
Abstract
Skin cutaneous melanoma (SKCM), a form of skin cancer, ranks among the most formidable and lethal malignancies. Exploring tumor microenvironment (TME)-based prognostic indicators would help improve the efficacy of immunotherapy for SKCM patients. This study analyzed SKCM scRNA-seq data to cluster non-malignant cells that could be used to explore the TME into nine immune/stromal cell types, including B cells, CD4 T cells, CD8 T cells, dendritic cells, endothelial cells, Fibroblasts, macrophages, neurons, and natural killer (NK) cells. Using data from The Cancer Genome Atlas (TCGA), we employed SKCM expression profiling to identify differentially expressed immune-associated genes (DEIAGs), which were then incorporated into weighted gene co-expression network analysis (WGCNA) to investigate TME-associated hub genes. Discover candidate small molecule drugs based on pivotal genes. Tumor immune microenvironment-associated genes (TIMAGs) for constructing TIMAS were identified and validated. Finally, the characteristics of TIAMS subgroups and the ability of TIMAS to predict immunotherapy outcomes were analyzed. We identified five TIMAGs (CD86, CD80, SEMA4D, C1QA, and IRF1) and used them to construct TIMAS. In addition, five potential SKCM drugs were identified. The results showed that TIMAS-low patients were associated with immune-related signaling pathways, high MUC16 mutation frequency, high T cell infiltration, and M1 macrophages, and were more favorable for immunotherapy. Collectively, TIMAS constructed by comprehensive analysis of scRNA-seq and bulk RNA-seq data is a promising marker for predicting ICI treatment outcomes and improving individualized therapy for SKCM patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The incidence of skin cutaneous melanoma (SKCM), a remarkably aggressive and lethal malignancy, is progressively escalating on a global scale [1]. The prognosis of individuals afflicted with SKCM is disheartening, with a mere 20% survival rate observed for those with distant metastases within a 5-year timeframe [2]. In recent times, the application of immune checkpoint inhibitors (ICI) targeting CTLA-4 (cytotoxic T lymphocyte antigen 4), PD-1 (programmed death 1), and PD-L1 (PD-1 ligand) has significantly enhanced the clinical prognosis of individuals afflicted with SKCM [3,4,5,6]. However, a mere fraction of approximately 30–40% of patients grappling with advanced melanoma can avail themselves of immunotherapy unhindered by innate resistance [6, 7]. In order to explore prognostic signatures that can significantly enhance the prognosis of SKCM patients, it is imperative to engage in pioneering searches for tumor immune microenvironment-associated genes (TIMAGs).
The efficacy of immunotherapy is intricately related to the composition of the tumor microenvironment (TME), which comprises a complex interplay of immune cells, stromal cells, and extracellular matrix molecules [8,9,10,11,12,13,14,15]. Within the TME, two distinct categories of immune cells can be distinguished: innate immune cells and acquired immune cells. Innate immune cells comprise neutrophils, dendritic cells, and macrophages, whereas acquired immune cells encompass T cells and B cells [14,15,16]. High levels of tumor-infiltrating T lymphocytes in SKCM patients have been linked to improved treatment outcomes [17, 18]. However, T cells cannot autonomously implement effector functions, such as the initiation, maintenance of T cell responses, and the development of long-term protective memory T cells and they all require the participation of innate immune cells [18]. Otherwise, stromal cells within the TME affect SKCM patients’ prognosis by participating in the processes of tumorigenesis, progression, and metastasis [19, 20]. For example, CXCL12 produced by carcinoma-associated fibroblasts (CAFs) can stimulate cancer cell proliferation while restricting T cell infiltration, and CAFs from the original tumor have been found in metastases [21, 22].
As a traditional transcriptomic analysis tool, bulk RNA sequencing (bulk RNAseq) solely enables the detection of the average expression level of genes within the cell population so that may cover up the heterogeneity information in the cell population, which is not conducive to our exploration of TME [23]. In recent times, the expeditious advancement of single-cell RNA sequencing (scRNA-seq) technology has made it an important tool to reveal information on cellular transcriptome heterogeneity [24, 25]. Therefore, conducting a thorough analysis of bulk RNA-seq and scRNA-seq to find TIMAGs to establish prognostic markers is the key to solving the current poor immune efficacy in melanoma patients.
