employed to improve production of microbial products. It was reported that production of FK506 was significantly improved by perturbation of multiple genes simultaneously [18]. The production of rapamycin was further promoted by co-expression of genes rapK and dahP. Furthermore, there existed synergistic effect between genes dahP and rapK in promoting the improvement of rapamycin. The differences of DAHP enzyme activity in S. hygroscopicus-D and S. hygroscopicus-DR further confirmed that DAHP synthase was subjected to feedback inhibition. In addition, the results gave us insight into causes of synergistic effect. As shown in Fig. 4, Shikimate can be transformed into chorismate which was a common precursor in synthesis of rapamycin and aromatic amino acids in Streptomyces hygroscopicus. Overexpression of chorismatase can promote the transformation from chorismate to structural unit of rapamycin in S. hygroscopicus-DR. More metabolic flux to DHCHC reduced the accumulation of aromatic amino acids and weakened the feedback inhibition to DAHP synthase, but provided more precursors for the synthesis of rapamycin. At last, the production of rapamycin reached 250.8 mg/L by co-expression of genes rapK and dahP in S. hygroscopicus-Δk. The production of rapamycin was significantly improved by combined genetic modifications.
In the present study, the GSMM-guided metabolic engineering strategy was employed to improve the rapamycin production of S. hygroscopicus ATCC 29253. Fermentation characterization of the engineered strains with pfk gene knockout and rapK, dahP overexpression showed the improved capacities of rapamycin production. The production of rapamycin was further improved through target gene pfk knock out and co-expression of target genes dahP and rapK simultaneously. The titer of rapamycin reached 250.8 mg/L in engineering strain S. hygroscopicus-Δk-DR compared with parent strain (103.5 mg/L). Our method for the industrial production of macrocyclic polyketide (rapamycin) is a successful example which can be applied to guide improvement of other secondary metabolites.
References
Alper H, Jin Y-S, Moxley J, Stephanopoulos G (2005) Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab Eng 7:155–164
Andexer JN, Kendrew SG, Nur-e-Alam M, Lazos O, Foster TA, Zimmermann A-S, Warneck TD, Suthar D, Coates NJ, Koehn FE (2011) Biosynthesis of the immunosuppressants FK506, FK520, and rapamycin involves a previously undescribed family of enzymes acting on chorismate. Proc Natl Acad Sci 108:4776–4781
Baranasic D, Gacesa R, Starcevic A, Zucko J, Blažič M, Horvat M, Gjuračić K, Fujs Š, Hranueli D, Kosec G (2013) Draft genome sequence of Streptomyces rapamycinicus strain NRRL 5491, the producer of the immunosuppressant rapamycin. Genom Announc 1:e00581–e00613
Becker J, Zelder O, Häfner S, Schröder H, Wittmann C (2011) From zero to hero—Design-based systems metabolic engineering of Corynebacterium glutamicum for l-lysine production. Metab Eng 13:159–168
Boghigian BA, Armando J, Salas D, Pfeifer BA (2012) Computational identification of gene over-expression targets for metabolic engineering of taxadiene production. Appl Microbiol Biotechnol 93:2063–2073
Borodina I, Krabben P, Nielsen J (2005) Genome-scale analysis of Streptomyces coelicolor A3 (2) metabolism. Genom Res 15:820–829
Borodina I, Siebring J, Zhang J, Smith CP, van Keulen G, Dijkhuizen L, Nielsen J (2008) Antibiotic overproduction in Streptomyces coelicolor A3 (2) mediated by phosphofructokinase deletion. J Biol Chem 283:25186–25199
Calne R, Lim S, Samaan A, Collier DSJ, Pollard S, White D, Thiru S (1989) Rapamycin for immunosuppression in organ allografting. Lancet 334:227
Chen X, Li M, Zhou L, Shen W, Algasan G, Fan Y, Wang Z (2014) Metabolic engineering of Escherichia coli for improving shikimate synthesis from glucose. Bioresour Technol 166:64–71
