
Abstract
The tumor microenvironment (TME) dynamically regulates cancer progression and affects clinical outcomes. This study aimed to identify molecular subtypes and construct a prognostic risk model based on TME-related signatures in skin cutaneous melanoma (SKCM) patients. We categorized SKCM patients based on transcriptome data of SKCM from The Cancer Genome Atlas (TCGA) database and 29 TME-related gene signatures. Differentially expressed genes were identified using univariate Cox regression and Lasso regression analysis, which were used for risk model construction. The robustness of this model was validated in independent external cohorts. Genetic landscape alterations, immune characteristics, and responsiveness to immunotherapy/chemotherapy were evaluated. Three TME-related subtypes were identified, and subtype C3 exhibited the most favorable prognosis, had enriched immune-related pathways, and possessed more infiltration of T_cells_CD8, T_cells_CD4_memory_activated, and Macrophages_M1 but a lower TumorPurity, whereas Macrophages_M2 were increased in subtype C1 and subtype C2. Subtype C1 was more sensitive to Cisplatin, subtype C2 was more sensitive to Temozolomide, and subtype C3 was more sensitive to Paclitaxel; 8 TME-related genes (NOTCH3, HEYL, ZNF703, ABCC2, PAEP, CCL8, HAPLN3, and HPDL) were screened for risk model construction. High-risk patients had dismal prognosis with good prediction performance. Moreover, low-risk patients were more sensitive to Paclitaxel and Temozolomide, whereas high-risk patients were more sensitive to Cisplatin. This risk model had robustness in predicting prognosis in SKCM patients. The results facilitate the understanding of TME-related genes in SKCM and provide a TME-related genes-based predictive model in prognosis and direction of personalized options for SKCM patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Skin cutaneous melanoma (SKCM) is one of the live-threaten malignant cancers of the skin. Accordingly, the estimated new diagnosed cases are 324,635, and the new deaths are 57,043 worldwide in 2020 (Sung et al. 2020). Early diagnosis of SKCM (Breslow thickness less than 0.8 mm) is essential to clinical outcome. Unfortunately, it is difficult to distinguish SKCM from benign pigmented lesions, leading to its exacerbation and metastasis (Santos, et al. 2018). Additionally, SKCM is a highly heterogeneous malignancy due to the complex and layered structure of human skin with a large body of specialized cells, which may result in drug resistance as well as a dismal prognosis (Thrane et al. 2018; Novotný et al. 2020). At present, although the traditional therapeutic strategies including surgery, radiotherapy, and chemotherapy have improved the prognosis of SKCM patients, only a few advanced SKCM patients can receive surgical intervention and drug resistance remains a great challenge. Meanwhile, novel-targeted therapy through mutant-BRAF inhibition, immunotherapies, and intratumoral application of immunomodulators exhibit potential for SKCM treatment (Kasakovski et al. 2021). However, the heterogeneity of SKCM affects the response to these therapeutic options. Thus, understanding the intricacy of SKCM is necessary for clinical classification, prognosis prediction, and patient-centered therapeutic interventions.
It has been acknowledged that tumor microenvironment (TME) is imperative in cancer progression and affects therapeutic response. TME generally comprises multiple cell types containing cancer cells and non-cancer cells including immune cells, extracellular matrix, heterogeneous stromal cells, blood, and lymphatic vascular and secreted factors. Their intricate interaction is crucial in the immortality of replication, invasion, metastasis, and immune escape (Najafi et al. 2019). Immune infiltrates are one of the most important components in the TME, and it has been reported that immune infiltrates are responsible for the development and progress of tumors as well as either success or failure of cancer-targeted therapies (Zhang and Zhang 2020). More CD8 + T-cell infiltration significantly prolongs survival in patients with SKCM (Zhu et al. 2021). Activated natural killer (NK) cell has been proven to be associated with the prognosis of SKCM patients (Cursons et al. 2019). Recently, several novel TICs genes such as CCL8, C1QA, C1QB, and GZMB have been identified as prognostic biomarkers for SKCM patients (Liang et al. 2022; Yang et al. 2021). We understand that deep machine learning could help predict immune response and prognosis in cancer (Jin et al. 2022; Zhao et al. 2022), including head and neck squamous cell carcinoma (Chi et al. 2022a; Chi et al. 2022b), glioblastoma, and Parkinson’s disease (Zhao et al. 2023). Recently, several prognostic signatures have been found to predict the prognosis and immune microenvironment of SKCM (Song et al. 2022a; Song et al. 2022b). An in-depth understanding of TME can reveal the mechanisms of TME in SKCM, facilitate clinical classification, identify predictive biomarkers, and improve anti-tumor therapies.
Hence, in this study, we identified three TME-related molecular subtypes based on RNA-Seq data of SKCM patients from The Cancer Genome Atlas (TCGA) database and 29 TME-related gene signatures, and their associations with clinicopathological features, genomic landscape, immune characteristics, and anti-tumor therapy response were evaluated. Furthermore, we established and validated a prognostic risk model that could predict prognosis and response to immunotherapy/chemotherapy. These results facilitate an understanding of the potential mechanisms of TME-related genes in SKCM and provide a direction to improve prognosis as well as personalized therapies for patients with SKCM.
Material and methods
Data collection and pre-processing
RNA-Seq data of SKCM patients was obtained in The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) database using Genomic Data Commons Application Programming Interface, including 354 metastatic tumor samples. Patients lacking clinical follow-up information and survival states were eliminated, while patients with survival time > 30 days were retained in this study. Next, FPKM was converted into TPM, and ensembl gene IDs were converted into gene symbol IDs. The gene with multiple gene symbols was expressed as a median. Additionally, the gene expression profiles of SKCM samples from GSE69504 (containing 186 samples) and GSE54467 (containing 79 samples) were downloaded from the Gene-Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database. We removed the probe matching to multiple genes. The gene with multiple probes was expressed as average. To investigate the TME-related gene signatures, we obtained 29 types of functional gene expression characteristics representing the main functional components of tumor, immune, matrix, and other cell populations from published research (Bagaev et al. 2021).