In this study, we clustered non-malignant cells in scRNA-seq into 9 immune/stromal cells as a basis for exploring the TME of melanoma. We separated TCGA-SKCM patients into two distinct immunity clusters based on the degree of infiltration of immune-related gene sets. We discerned the differentially expressed immune-associated genes (DEIAGs) within patients exhibiting distinct immune infiltration profiles, subsequently integrating them into weighted gene co-expression network analysis (WGCNA) to uncover hub genes associated with the melanoma TME. Based on the hub genes, potential drugs which could assist in treating patients of SKCM were explored. We then constructed and validated a tumor immune microenvironment-associated signature (TIMAS) consisting of five TIMAGs. We elucidated the molecular and immunological characteristic of TIMAS and compared the predictive ability for immunotherapy outcomes of patients among TIMAS, tumor immune dysfunction and exclusion (TIDE) as well as tumor inflammatory markers (TIS). The findings suggested that TIMAS developed in this study was a promising prognostic signature in SKCM immunotherapy.
Methods
Acquisition of SKCM patient dataset
Bulk RNA-seq data for 457 SKCM patients and clinical case information and somatic mutation data for 466 patients were made available through the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/). Single-cell RNA-seq dataset GSE115978, encompassing a table comprising 7186 cells extracted from 31 SKCM patients along with their respective annotations, was procured from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The GSE65904 dataset, comprising 214 samples obtained from the GEO database, served as the external validation data for this study. To assess the immunotherapy predictive capacity of TIMAS, both the phs000452.v2.p1 and GSE78220 datasets were examined.
Single-cell analysis
The conversion of non-malignant cells from GSE115978 to Seurat objects was performed by the Seurat R package. We filtered out cells with less than 2500 genes and normalized the selected cells using the LogNormalize method. Based on the FindVariableFeatures function, we screened for highly variable genes between cells. Subsequently, principal component analysis (PCA) was employed for dimensionality reduction of the single-cell samples, and the available dimensions of samples were determined by combining ElbowPlot and JackStrawPlot. Next, we visualized the selected principal components (PCs) using the umap algorithm [26] and clustered the cell samples by SingleR [27]. Finally, the cells were annotated based on HumanPrimaryCellAtlasData, BlueprintEncodeData, MonacoImmuneData in Celldex.
Immunity subtype clustering and cell subgroups analysis
Quantification of immune infiltration status in TCGA-SKCM patients was achieved by single-sample gene set enrichment analysis (ssGSEA) based on the degree of enrichment of 29 immune-related gene sets [28]. The TCGA-SKCM samples were then hierarchically clustered according to immunity by the sparcl R package. We then explored differences in prognosis between different immunity subtypes. Next, we calculated the expression levels of cell subgroups in different immunity clusters based on the TCGA dataset. Furthermore, we investigated the biological processes associated with the differentially expressed genes (DEGs) among cell subtypes using Gene Ontology (GO) analysis.
TIMAGs and potential small molecule drugs identification
Based on LIMMA, DEGs satisfying |logFC| greater than 1 and false discovery rate (FDR) less than 0.01 were obtained in different immunity clusters. To identify DEIAGs, DEGs were then included in a comprehensive consideration with immune-associated genes in ImmPort (https://www.immport.org/shared/home) and InnateDB (https://www.innateDBdb.com/). We performed WGCNA analysis and constructed the network by selecting genes (weight > 0.2) from the significant correlation module. We conducted enrichment analysis of genes in significantly correlated modules using GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) [29]. As hub genes, the network’s genes with degrees greater than 20 were chosen.
Our training set was derived from a random allocation of two-thirds of the TCGA-SKCM samples (n = 306), and the remaining third as the test set (n = 151). Then, to identify TIMAGs from the hub genes, we employed univariate Cox regression and LASSO regression analyses [30]. By GEPIA (http://gepia.cancer-pku.cn/), the relationship between TIMAGs and overall survival (OS) in SKCM patients was explored. Biological characterization of hub genes was performed in Metasscape (https://metascape.org) [31]. The CMap information portal (http://www.broadinstitute.org/) was used to explore potential SKCM drugs related to the hub genes [32]. Finally, the 3D structures of the potential medication candidates were obtained by accessing the PubChem database (https://pubchem.ncbi.nlm.nih.gov/).
TIMAS design and validation
Cistrome Cancer database (http://cistrome.org/CistromeCancer/CancerTarget/) provided 318 transcription factors (TFs) [33], and we then chose differentially expressed TFs from DEIAGs. Using Pearson correlation coefficient analysis, a regulatory network based on TFs and five TIMAGs was created (coefficient of correlation > 0.4, P-value < 0.05).