Chen X, Wei P, Fan L, Yang D, Zhu X, Shen W, Xu Z, Cen P (2009) Generation of high-yield rapamycin-producing strains through protoplasts-related techniques. Appl Microbiol Biotechnol 83:507–512
Douros J, Suffness M (1981) New antitumor substances of natural origin. Cancer Treat Rev 8:63–87
Dubois M, Gilles KA, Hamilton JK, Rebers P, Smith F (1956) Colorimetric method for determination of sugars and related substances. Anal Chem 28:350–356
Fang A, Demain A (1995) Exogenous shikimic acid stimulates rapamycin biosynthesis in Streptomyces hygroscopicus. Folia Microbiol 40:607–610
Fowler ZL, Gikandi WW, Koffas MA (2009) Increased malonyl coenzyme A biosynthesis by tuning the Escherichia coli metabolic network and its application to flavanone production. Appl Environ Microbiol 75:5831–5839
Gunnarsson N, Eliasson A, Nielsen J (2004) Control of fluxes towards antibiotics and the role of primary metabolism in production of antibiotics. In Molecular biotechnology of fungal beta-lactam antibiotics and related peptide synthetases. Springer, Berlin, pp 137–178
He J, Magarvey N, Piraee M, Vining L (2001) The gene cluster for chloramphenicol biosynthesis in Streptomyces venezuelae ISP5230 includes novel shikimate pathway homologues and a monomodular non-ribosomal peptide synthetase gene. Microbiology 147:2817–2829
Helmstaedt K, Strittmatter A, Lipscomb WN, Braus GH (2005) Evolution of 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase-encoding genes in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA 102:9784–9789
Huang D, Li S, Xia M, Wen J, Jia X (2013) Genome-scale metabolic network guided engineering of Streptomyces tsukubaensis for FK506 production improvement. Microb Cell Fact 12:1
Huang D, Xia M, Li S, Wen J, Jia X (2013) Enhancement of FK506 production by engineering secondary pathways of Streptomyces tsukubaensis and exogenous feeding strategies. J Ind Microbiol Biotechnol 40:1023–1037
Jin Y-S, Stephanopoulos G (2007) Multi-dimensional gene target search for improving lycopene biosynthesis in Escherichia coli. Metab Eng 9:337–347
Juneja P, Hubrich F, Diederichs K, Welte W, Andexer JN (2014) Mechanistic implications for the chorismatase FkbO based on the crystal structure. J Mol Biol 426:105–115
Jung WS, Yoo YJ, Park JW, Park SR, Han AR, Ban YH, Kim EJ, Kim E, Yoon YJ (2011) A combined approach of classical mutagenesis and rational metabolic engineering improves rapamycin biosynthesis and provides insights into methylmalonyl-CoA precursor supply pathway in Streptomyces hygroscopicus ATCC 29253. Appl Microbiol Biotechnol 91:1389–1397
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical streptomyces genetics. John Innes Foundation, Norwich
Kim YH, Park BS, Bhatia SK, Seo H-M, Jeon J-M, Kim H-J, Yi D-H, Lee J-H, Choi K-Y, Park H-Y (2014) Production of rapamycin in Streptomyces hygroscopicus from glycerol-based media optimized by systemic methodology. J Microbiol Biotechnol 24:1319–1326
Kosec G, Goranovič D, Mrak P, Fujs Š, Kuščer E, Horvat J, Kopitar G, Petković H (2012) Novel chemobiosynthetic approach for exclusive production of FK506. Metab Eng 14:39–46
Kumar VS, Dasika MS, Maranas CD (2007) Optimization based automated curation of metabolic reconstructions. BMC Bioinform 8:1
Kuščer E, Coates N, Challis I, Gregory M, Wilkinson B, Sheridan R, Petković H (2007) Roles of rapH and rapG in positive regulation of rapamycin biosynthesis in Streptomyces hygroscopicus. J Bacteriol 189:4756–4763
Lee SJ, Lee D-Y, Kim TY, Kim BH, Lee J, Lee SY (2005) Metabolic engineering of Escherichia coli for enhanced production of succinic acid, based on genome comparison and in silico gene knockout simulation. Appl Environ Microbiol 71:7880–7887
Liu Y-J, Li P-P, Zhao K-X, Wang B-J, Jiang C-Y, Drake HL, Liu S-J (2008) Corynebacterium glutamicum contains 3-deoxy-d-arabino-heptulosonate 7-phosphate synthases that display novel biochemical features. Appl Environ Microbiol 74:5497–5503