The association between TME gene signatures and clinical characteristics in TCGA-SKCM
Based on 29 TME-related gene signatures, we score the TME using single sample gene set enrichment analysis (ssGSEA), and the differences of TME between primary solid tumor samples and metastatic samples were analyzed using the Kruskal test. P < 0.05 was considered statistically significant. The correlation of TME with the primary solid tumor and the metastatic tumor was analyzed by the “ggcorrplot” package (Kassambara 2019). Univariate Cox regression analysis was conducted to investigate the relationship between TME and prognosis. Next, we compared the TME in different T stage, N stage, M stage, stage, age, and gender.
Identification of TME-related subtypes
On the basis of the TME gene signatures score, a consensus clustering analysis was conducted to categorize TCGA-SKCM patients using the “ConsensusClusterPlus” package (Wilkerson et al. 2013), with pam algorithm and 1-Pearson correlation, processing with 500 bootstraps containing 80% TCGA-SKCM patients; 2–10 clusters were tested, and the cumulative distribution function (CDF) and consensus matrix were employed to determine the optimal subtypes. Kaplan–Meier curves were generated in the TCGA-SKCM cohort, and the log-rank test was used to determine the significance of differences. The “ComplexHeatmap” package (Gu 2022) was employed to visualize the distributions of TME-related signatures among the three subtypes.
Construction and validation of the prognostic model
Differential expression analysis among molecular subtypes was performed using the “limma” package (Ritchie et al. 2015) to identify differentially expressed genes (DEGs). Next, univariate Cox regression analysis was used to screen genes that had a greater impact on prognosis with P < 0.01. In order to reduce the number of genes, we performed LASSO Cox regression using the “glmnet” package (Hastie et al. 2021), along with stepwise multivariate regression analysis with stepwise Akaike information criterion (stepAIC). The risk score was calculated using the following formula: RiskScore = + 0.131*NOTCH3 + 0.093*HEYL + 0.084*ZNF703 + 0.091*ABCC2 + 0.046*PAEP-0.147*CCL8-0.255*HAPLN3 + 0.087*HPDL. According to the above formula, we calculated the risk score in the TCGA-SKCM cohort. Receiver operating characteristic (ROC) analysis was conducted using the “timeROC” package (TimeROC 2015), followed by areas under the ROC curve (AUCs) for 1 year, 3 years, and 5 years. The optimal cutoff was determined by the “survminer” package (Kassambara et al. 2017). Kaplan–Meier curves were generated between high- and low-risk groups, and the log-rank test was used to determine the significance of differences. Furthermore, we validated the robustness of this prognostic model in the GSE65904 and GSE54467 cohorts.
Genetic landscape alterations
To assess the differences in genomic changes, we downloaded Simple Nucleotide Variation (SNV) dataset at the level4 of TCGA-SKCM samples processed by MuTect2 software and calculated the tumor mutation burden (TMB), mutant-allele tumor heterogeneity (MATH), and homologous recombination defects (HRD) using “maftools” package (version 2.8.05) (Mayakonda et al. 2018) in R. From the previous study (Akbani et al. 2015), we acquired other subtypes and analyzed the distribution of reported subgroups in this TME-related molecular subtypes. Furthermore, a somatic mutation was evaluated using the Chi-square test among molecular subtypes. Comparisons were performed by The Kruskal test. P < 0.05 was considered statistically significant. The correlation between risk score and 22 immune cells as well as 29 gene signatures was analyzed by the “ggcorrplot” package (Kassambara 2019).
Gene set enrichment analysis and functional annotation
To evaluate the different biological processes among molecular subtypes, we performed gene set enrichment analysis (GSEA) using all candidate gene sets in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database with the threshold of false discovery rate (FDR) < 0.05. Functional gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed using the “clusterProfiler” package (Yu et al. 2012). The GO functional enrichment included biological process (BP), cellular component (CC), and molecular function (MF) categories. Meanwhile, ssGSEA was conducted using the “GSVA” package (Hänzelmann et al. 2013) using Hallmark gene sets to calculate each sample on different functions, and the Kruskal test was used to perform statistics. P < 0.05 was considered statistically significant.
Assessment of immune characteristics
Subsequently, immune cell infiltration was assessed in the TCGA-SKCM cohort using the ESTIMATE algorithm (Yoshihara et al. 2013) among molecular subtypes containing StromalScore, ImmuneScore, ESTIMATEScore, and TumorPurity. Besides, CIBERSORT (https://cibersort.stanford.edu/) (Chen et al. 2018) was employed to evaluate the abundance of 22 immune cells. Comparisons were analyzed using the Kruskal test. Distributions of ESTIMATE score and immune cells among molecular subtypes as well as 29 TME-related gene signatures between risk groups were visualized using the “ComplexHeatmap” package (Gu 2022).
Prediction of responsiveness to immunotherapy/chemotherapy
To evaluate the responsiveness to chemotherapy, we analyzed the half-maximal inhibitory concentration (IC50) values of traditional chemotherapeutic drugs including Paclitaxel, Cisplatin, Vinblastine, and Temozolomide using the “pRRophetic” package (Geeleher et al. 2014) in R. Moreover, we assessed the predictive potential of different subtypes of immunotherapy using a T-cell-inflamed GEP score. In addition, we scored cytolytic activity (CYT) as well as Th1/IFNγ gene signature (Danilova et al. 2019) with ssGSEA. Furthermore, we assessed some presentative immune checkpoints based on a published study (Auslander et al. 2018). Differential expressed immune checkpoint genes were screened using the Kruskal test, and FDR < 0.05 was considered statistically significant. Correlation analysis was performed between risk score and immune checkpoints and visualized using the “ggcorrplot” package.