The TIMAGs were added to the formula below to get the TIMAS for each patient:
The β in the formula is derived from the coefficient corresponding to each TIMAGs in the LASSO regression analysis.
We grouped patients based on their median TIMAS, and then explored differences in OS among patients in different TIMAS subgroups. Three time periods of 1, 3, and 5 years were set to analyze the time-dependent ROC of TIMAS [34]. The TIMAS formulation was applied to the internal and external validation sets, repeated the above Kaplan-Meier and ROC curve analysis. The independence of TIMAS was then verified by univariate and multivariate Cox analyses, and factors from the Cox regression model were incorporated into the nomogram.
Characteristic analysis of TIMAS
In signaling pathway analysis, DEGs (P-value < 0.05) between TIMAS subgroups were involved in gene set enrichment analysis (GSEA). In addition, we performed Signaling pathways and tumor mutational burden (TMB) analysis of patients in different TIMAS subgroups.
The immune characteristics of different TIMAS subgroups in patients were assessed by analyzing the composition of 22 immune cells through the utilization of CiberSort [35]. Furthermore, we compared the ability of TIMAS, TIDE, and TIS to predict immunotherapy outcome by time-dependent ROC curves.
Statistical analysis
GSE65904 data were normalized and transformed based on log2 by the limma R package. The association of the two datasets was estimated by the Pearson correlation coefficient. The calculation of the TIDE score was finished on the TIDE database (http://tide.dfci.atherard.edu/). The TIS score was computed by averaging the expression levels of six characteristic genes [36]. This study was based on R (version 4.1.2) for data analysis, model construction and validation.
Results
Identification of cell subgroups in SKCM single-cell RNA-seq
After excluding non-compliant cells, a total of 4569 cells were collected (Fig. 1a). We identified 2000 highly variable genes and annotated the top 10 genes (Fig. 1b). After performing dimensionality reduction on single-cell samples using PCA, we comprehensively analyze JackStrawPlot and ElbowPlot to determine the actual number of PCs available. The JackStrawPlot showed that from the 15th PC onwards, the P-values for the PCs drop significantly (Fig. 1c). It could be seen from the ElbowPlot that the inflection point of the curve appeared near PC14 (Fig. 1d). These results implied that the first 14 PCs were actually usable. Then we visualized the selected 14 PCs using umap, which were clearly divided into 14 clusters (Fig. 1e). Based on the SingleR package and Celldex package, we successfully clustered single-cell samples into 9 cell subgroups. After annotation, nine types of immune/stromal cells including B cells, CD4 T cells, CD8 T cells, dendritic cells, endothelial cells, fibroblasts, macrophages, neurons, and natural killer (NK) cells were obtained (Fig. 1f).

Clustering of cell subtypes in SKCM scRNA-seq data. a Quality-compliant cell information visualization. b Highly variable genes in scRNA-seq. c JackStrawPlot identified PCs available in samples. d ElbowPlot identified PCs available in samples. e Visualization of 14 PCs clustering. f Visualization of cell subgroups in scRNA-seq
Relationship between immunity subtypes and cell subgroups
Tumor immunotherapy is affected by the state of immune cell infiltration within the TME. The samples were stratified into immunity-high (n = 342) and immunity-low (n = 115) groups based on ssGSEA scores. Survival analysis results showed that a good immune infiltration state could improve the OS of patients (Fig. 2a). We found that high expression of immune/stromal cells in SKCM patients within the TCGA dataset was strongly associated with high immune infiltration (Fig. 2b). In addition, we performed GO analysis on DEGs between 9 kinds of immune/stromal cells, and the analysis revealed a significant enrichment of DEGs in immune-related pathways, including neutrophil activation, immune response involving neutrophil activation, and neutrophil-mediated immunity (Fig. 2c). The above results demonstrated the accuracy of the clustering method and the significant correlation of immunophenotype with SKCM progression and patient prognosis.