Park JH, Lee KH, Kim TY, Lee SY (2007) Metabolic engineering of Escherichia coli for the production of l-valine based on transcriptome analysis and in silico gene knockout simulation. Proc Natl Acad Sci 104:7797–7802
Park JM, Park HM, Kim WJ, Kim HU, Kim TY, Lee SY (2012) Flux variability scanning based on enforced objective flux for identifying gene amplification targets. BMC Syst Biol 6:106
Patsenker E, Schneider V, Ledermann M, Saegesser H, Dorn C, Hellerbrand C, Stickel F (2011) Potent antifibrotic activity of mTOR inhibitors sirolimus and everolimus but not of cyclosporine A and tacrolimus in experimental liver fibrosis. J Hepatol 55:388–398
Pierson LS, Gaffney T, Lam S, Gong F (1995) Molecular analysis of genes encoding phenazine biosynthesis in the biological control bacterium Pseudomonas aureofaciens 30–84. FEMS Microbiol Lett 134:299–307
Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S (2011) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2. 0. Nat Protoc 6:1290–1307
Segre D, Vitkup D, Church GM (2002) Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci 99:15112–15117
Sehgal S, Baker H, Vézina C (1975) Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot 28:727–732
Sharma B (2011) Modulation of phosphofructokinase (PFK) from Setaria cervi, a bovine filarial parasite, by different effectors and its interaction with some antifilarials. Parasit Vectors 4:1
Sinha R, Singh S, Srivastava P (2014) Studies on process optimization methods for rapamycin production using Streptomyces hygroscopicus ATCC 29253. Bioprocess Biosyst Eng 37:829–840
Thiele I, Palsson BØ (2010) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc 5:93–121
Umbarger HE (1978) Amino acid biosynthesis and its regulation. Annu Rev Biochem 47:533–606
Wilkinson CJ, Hughes-Thomas ZA, Martin CJ, Bohm I, Mironenko T, Deacon M, Wheatcroft M, Wirtz G, Staunton J, Leadlay PF (2002) Increasing the efficiency of heterologous promoters in actinomycetes. J Mol Microbiol Biotechnol 4:417–426
Wood CE, Giroux D, Gridley K (2003) Fetal brain regional responses to cerebral hypoperfusion: modulation by estrogen. Brain Res 993:84–89
Xu P, Ranganathan S, Fowler ZL, Maranas CD, Koffas MA (2011) Genome-scale metabolic network modeling results in minimal interventions that cooperatively force carbon flux towards malonyl-CoA. Metab Eng 13:578–587
Xu ZN, Shen WH, Chen XY, Lin JP, Cen PL (2005) A high-throughput method for screening of rapamycin-producing strains of Streptomyces hygroscopicus by cultivation in 96-well microtiter plates. Biotechnol Lett 27:1135–1140
Yim H, Haselbeck R, Niu W, Pujol-Baxley C, Burgard A, Boldt J, Khandurina J, Trawick JD, Osterhout RE, Stephen R (2011) Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol 7:445–452
Yu T-W, Müller R, Müller M, Zhang X, Draeger G, Kim C-G, Leistner E, Floss HG (2001) Mutational analysis and reconstituted expression of the biosynthetic genes involved in the formation of 3-amino-5-hydroxybenzoic acid, the starter unit of rifamycin biosynthesis in Amycolatopsis mediterranei S699. J Biol Chem 276:12546–12555
Zhao S, Huang D, Qi H, Wen J, Jia X (2013) Comparative metabolic profiling-based improvement of rapamycin production by Streptomyces hygroscopicus. Appl Microbiol Biotechnol 97:5329–5341
Acknowledgements
This work was financially supported by the National 973 Project of China (No. 2013CB733600), the Key Program of National Natural Science Foundation of China (No. 21236005), and the National Natural Science Foundation of China (No. 21376171).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dang, L., Liu, J., Wang, C. et al. Enhancement of rapamycin production by metabolic engineering in Streptomyces hygroscopicus based on genome-scale metabolic model. J Ind Microbiol Biotechnol 44, 259–270 (2017). https://doi.org/10.1007/s10295-016-1880-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-016-1880-1