Clinical performance and prognosis analysis of risk score
To test the relationship between risk score and clinicopathological features of TCGA-SKCM patients, the distribution of risk scores among different clinicopathological features including T stage, N stage, M stage, stage, age, gender, and three molecular subtypes and differences were analyzed using the Kruskal test. Sankey diagram detailed the distribution of three subtypes between high- and low-risk groups. Kaplan–Meier curves were generated between high- and low-risk groups among different clinicopathological features.
Results
Association of TME-related gene signatures and clinicopathologic features in TCGA-SKCM
To assess the difference in TME between primary solid tumor samples and metastatic samples, we scored 29 TME-related gene signatures and found that most TME-related gene signatures such as angiogenesis, endothelium, cancer-associated fibroblasts, tumor-associated macrophages, immune suppression by myeloid cells, MHCII, and anti-tumor cytokines had higher ssGSEA scores in metastatic samples than that of primary solid tumor samples; meanwhile, immune cells in the metastatic samples were much more abundant (Fig. 1A). Figure 1B revealed more correlations between TMEs in metastatic samples. Univariate Cox regression analysis showed that TME was significantly associated with the survival of patients (Fig. 1C). Besides, tumor-associated macrophages, NK cells, immune suppression by myeloid cells, anti-tumor cytokines, M1 signature, and MHCII were distinctly increased in female (Fig. 1D).

Association of TME-related gene signatures and clinical characteristics in TCGA-SKCM patients. A, Difference of TME between primary solid tumor samples and metastatic samples. B, Correlation analysis of TMEs in metastatic samples. C, Forest plots of univariate Cox regression analysis. D, Differences of TME among clinicopathologic features (TNM stage, stage, age, and gender). ns represents P > 0.05; *P < 0.05, **P < 0.01, ***P < 0.001, and.****P < 0.0001
Three TME-related molecular subtypes were identified
Based on the TME gene signatures score, we performed a consensus clustering analysis to categorize TCGA-SKCM patients. The optimal number of clusters was determined according to the CDF. CDF Delta area displayed a relatively stable clustering effect when cluster = 3 (Supplementary Fig. S1A,B). Finally, we identified three subtypes when consensus matrix k = 3 (Fig. 2A). Principal components analysis (PCA) showed a distinct separation among the three subtypes (Fig. 2B). Subsequently, survival analysis deciphered subtype C3 had the highest survival probability while subtype C1 exhibited the dismal prognosis in TCGA-SKCM cohort (P < 0.0001) (Fig. 2C). Furthermore, Fig. 2D displayed that these TME signatures had significant differences among three molecular subtypes except for the neutrophil signature. Figure 2E depicted the distributions of TME-related signatures among the three subtypes. Immune-related signatures were remarkably increased in subtype C3. Matrix-related signatures including angiogenesis, endothelium, cancer-associated fibroblasts, matrix, matrix remodeling, and EMT signature were enriched in subtype C2, while tumor proliferation rate was enriched in subtype C1.

Identification of TME-related molecular subtypes in TCGA-SKCM patients. A, Consensus clustering heatmap when consensus k = 3. B, Principal components analysis showing a distinct separation among the three subtypes. C, Kaplan–Meier curves of OS among the three subtypes. D, Box plots of TME signatures among the three subtypes. E, Heatmap diagram of the distribution of TME signatures among the three subtypes. ns represents P > 0.05; **P < 0.01, and.****P < 0.0001
Differences in clinicopathologic features among three subtypes
Furthermore, we compared the clinicopathological differences (T stage, N stage, M stage, stage, age, gender, and survival status) between the three molecular subtypes in the TCGA-SKCM cohort. As shown in Fig. 3, patients with subtype C1 exhibited a higher proportion of advanced T stage and a number of males.

Differences of clinicopathologic features among three subtypes. Distributions of clinicopathological differences including T stage (A), N stage (B), M stage (C), stage (D), age (E), gender (F), and survival status (G)
Genetic landscape alterations among three subtypes
To evaluate genetic landscape alterations among three subtypes, we analyzed the difference of TMB, MATH, and HRD and found that there was no significant difference in TMB among the three subtypes (P > 0.05), while subtype C1 possessed the lowest MATH and the highest HRD score than that of others (Fig. 4A–C). Besides, we found subtype C2 had a higher proportion of BRAF_Hotspot_Mutants (Fig. 4D). Moreover, we assessed the somatic mutation among three subtypes in the TCGA-SKCM cohort. Figure 4E revealed that TACC2 (32.0%), SLCO1B3 (24.4%), and MDN1 (16.2%) were the most frequently mutated genes in the TCGA-SKCM cohort.