Immunity subtypes and cell subgroups a Kaplan-Meier analysis of OS for the immunity subtypes. b Expression levels of cell subgroups on different immunity subtypes. c Enrichment analysis of DEGs among 9 kinds of immune/stromal cells
TIMAGs and potential small molecule drugs
We identified the intersection of DEGs (n = 1049) and immune-related gene lists (n = 1520), a total of 192 eligible DEIAGs were included in the following study (186 genes upregulated, 6 genes downregulated). We performed WGCNA analysis on DEIAGs using weighted gene co-expression network (Fig. 3a, b). Finally, five modules are determined based on the mean connectivity and the optimal soft threshold power (Fig. 3c). Genes within the blue and turquoise modules, which exhibited strong associations with SKCM TME, were selected according to the modules and immune/stromal cells (Fig. 3d). The module networks with threshold weight > 0.2 were shown in Supplementary Fig. 1. Supplementary Fig. 2a-c presented the results of the GO and KEGG enrichment analyzes conducted on the genes belonging to blue and turquoise module genes. We obtained 37 hub genes (degree > 20).

Identification of TIMAGs. a, b Estimation of the optimal soft-thresholding power in the WGCNA analysis. c WGCNA analysis of DEIAGs with soft threshold β = 4. d Gene modules and immune/stromal cells correlation analysis. e Univariate Cox regression analysis of hub genes. f Lasso Cox regression analysis of 12 prognosis-related hub genes
We used univariate Cox regression analysis on 37 hub genes based on the training set and obtained 12 hub genes with prognostic features (Fig. 3e). We then performed Lasso Cox regression on 12 prognosis-related hub genes and identified five TIMAGs (CD86, CD80, SEMA4D, C1QA, IRF1), which were most significant in terms of prognosis in SKCM patients (Fig. 3f). The GEPIA results revealed a favorable association between the expression levels of the five TIMAGs and the patients’ prognosis (Supplementary Fig. 3).
We investigated the biological information of hub genes, including functions and potential pathways (Fig. 4a, b). We selected five compounds with connectivity scores less than − 99 as five candidate small molecule drugs (Table 1) and obtained their 3D conformations from the PubChem database (Fig. 4c).

Analyses of the candidate small molecule drugs. a Hub gene enrichment analysis. b The hub gene interaction network. c 3D conformers of five potential SKCM drugs
Survival analysis of TIMAS in the training set
We built TIMAS based on five TIMAGs with independent regression coefficients. The formula for constructing the prognostic model:
Using the median TIMAS of SKCM patients as a grouping basis, we obtained TIMAS-low patients (n = 229) and TIMAS-high patients (n = 228).
According to a heatmap we created of five TIMAGs in TIMAS subgroups, patients with TIMAS-low had higher expression levels of the five TIMAGs than those with TIMAS-high. (Fig. 5a). In the regulatory mechanism analysis of 5 TIMAGs, we selected six TFs from the DEIAGs of the immunity subgroup, of which 24 pairs of TIMAGs interacting with transcription factors were significantly correlation. We constructed regulatory networks and draw Sankey diagrams to show the relationship between five TIMAGs and TFs (Fig. 5b). We conducted an analysis of the OS among the patients and found that TIMAS-low patients lived longer and had a better survival probability than TIMAS-high patients (P-value < 0.001, Fig. 5c). We explored the distribution and survival status of patients based on TIMAS built on the training set (Fig. 5d). Their survival status showed a clear separation between TIMAS subgroups, with more patients surviving in the TIMAS-low group. The area under the time-dependent ROC curve (AUC) of TIMAS in the three time periods reached above 0.6, indicated that TIMAS had acceptable sensitivity and specificity for survival prediction (Fig. 5e).

Characterization of TIMAGs and construction of TIMAS. a Heatmap of five TIMAGs in different TIMAS subgroups. b Sankey diagram of the regulatory network of five TIMAGs and TFs. c Survival prognostic analysis of different TIMAS subgroups of patients in the training set. d TIMAS distribution (top) and survival status plot of patients (bottom) in TIMAS subgroups. e Time-dependent ROC curves of TIMAS
Stability verification of TIMAS
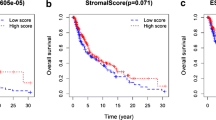
In the validation set, it was observed that the survival curves of patients in the TIMAS-low subgroup were all above those of patients in the TIMAS-high subgroup, indicating that TIMAS-low patients had better OS (Fig. 6a, b). The patient distribution and survival status of TIMAS in two validation cohorts demonstrated TIMAS built using the TCGA training set to be reliable. (Fig. 6c-f). Figures 4h and 6g showed the AUC of TIMAS in 1, 3, and 5 years in the TCGA test set and GSE65904.