Genetic landscape alterations among three subtypes in the TCGA-SKCM cohort. A–C, Changes of TMB, MATH, and HRD score among three subtypes. D, Distribution of reported subgroups among three subtypes. E, Alteration of somatic mutation among three subtypes (Chi-square test)
Biological pathway characteristics among three subtypes
Next, the biological pathway characteristics were evaluated among three subtypes in the TCGA-SKCM cohort. Figure 5A displayed that KEGG_ AMNOACYL_TRNA_BOSYNTHEBIS, KEGG_CITRATE_CYCLE_TCA_CYCLE, and KEGG_TERPENOD_BACKBONE_BIOSYNTHESES were significantly enriched in subtype C1. EMT-related pathways such as KEGG_TGF_BETA_SIGNALING_PATHWAY and KEGG_WNT_SIGNALING_PATHWAY were enriched in subtype C2 (Fig. 5B). Some immune-related pathways such as KEGG_CHEMOKINE_SIGNALING_PATHWAY, KEGG_CYTOKINE_CYTOKINE_RECEPTOR_INTERACTION, KEGG_NATURAL_KILLER_CELL_MEDIATED_CYTOTOXICITY, and KEGG_TOLL_LIKE_RECEPTOR_SIGNALING_PATHWAY were enriched in subtype C3 (Fig. 5C). Meanwhile, we found that cell cycle-related pathways including HALLMARK_MYC_TARGETS_V1, HALLMARK_G2M_CHECKPOINT, HALLMARK_E2F_TARGETS, HALLMARK_DNA_REPAIR, and HALLMARK_MYC_TARGETS_V2 were observed in subtype C1. Similarly, EMT-related pathways HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION, HALLMARK_WNT_BETA_CATENIN_SIGNALING, and HALLMARK_NOTCH_SIGNALING were found in subtype C2. Immune-related pathways were also observed in subtype C3 (Fig. 5D, E).

Biological pathway characteristics among three subtypes in the TCGA-SKCM cohort. Results of GSEA between subtype C1 vs. other subtypes (A), C2 vs. other subtypes (B), and C3 vs. other subtypes (C). D, Heatmap diagram of pathway enrichment among three subtypes. E, Box plots of pathway enrichment scores among three subtypes. **P < 0.01, ***P < 0.001, and ****P < 0.0001
Changes in immune characteristics among three subtypes
In order to clarify the difference in immune microenvironment among different molecular subtypes, we evaluated the degree of immune cell infiltration of TCGA-SKCM patients using the gene expression levels of immune cells. As displayed in Fig. 6A–D, subtype C3 possessed higher ImmuneScore and ESTIMATEScore but a lower TumorPurity than that of other subtypes. We further assessed the relative abundance of 22 immune cells (Fig. 6E, F). T_cells_CD8, T_cells_CD4_memory_activated, and Macrophages_M1 were highly expressed in subtype C3, whereas T_cells_CD4_memory_resting, Macrophages_M0, and Macrophages_M2 were increased in subtype C1 and subtype C2.

Immune characteristics among three subtypes in the TCGA-SKCM cohort. A–D, ESTIMATE evaluating ImmuneScore, ImmuneScore, ESTIMATEScore, and TumorPurity among three subtypes. E, Box plots of relative abundance of 22 immune cells among three subtypes. F, Heatmap diagram of the abundance of 22 immune cells among three subtypes. ns represents P > 0.05; *P < 0.05, **P < 0.01, ***P < 0.001, and. ****P < 0.0001
Prediction of responsiveness to immunotherapy/chemotherapy among three subtypes
To evaluate the response of three subtypes to anti-tumor therapies, we analyzed the response of three subtypes to traditional chemotherapeutic drugs and found that subtype C1 was more sensitive to Cisplatin, subtype C2 was more sensitive to Temozolomide, and subtype C3 was more sensitive to Paclitaxel (Fig. 7A). Meanwhile, we assessed the predictive potential of three subtypes to immunotherapy using T-cell-inflamed GEP (Fig. 7B). T-cell–inflamed GEP score was remarkably elevated in subtype C3. Subtype C3 also exhibited Th1/IFNγ gene signature (Fig. 7C) and more CYT score (Fig. 7D). Figure 7E displayed that most of the immune checkpoints that activate immune and inhibit immune were significantly upregulated in subtype C3. Furthermore, we screened 28 differential expressed immune checkpoint genes (Fig. 7F).

Prediction of responsiveness to immunotherapy/chemotherapy among three subtypes in the TCGA-SKCM cohort. A, Box plots of the estimated IC50 for Paclitaxel, Cisplatin, Vinblastine, and Temozolomide. B, Difference of T cell inflamed GEP score among three subtypes. C, Difference of Th1/IFNγ gene signature among three subtypes. D, Difference of cytolytic activity among three subtypes. E, Heatmap diagram of immune checkpoints among three subtypes. F, Differential expression analysis of 28 immune checkpoints among three subtypes
Identification of differentially expressed genes
Furthermore, differential expression analysis among molecular subtypes was performed. We identified 1054 DEGs between subtype C1 vs C2 (107 upregulated and 947 downregulated DEGs), 669 DEGs between subtype C2 vs. C3 (378 upregulated and 291 downregulated DEGs), and 762 DEGs between subtype C1 vs. C3 (22 upregulated and 740 downregulated DEGs) (Supplementary Fig. S2A–C). Supplementary Fig. S2D showed there were 77 DEGs among the three subtypes, which were used for enrichment analysis (Supplementary Fig. S3). Several immune-related pathways were significantly enriched.
Construction and validation of the prognostic model
Based on differential analysis among the three subtypes, 1552 DEGs were screened and used for univariate Cox regression analysis. With P < 0.05, 883 genes that have a greater impact on prognosis were identified, including 74 risk genes and 809 protective genes. Through LASSO Cox regression analysis, the number of genes was reduced. The number of independent variable coefficients tending to zero increased gradually with the gradual increase of lambda (Fig. 8A); tenfold cross-validation was utilized, and the confidence interval under each lambda was shown in Fig. 8B; 14 genes were selected when lambda = 0.0981. Furthermore, stepwise multivariate regression analysis with stepAIC was performed, and 8 TME-related genes affecting prognosis were identified (NOTCH3, HEYL, ZNF703, ABCC2, PAEP, CCL8, HAPLN3, and HPDL) (Fig. 8C).

Construction and validation of the prognostic model. A, Independent variable coefficients changed with lambda increase. B, tenfold cross-validation determining the confidence interval under each lambda. C, 8 TME-related genes were identified. D, E, ROC curves with AUCs for 1 year, 3 years, and 5 years and Kaplan–Meier curves of high- and low-risk patients in TCGA-SKCM cohort. F, G, ROC curves with AUCs for 1 year, 3 years, and 5 years and Kaplan–Meier curves of high- and low-risk patients in GSE65904. H, I, ROC curves with AUCs for 1 year, 3 years, and 5 years and Kaplan–Meier curves of high- and low-risk patients in GSE54467
Accordingly, the risk score was calculated as follows: risk score = + 0.131*NOTCH3 + 0.093*HEYL + 0.084*ZNF703 + 0.091*ABCC2 + 0.046*PAEP-0.147*CCL8-0.255*HAPLN3 + 0.087*HPDL. Next, the risk score of each sample was calculated in the TCGA-SKCM cohort. Survival analysis in the TCGA-SKCM cohort showed that high-risk patients had a dismal prognosis than that of low-risk patients (P < 0.0001) with 1-year AUC of 0.76, 3-year AUC of 0.73, and 5-year AUC of 0.75 (Fig. 8D, E). To validate the robustness of this prognostic model, survival analysis was also performed in GSE65904 and GSE54467. The results displayed that high-risk patients possessed better prognosis with good prediction performance (Fig. 8F–I).
Clinical performance and prognosis analysis of risk score
To clarify the performance of risk scores in clinical practice, the distribution of risk scores among clinicopathological features (TMN stage, stage, age, gender, and three subtypes) was examined in the TCGA-SKCM cohort. The results showed that patients with advanced stage had higher risk scores (Fig. 9A), and patients with subtype C3 possessed the lowest risk score (Fig. 9B). Besides, the Sankey diagram demonstrated high-risk patients mainly belonged to subtype C1 and subtype C2 (Fig. 9C). Meanwhile, Kaplan–Meier curves were generated between high- and low-risk groups among different clinicopathological features. We found that high-risk patients with different clinicopathological features also displayed poor outcomes (Fig. 9D).

Clinical performance and prognosis analysis of risk score in TCGA-SKCM cohort. A, Violin plots of the risk score in TMN stage, stage, age, and gender. B, Violin plots of the risk score in three subtypes. C, Sankey diagram showing the distribution of three subtypes in high- and low-risk groups. D, Kaplan–Meier curves of high- and low-risk patients in different clinicopathological features
Differences in immune microenvironment between risk groups
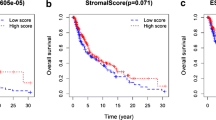
Furthermore, we assessed the alteration of the immune microenvironment between the two risk groups. Figure 10A revealed that low-risk patients had increased StromalScore, ImmuneScore, and ESTIMATEScore, whereas high-risk patients possessed higher TumorPurity scores. Additionally, some immune cells such as T_cells_CD8, T_cells_CD4_memory_activated, NK_cells_activated, and Macrophages_M1 were significantly elevated in low-risk patients, while Macrophages_M2 and Mast_cells_resting were increased in high-risk patients (Fig. 10B). We also compared the enrichment score of 29 TME-related gene signatures in high- and low-risk groups, and Fig. 10C, D deciphered that most TME-related gene signatures were higher in the low-risk group. Moreover, the risk score was positively correlated with B_cells_naïve, T_cells_CD4_memory_resting, NK_cells_resting, Macrophages_M0, Macrophages_M2, and Tumor proliferation rate, while negatively correlated with some anti-tumor signatures such as M1 signature, MHCII, anti-tumor cytokines, and co-activation molecules (Fig. 10E).

Differences in immune microenvironment between risk groups in the TCGA-SKCM cohort. A, ESTIMATE evaluating ImmuneScore, ImmuneScore, ESTIMATEScore, and TumorPurity between risk groups. B, Box plots of relative abundance of 22 immune cells between risk groups. C, Heatmap of 29 TME-related gene signatures in high- and low-risk groups. D, Box plots of TME-related gene signatures. E, Correlation analysis between risk score and 29 TME-related signatures. ns represents P > 0.05; *P < 0.05, **P < 0.01, ***P < 0.001, and.****P < 0.0001
Differences in immunotherapy between risk score groups
As shown in Fig. 11A, the IC50 values of Paclitaxel (P = 2.8e-09) and Temozolomide (P = 0.049) were distinctly decreased in the low-risk group, while the IC50 value of Cisplatin was lower in the high group, indicating that low-risk patients were more sensitive to Paclitaxel and Temozolomide, whereas high-risk patients were more sensitive to Cisplatin (P = 0.00082). Furthermore, we found that low-risk patients exhibited increased TMB (P = 0.019) and intra-tumor genetic heterogeneity (P = 0.031). The risk score was negatively correlated with TMB and intra-tumor genetic heterogeneity (Fig. 11B–D). Figure 11E–G demonstrated that T-cell–inflamed GEP score, Th1/IFNγ gene signature, and CYT score were remarkably increased in low-risk patients, which were negatively correlated with a risk score. Moreover, we also evaluated the distribution of immune checkpoints and their correlations with two risk groups. Figure 11H, I revealed that most immune checkpoints were significantly highly expressed in the low-risk group, and risk score was negatively correlated with immune checkpoints.