TIMAS in validation set. a Kaplan-Meier analysis of TIMAS in testing set. b Survival prognostic analysis of TIMAS in GSE65904. c-f The distribution (c, d) and survival state (e, f) of patients for TIMAS in internal and external cohorts. g Time-dependent ROC analysis of TIMAS in testing set. h Time-dependent ROC curves of TIMAS in GSE65904 cohort
Age, Breslow depth, Stage, Malignant neoplasm mitotic count rate, Melanoma Clark level, T, N, and TIMAS were all significantly correlated with patients’ overall survival, according to a univariate Cox analysis of SKCM patients (Supplementary Fig. 4a). Next, we used multivariate Cox analysis on the variables that were significant in the univariate Cox model. TIMAS was found to be significantly associated with patients’ OS in both Cox models, implying that TIMAS was an indicator that could independently predict the patient’s OS (Supplementary Fig. 4b). Furthermore, we constructed nomograms for OS prediction in SKCM patients based on significant prognostic factors in multivariate Cox analysis and 1-year, 3-year and 5-year patient survival (Supplementary Fig. 4c). The above results implied the validity of TIMAS for predicting survival outcomes in SKCM patients.
Signaling pathways and tumor mutational burden (TMB) in different TIMAS subgroups
Among the signaling pathways we identified to be enriched in different TIMAS subgroups, 37 KEGG pathways and 14 HALLMARK pathways were included. The signaling pathway information of patients with different TIMAS subgroups in GSEA was summarized in Supplementary Tables 1 and Supplementary Tables 2, and representative pathways in TIMAS-low patients with SKCM were shown in Fig. 7a, b. Signaling pathways in TIMAS-low patients are immune-related, including interferon gamma (IFN γ) response, cytokine and cytokine receptor interaction, IL2 STAT5 signaling pathway, while signaling pathways in TIMAS-high patients are tumor-related, such as MYC targeting V1 MYC targets V2, oxidative phosphorylation, and ribosomes.

GSEA analysis of TIMAS-low subgroups. a KEGG pathway enrichment analysis. b HALL-MARK pathway enrichment analysis
Moreover, we found that missense mutations and single nucleotide polymorphism (SNP) accounted for the highest proportion in different classification methods (Fig. 8a, b). Single nucleotide variants (SNVs) mainly appeared in the form of C > T (Fig. 8c). The mutation counts for each sample and the corresponding mutation type were shown in Fig. 8d, e. Among the ten most frequently mutated genes, TTN, MUC16, BRAF, DNAH5, PCLO, ADGRV1, LRP1B, and RP1 had mutation rates above 30% in both subgroups (Fig. 8f). Whereas HYDIN and XIRP2 mutations were more common in TIMAS-high patients, DNAH7 and THSD7B mutations were more common in TIMAS-low patients.

Summary of TMB analysis of different TIMAS groups. a, b Mutation classification of TIMAS subgroups under different classification categories. c SNVs class in different TIMAS subgroups. d Mutation counts per sample in different TIMAS subgroups. e Mutation classification per sample in different TIMAS subgroups. f Top 10 mutated genes in different TIMAS subgroups
The immune microenvironment of different TIMAS subgroups
Upon analyzing the content of 22 immune cells in different TIMAS subgroups, it was discovered that the relative proportions of memory B cells, plasma cells, CD8 T cells, activated memory CD4 T cells, and M1 macrophages were significantly greater in the TIMAS-low subgroup compared to the TIMAS-high subgroup. The TIMAS-high subgroup was enriched in resting memory CD4 T cells, M0 macrophages, M2 macrophages and resting mast cells (Fig. 9a). Figure 9b displayed the clinicopathological characteristics of the TIMAS subgroups along with other immune landscape-related factors.

TME landscape and immune features in different TIMAS subgroups. a Content of 22 immune cells within the TME in different TIMAS subgroups (ns: not significant, **: P-value < 0.01, ***: P-value < 0.001). b Immune landscape of TIMAS subgroups
Compared with TIMAS-high patients, TIMAS-low patients had lower TIDE and T-cell exclusion score, higher T-cell dysfunction score and TIS score, implied that TIMAS-low patients performed better on ICI therapy (Fig. 10a, b). We explored the association of TIMAS with key immune checkpoints genes (Fig. 10c). We compared the time-dependent ROC curves among TIMAS, TIDE, and TIS three algorithms in 6, 12, and 24 months, and found that the performance of TIMAS is the best (Fig. 10d-f). The above results implied that this TIMAS might have potential value in predicting SKCM patients’ survival prognosis and the effect of ICI treatment based on the immune status of patients’ TME.

The value of TIMAS in immunotherapy. a Analysis results of TIDE for different TI-MAS subgroups (TIDE score, exclusion score, dysfunction score). b TIS score in different TIMAS subgroups. c Chord diagram of TIMAS and immune checkpoint-related genes. d-f TIDE, TIS, TIMAS time-dependent ROC curves in 6, 12, 24 months
Discussion
Compared with traditional therapy, ICI therapy is considered as a promising treatment option that has greatly improved survival in patients with advanced (metastatic) SKCM [3,4,5]. Taking difference between the outcomes of immunotherapy in patients with SKCM into consideration [6, 7], understanding and elucidating the TME of SKCM will help to identify a biomarker that can predict immunotherapy response and OS. The scRNA-seq can be used to define cell subgroups, providing new avenues to reveal the impact of TME on ICI treatment [37]. Therefore, we comprehensively analyzed bulk RNA-seq and scRNA-seq to identify IAGs closely related with TME in SKCM and to establish a robust signature that characterizes the tumor immune microenvironment and guides immunotherapy strategies for patients with SKCM.
In our study, we clustered 4569 cells in scRNA-seq into 9 non-malignant cell subgroups composed of immune cells and stromal cells (B cells, CD4 T cells, CD8 T cells, dendritic cells, endothelial cells, fibroblasts, macrophages, neurons, and NK cells) to explore the TME that could affect ICI therapy. Subsequently, we divided SKCM patients into immunity-high and immunity-low groups based on their level of immune infiltration and calculated the expression levels of 9 immune/stromal cells in different immunity subtypes and found that these cells were highly expressed in patients with high immune infiltration. We also found that DEGs between immune/stromal cells were enriched in pathways related to immune cells such as neutrophils. The above results indicated that the heterogeneity of these TME cells in tumor immune infiltration was closely related to the effect of ICI therapy [38].
To further identify the genes that affect the TME, we conducted WGCNA to screen hub genes closely associated with 9 immune/stromal cells. Based on these hub genes, we found five potential drugs (GW-501,516, didanosine, Tyrphostin-AG-1296, SDZ-205-557 and LY-456,236) that might alter the state of low immune infiltration in SKCM patients using the CMap database. It has been reported that GW-501,516 could inhibit the development and metastasis of melanoma by inhibiting cathepsin B [39, 40]. Previous study has found that the co-application of low-dose didanosine and azidothymidine could play an anti-tumor effect [41]. Tyrphostin-AG-1296 was found to potently inhibit the viability of melanoma cells, and its combination with PLX4032 had an additive effect in reducing cell viability [42]. High expression of serotonin receptors may promote tumor progression, SDZ-205-557 may have antitumor potential as a serotonin receptor antagonist [43]. Metabotropic gluta-mate receptor 1 (GRM1) has been reported to affect tumor cell survival [44]. LY-456,236 as a glutamate receptor antagonist targeting GRM1 might have antitumor potential. Next, we identified five TIMAGs from the hub genes and built TIMAS. We grouped patients according to TIMAS. Patients with TIMAS-low demonstrated significantly enhanced rates of survival compared to those with TIMAS-high in both the TCGA and GEO datasets. Univariate and multivariate Cox regression analyses revealed that TIMAS was an independent prognostic factor for OS in SKCM patients. We then constructed a nomogram which consists of TIMAS and clinical factors had excellent predictive ability. The above results showed that our TIMAS could accurately predict OS in SKCM patients.