Differences in immunotherapy between risk score groups. A, Box plots of the estimated IC50 for Paclitaxel, Cisplatin, Vinblastine, and Temozolomide in the TCGA-SKCM cohort. B–D, Violin plots of TMB, intra-tumor genetic heterogeneity, and HRD score in high- and low-risk groups, along with their correlation with a risk score. E–G, Violin plots of T cell inflamed GEP score, Th1/IFNγ gene signature, and cytolytic activity in high- and low-risk groups, and correlation analysis between these parameters and risk score. H, Correlation analysis between immune checkpoints and risk score. I, Distribution of immune checkpoints in high- and low-risk groups
Discussion
TME plays a pivotal role in the biological process of tumors, and the tumor immune microenvironment is strongly associated with the clinical outcome and anti-tumor therapy responses. In the present study, transcriptome data of SKCM was used to identify three TME-related subtypes based on 29 TME-related gene signatures. We confirmed that subtype C3 had the most favorable prognosis, enriched in immune-related pathways, and possessed higher ImmuneScore and ESTIMATEScore but a lower TumorPurity. Next, we constructed and validated a prognostic risk model based on 8 TME-related genes, which could predict prognosis, immune microenvironment, and immunotherapy response between high- and low-risk SKCM patients.
Increasing pieces of evidence have demonstrated a close correlation between CD8 + T cell infiltration in TME and good prognosis in the tumor. Tumor cells can be destroyed and eliminated when cytotoxic CD8 + T cells are activated and infiltrate into the tumor site (Maimela et al. 2019). However, suppression of CD8 + T cell infiltration in the TME contributes to the underlying mechanism of resistance to radiotherapy (Chen et al. 2018). A previous study has confirmed that decreased CD8 + T cell infiltration in TME and increased PD-L1 expression are related to CD44 + /CD133 + cancer stem cells and dismal outcomes in pancreatic cancer (Hou et al. 2019). Macrophages occupy the predominant position in the TME. Under the TME, macrophages polarize into macrophages M1 exhibiting anti-tumor effect and macrophages M2 possessing anti-inflammatory and oncogenic features. Additionally, evaluating the ratio of M1 to M2 macrophages can serve as a prevailing approach for clinical application in cancer patients (Jayasingam et al. 2020). In this study, we found that subtype C3 possessed higher ImmuneScore and ESTIMATEScore but a lower TumorPurity, indicating that patients with subtype C3 had more immunity. Besides, T_cells_CD8, T_cells_CD4_memory_activated, and Macrophages_M1 were highly expressed in subtype C3, whereas Macrophages_M2 were increased in subtype C1 and subtype C2. These suggested that more CD8 T cells infiltration into TME and increased polarization of Macrophages into M2 phenotypes were a benefit for favorable prognosis of SKCM patients. Meanwhile, a previous study has reported that the expression of Th1/IFNγ genes in the TME is associated with prognosis in melanoma patients, implying Th1/IFNγ-related signatures may be responsible for the progression of melanoma (Dizier et al. 2020). Therefore, increased T-cell-inflamed GEP, Th1/IFNγ gene signature, and CYT score also contributed to the good prognosis of subtype C3 in this study.
A previous study has reported that gene signatures could predict prognosis, immune status, and chemosensitivity in cancers using various machine learning (Peng et al. 2022; Chi et al. 2022; Chi et al. 2022c; Chi et al. 2023). A recent study has generated 6 TME-related signatures (EDN3, CLEC4E, SRPX2, KIR2DL4, UBE2L6, and IFIT2) based on 471 SKCM patients from the TCGA dataset and found that these genes have good predictive values in the prognosis of patients with SKCM (Zhou et al. 2021). In this study, we identified 8 TME-related genes affecting prognosis (NOTCH3, HEYL, ZNF703, ABCC2, PAEP, CCL8, HAPLN3, and HPDL) using 354 metastatic tumor samples from the TCGA dataset, and these TME-related genes could predict the clinical outcome. Also, we revealed that low-risk patients were more sensitive to Paclitaxel and Temozolomide, while high-risk patients were more sensitive to Cisplatin, which indicated therapeutic options for patients with SKCM. The different TME-related genes identified in the two studies may be due to the selection of SKCM samples. These 8 TME-related genes in our study can be used as supplements for biomarkers of SKCM and provide novel directions for the precision treatment of SKCM.
NOTCH signaling has been proven to modulate various aspects of cancer biology, and NOTCH shapes the TME in the tumor through Juxtacrine and Paracrine Signaling between the diverse compartments of TME (Meurette and Mehlen 2018). It has been demonstrated that NOTCH3 could promote tumor growth via provoking the infiltration of immunosuppressive cells in TME of colorectal cancer (Huang et al. 2022). HEYL has been identified as a novel prognostic biomarker of hypoxic gastrointestinal cancer exhibiting more immune and stromal infiltration (Hu et al. 2022). ZNF703 belongs to the zinc finger protein family, which is the largest transcription factor family in the human genome, and it is a common luminal B breast cancer oncogene. ZNF703 has been reported to be a progression-related gene that is associated with M2 macrophage infiltration in colon cancer (Xu et al. 2021). ABCC2 is one of the ATP-binding cassette transporters and is implicated in clinical drug resistance modulated by TNF-α in TME of tumors (Alamolhodaei et al. 2020). PAEP has been identified as an immune-related gene predicting prognosis in lung squamous cell carcinoma (Wu et al. 2021). Besides, CCL8 is associated with immune cell infiltration of SKCM, which can be used as a prognostic biomarker for survival and therapy (Yang et al. 2021). HAPLN3 exerts potential immunosuppressive in malignant melanoma patients. Recently, based on the immune subtype classification of melanoma, Mei and colleagues have identified 4 genes including SEL1L3, HAPLN3, BST2, and IFITM1, which could predict prognosis and responses to anti-CTLA4 immunotherapy (Mei et al. 2021). A previous study has revealed that elevated HPDL is remarkably positively correlated with the infiltration of immune cells and critical immune checkpoints, which lead to immunosuppressive TME and an unfavorable outcome (Jiang et al. 2022). In the present study, the identified 8 TME-related signatures might be involved in the development and progression of SKCM and considered as prognostic biomarkers for outcome and response to clinical therapy. We also validated the robustness of this risk model with good prediction performance for SKCM in independent external GEO cohorts.