TIMAS consisted of 5 TIMAGs significantly associated with OS (CD86, CD80, SEMA4D, C1QA, IRF1). CD80 (B7-1) and CD86 (B7-2) are proteins belonging to the B7 family, expressed on antigen-presenting cells (APCs), that actively participate in the stimulation of T cell activation via costimulatory signaling [45, 46]. Moreover, CD80/CD86 has been observed to correlate with elevated levels of M1 macrophage expression [13]. Ipilimumab has clinically achieved favorable immunological efficacy by inhibiting the interaction between CTLA-4 and CD80/CD86 on APCs, which indicated the importance of CD80/CD86 in anti-CTLA-4 immunotherapeutic strategies [47, 48]. SEMA4D (CD100) is a class IV semaphores that inhibits tumor cell invasion in SKCM by inhibiting activation of the tyrosine kinase receptor Met induced by hepatocyte growth factor (HGF) and Plexin-B1 expression [49, 50]. C1QA is the gene encoding the A chain of the complement subcomponent C1q, patients with high C1QA expression have a better prognosis. This may be due to the fact that SKCM belongs to cancers with protective complement, and high expression of genes encoding classical and alternative pathways in such cancers leads to good prognosis [51]. IRF1, also known as IFN-regulatory factor 1, involved in the transcription of genes related with the body’s response to viruses and associated with tumor suppression, innate and acquired immune responses [52]. Overall, we constructed TIMAS consisting of five TIMAGs, and found that patients with higher expression of the five TIMAGs had lower TIMAS and improved prognoses.
In order to investigate its immunological properties, we analyzed gene mutations in different TIMAS subgroups of SKCM patients. Missense mutations, SNP, and C > T mutations accounted for the largest percentages in their respective classification categories [53]. The greatest variation in mutation frequency between risk subgroups was MUC16, which was more frequently mutated in low-risk patients [54]. Higher TMB and better OS were seen in patients with MUC16 mutations. In addition, it was discovered that patients with MUC16 mutations had considerably higher levels of CD8 T cells, M1 macrophages, IFN γ, and T cell inflammatory signals [55]. Therefore, TIMAS-low patients with high MUC16 mutations have high immune infiltration, good survival outcomes and immunotherapy efficacy, suggesting that our findings were accurate.
Considering the influence of TME on SKCM immunotherapy, we further analyzed the immune cell composition in different TIMAS subgroups of patients. The results showed that immune cell composition is different in patients with different TIMAS subgroups. TIMAS-low patients had higher proportions of CD8 T cells, activated memory CD4 T cells, and M1 macrophages in immune cells, while TIMAS-high patients had more abundant resting memory CD4 T cells, M2 macrophages in the TME. Numerous studies have shown that therapeutic efficacy is dependent on the degree of infiltration of T cells, especially CD8 T cells [56]. M1 macrophages are pro-inflammatory macrophages that respond defensively to tumors and are abundant in primary and thin melanomas and decreased in advanced (metastatic) melanomas [13, 57]. In contrast, M2 macrophages are an anti-inflammatory macrophage and were found to promote melanoma tumor angiogenesis and tumor growth [57]. These findings indicated that TIMAS has the potential to predict immune infiltration in SKCM patients.
To compare the predictive ability of TIMAS with known immunotherapy predictive biomarkers (TIDE, TIS), we analyzed their performance in predicting anti-CTLA-4 and anti-PD-1/PD-L1 immunotherapy. Ipilimumab, a CTLA-4 inhibitor that blocks the suppression of activated T cells, was the first medication to demonstrate an improvement in overall survival. At the same time, Wbber et al. found that the PD-1 inhibitor nivolumab was also applicable to patients with advanced melanoma [3, 58]. TIDE and TIS signatures are commonly used to predict patient response to ICI therapy, TIDE has been shown to accurately predict response in melanoma patients receiving immuno-therapy (anti-PD-1 therapy or anti-CTLA-4 therapy), and TIS can predict an-ti-PD-1/PD-L1 therapy outcomes [36, 59]. Differences in patient TIMAS might reflect different treatment outcomes for patients treated with anti-PD1 or anti-CTLA4 immunotherapy. We found that TIMAS-high patients had higher TIDE scores, T cell exclusion scores, and lower TIS scores compared with TIMAS-low patients. The higher the T cell exclusion score, the lower the infiltration of cytotoxic T lymphocytes (CTL), and tumors with low CTL were not conducive to T cell infiltration, which might cause immune evasion and lead to poor ICI response [59]. Unfortunately, comparing TIMAS, TIDE and TIS were more biased to reflect the function and status of T cells, rather than considering the important role of TME in immunotherapy response. In addition, these two markers were more focused on predicting the immune efficacy of patients, which did not include the survival time of patients. In our study, TIMAS had the ability to predict immunotherapy not inferior to TIDE and TIS, TIMAS could much fully reflect the impact of TME in immunotherapy and better predict the long-term OS of patients, and also showed excellent accuracy in time-dependent ROC analysis.
In conclusion, the tumor immune microenvironment-related signature TIMAS had potential. Grouping patients according to their TIMAS might help differentiate immune and molecular signatures, predict patient outcomes. Nonetheless, the sample cohort we used to construct TIMAS was retrospective, so further experimental studies of the predictive power of TIMAS are needed.