Data used in this study was obtained from the TCGA database, while more convincing prospective studies should be performed. Besides, the underlying mechanism of these TME-related genes in SKCM warrants further investigation.
Conclusion
In conclusion, we identify three TME-related subtypes and provided a supplement to understanding the heterogeneity of SKCM. Moreover, TME-related genes can predict prognosis and evaluate the response to immunotherapy in SKCM patients.
Data availability
The data supporting the conclusions of this article can be obtained in The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) database and the Gene-Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database.
References
Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB et al (2015) Genomic classification of cutaneous melanoma. Cell 161(7):1681–1696
Alamolhodaei NS, Rashidpour H, Gharaee ME, Behravan J, Mosaffa F (2020) Overexpression of ABCC2 and NF-Κβ/p65 with reduction in cisplatin and 4OH-Tamoxifen sensitivity in MCF-7 breast cancer cells: the influence of TNF-α. Pharmaceutical Sciences 26(2):150–158
Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T et al (2018) Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med 24(10):1545–1549
Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O et al (2021) Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer cell 39(6):845–65.e7
Blanche P. (2015) TimeROC: time-dependent ROC curve and AUC for censored survival data. R package version 2
Chen H-y, Xu L, Li L-f, Liu X-x, Gao J-x, Bai Y-r (2018) Inhibiting the CD8+ T cell infiltration in the tumor microenvironment after radiotherapy is an important mechanism of radioresistance. Scientific reports 8(1):1–10
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA (2018) Profiling tumor infiltrating immune cells with CIBERSORT. Springer, Cancer systems biology, pp 243–259
Chi H, Jiang P, Xu K, Zhao Y, Song B, Peng G et al (2022a) A novel anoikis-related gene signature predicts prognosis in patients with head and neck squamous cell carcinoma and reveals immune infiltration. Front Genet 13:984273
Chi H, Xie X, Yan Y, Peng G, Strohmer DF, Lai G et al (2022b) Natural killer cell-related prognosis signature characterizes immune landscape and predicts prognosis of HNSCC. Front Immunol 13:1018685
Chi H, Peng G, Yang J, Zhang J, Song G, Xie X et al (2022c) Machine learning to construct sphingolipid metabolism genes signature to characterize the immune landscape and prognosis of patients with uveal melanoma. Front Endocrinol 13:1056310
Chi H, Yang J, Peng G, Zhang J, Song G, Xie X et al (2023) Circadian rhythm-related genes index: a predictor for HNSCC prognosis, immunotherapy efficacy, and chemosensitivity. Front Immunol 14:1091218
Chi H, Peng G, Wang R, Yang F, Xie X, Zhang J, et al. (2022) Cuprotosis programmed-cell-death-related lncRNA signature predicts prognosis and immune landscape in PAAD patients. Cells 11 21
Cursons J, Souza-Fonseca-Guimaraes F, Foroutan M, Anderson A, Hollande F, Hediyeh-Zadeh S et al (2019) A Gene signature predicting natural killer cell infiltration and improved survival in melanoma patientsa gene signature for NK infiltration and melanoma survival. Cancer Immunol Res 7(7):1162–1174
Danilova L, Ho WJ, Zhu Q, Vithayathil T, De Jesus-Acosta A, Azad NS et al (2019) Programmed cell death ligand-1 (PD-L1) and CD8 expression profiling identify an immunologic subtype of pancreatic ductal adenocarcinomas with favorable survival. Cancer Immunol Res 7(6):886–895
Dizier B, Callegaro A, Debois M, Dreno B, Hersey P, Gogas HJ et al (2020) A Th1/IFNγ gene signature is prognostic in the adjuvant setting of resectable high-risk melanoma but not in non–small cell lung cancerprognostic biomarkers in metastatic melanoma and NSCLC. Clin Cancer Res 26(7):1725–1735
Geeleher P, Cox N, Huang RS (2014) pRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9(9):e107468
Gu Z (2022) Complex Heatmap Visualization Imeta 1(3):e43
Hänzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14(1):1–15
Hastie T, Qian J,Tay K. An introduction to glmnet. CRAN R Repositary
Hou Y-C, Chao Y-J, Hsieh M-H, Tung H-L, Wang H-C, Shan Y-S (2019) Low CD8+ T cell infiltration and high PD-L1 expression are associated with level of CD44+/CD133+ cancer stem cells and predict an unfavorable prognosis in pancreatic cancer. Cancers 11(4):541
Hu J, Hu W, Yang Y. (2022) Hypoxia Confers Tumor with a higher immune infiltration but lower mutation burden in gastrointestinal cancer. Journal of oncology
Huang K, Luo W, Fang J, Yu C, Liu G, Yuan X, et al. (2022) Notch3 signaling promotes colorectal tumor growth by enhancing immunosuppressive cells infiltration in the microenvironment
Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, Ch’ng ES (2020) Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front Oncol 9:1512
Jiang N, Zhang X, Chen Q, Kantawong F, Wan S, Liu J, et al. (2022) Identification of a mitochondria-related gene signature to predict the prognosis in AML. Frontiers in oncology 12
Jin W, Yang Q, Chi H, Wei K, Zhang P, Zhao G et al (2022) Ensemble deep learning enhanced with self-attention for predicting immunotherapeutic responses to cancers. Front Immunol 13:1025330
Kasakovski D, Skrygan M, Gambichler T, Susok L (2021) Advances in Targeting Cutaneous Melanoma Cancers 13(9):2090
Kassambara A. 2019 ggcorrplot: visualization of a correlation matrix using ggplot2 (0.1. 3)[Computer software]
Kassambara A, Kosinski M, Biecek P, Fabian S. (2017) survminer: drawing survival curves using ‘ggplot2’. R package version 03 1
Liang Z, Pan L, Shi J, Zhang L (2022) C1QA, C1QB, and GZMB are novel prognostic biomarkers of skin cutaneous melanoma relating tumor microenvironment. Sci Rep 12(1):1–15
Maimela NR, Liu S, Zhang Y (2019) Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J 17:1–13
Mayakonda A, Lin D-C, Assenov Y, Plass C, Koeffler HP (2018) Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 28(11):1747–1756
Mei Y, Chen M-JM, Liang H, Ma L (2021) A four-gene signature predicts survival and anti-CTLA4 immunotherapeutic responses based on immune classification of melanoma. Communications biology 4(1):1–12
Meurette O, Mehlen P (2018) Notch signaling in the tumor microenvironment. Cancer Cell 34(4):536–548
Najafi M, Goradel NH, Farhood B, Salehi E, Solhjoo S, Toolee H et al (2019) Tumor microenvironment: interactions and therapy. J Cell Physiol 234(5):5700–5721
Novotný J, Strnadová K, Dvořánková B, Kocourková Š, Jakša R, Dundr P et al (2020) Single-cell RNA sequencing unravels heterogeneity of the stromal niche in cutaneous melanoma heterogeneous spheroids. Cancers 12(11):3324
Peng G, Chi H, Gao X, Zhang J, Song G, Xie X et al (2022) Identification and validation of neurotrophic factor-related genes signature in HNSCC to predict survival and immune landscapes. Front Genet 13:1010044
Ritchie ME,Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research 43 7 e47-e
Santos P, I, van Doorn R, Caspers PJ, Bakker Schut TC, Barroso EM, Nijsten TE, et al (2018) Improving clinical diagnosis of early-stage cutaneous melanoma based on Raman spectroscopy. British journal of cancer 119(11):1339–46
Song B, Chi H, Peng G, Song Y, Cui Z, Zhu Y et al (2022a) Characterization of coagulation-related gene signature to predict prognosis and tumor immune microenvironment in skin cutaneous melanoma. Front Oncol 12:975255
Song B, Wu P, Liang Z, Wang J, Zheng Y, Wang Y et al (2022b) A novel necroptosis-related gene signature in skin cutaneous melanoma prognosis and tumor microenvironment. Front Genet 13:917007
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A et al (2020) Global cancer statistics 2020 GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers a cancers in 185 countries. CA a cancer journal for clinicians 71(3):209–49
Thrane K, Eriksson H, Maaskola J, Hansson J, Lundeberg J (2018) Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Can Res 78(20):5970–5979
Wilkerson M,Waltman P, Wilkerson MM. 2013 Package ‘ConsensusClusterPlus’
Wu H, Huang S, Zhuang W, Qiao G (2021) Prognostic significance of eight immune-related genes on survival in patients with lung squamous cell carcinoma. Biomark Med 15(4):295–306
Xu B, Peng Z, Yan G, Wang N, Chen M, Yao X, et al. (2021) Establishment and validation of a genetic label associated with M2 Macrophage infiltration to predict survival in colon cancer patients and assist immunotherapy
Yang P, Chen W, Xu H, Yang J, Jiang J, Jiang Y et al (2021) Correlation of CCL8 expression with immune cell infiltration of skin cutaneous melanoma: potential as a prognostic indicator and therapeutic pathway. Cancer Cell Int 21(1):1–11
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W et al (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4(1):1–11
Yu G, Wang L-G, Han Y, He Q-Y. (2012) clusterProfiler an R package for comparing biological themes among gene clusters Omics a journal of integrative biology 16 5 284-7
Zhang Y, Zhang Z (2020) The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol 17(8):807–821
Zhao Y, Wei K, Chi H, Xia Z, Li X (2022) IL-7: a promising adjuvant ensuring effective T cell responses and memory in combination with cancer vaccines? Front Immunol 13:1022808
Zhao S, Chi H, Yang Q, Chen S, Wu C, Lai G et al (2023) Identification and validation of neurotrophic factor-related gene signatures in glioblastoma and Parkinson’s disease. Front Immunol 14:1090040
Zhou S, Sun Y, Chen T, Wang J, He J, Lyu J et al (2021) The landscape of the tumor microenvironment in skin cutaneous melanoma reveals a prognostic and immunotherapeutically relevant gene signature. Frontiers in Cell and Developmental Biology 9:739594
Zhu G, Su H, Johnson CH, Khan SA, Kluger H, Lu L (2021) Intratumour microbiome associated with the infiltration of cytotoxic CD8+ T cells and patient survival in cutaneous melanoma. Eur J Cancer 151:25–34
Funding
This study was funded by Xiang’an Hospital of Xiamen University and the Guidance in Medical and Health Program of Xiamen, China (3502Z20224ZD1148).
Author information
Authors and Affiliations
Contributions
All authors wrote, revised, and approved the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
The study was conducted in compliance with the ethical principles of the Declaration of Helsinki and its later amendments. The Ethics Committee of Xiang’an Hospital of Xiamen University approved our study protocol (approval no. XAHLL2022046).
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lian, W., Zheng, X. Identification and validation of TME-related signatures to predict prognosis and response to anti-tumor therapies in skin cutaneous melanoma. Funct Integr Genomics 23, 153 (2023). https://doi.org/10.1007/s10142-023-01051-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10142-023-01051-x