Data availability
The data presented in this study are available on request from the corresponding author.
References
Liu Y, Zhang H, Hu D, Liu S (2023) New algorithms based on autophagy-related lncRNAs pairs to predict the prognosis of skin cutaneous melanoma patients. Arch Dermatol Res 9:1–6. https://doi.org/10.1007/s00403-022-02522-0
Carr S, Smith C, Wernberg J (2020) Epidemiology and risk factors of melanoma. Surg Clin North Am 100:1–2. https://doi.org/10.1016/j.suc.2019.09.005
Bagge RO, Ny L, Ascierto PA, Hodi FS, Larkin J, Robert C, Schachter J, Weber JS, Long GV, Van Akkooi AC (2021) The efficacy of immunotherapy for in-transit metastases of melanoma: an analysis of randomized controlled trials. Melanoma Res 31:181–185. https://doi.org/10.1097/CMR.0000000000000719
Duan J, Cui L, Zhao X, Bai H, Cai S, Wang G, Zhao Z, Zhao J, Chen S, Song J, Qi C (2020) Use of immunotherapy with programmed cell death 1 vs programmed cell death ligand 1 inhibitors in patients with cancer: a systematic review and meta-analysis. JAMA Oncol 6:375–384. https://doi.org/10.1001/jamaoncol.2019.5367
Nakamura K, Ashida A, Kiniwa Y, Okuyama R (2022) Chemokine level predicts the therapeutic effect of anti-PD-1 antibody (nivolumab) therapy for malignant melanoma. Arch Dermatol Res 314:887–895. https://doi.org/10.1007/s00403-021-02305-z
Hugo W, Zaretsky JM, Sun LU, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E (2016) Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165:35–44. https://doi.org/10.1016/j.cell.2016.02.065
Schoenfeld AJ, Hellmann MD (2020) Acquired resistance to immune checkpoint inhibitors. Cancer Cell 37:443–455. https://doi.org/10.1016/j.ccell.2020.03.017
Ruocco MR, Lamberti A, Serrano MJ, Fiume G, Arcucci A (2022) Tumor Microenvironment and Cancer Cell interactions in solid Tumor Growth and Therapy Resistance. Front Cell Dev Biol 10:896194. https://doi.org/10.3389/fcell.2022.896194
Woods DM, Laino AS, Winters A, Alexandre J, Freeman D, Rao V, Adavani SS, Weber JS, Chattopadhyay PK (2020) Nivolumab and ipilimumab are associated with distinct immune landscape changes and response-associated immunophenotypes. JCI Insight 5:e137066. https://doi.org/10.1172/jci.insight.137066
Avagliano A, Fiume G, Pelagalli A, Sanità G, Ruocco MR, Montagnani S, Arcucci A (2020) Metabolic plasticity of melanoma cells and their crosstalk with tumor microenvironment. Front Oncol 10:722. https://doi.org/10.3389/fonc.2020.00722
Hei Y, Chen Y, Li Q, Mei Z, Pan J, Zhang S, Xiong C, Su X, Wei S (2022) Multifunctional immunoliposomes enhance the Immunotherapeutic effects of PD-L1 antibodies against Melanoma by reprogramming immunosuppressive Tumor Microenvironment. Small 18:2105118. https://doi.org/10.1002/smll.202105118
Wang J, Lin F, Wan Z, Sun X, Lu Y, Huang J, Wang F, Zeng Y, Chen YH, Shi Y, Zheng W (2018) Profiling the origin, dynamics, and function of traction force in B cell activation. Sci Signal 11:eaai9192. https://doi.org/10.1126/scisignal.aai9192
Kuraitis D, Rosenthal N, Boh E, McBurney E (2022) Macrophages in dermatology: pathogenic roles and targeted therapeutics. Arch Dermatol Res 1:1–8. https://doi.org/10.1007/s00403-021-02207-0
Anderson NM, Simon MC (2020) The tumor microenvironment. Curr Biol 30:R921–R925. https://doi.org/10.1016/j.cub.2020.06.081
Hei Y, Teng B, Zeng Z, Zhang S, Li Q, Pan J, Luo Z, Xiong C, Wei S (2020) Multifunctional immunoliposomes combining catalase and PD-L1 antibodies overcome tumor hypoxia and enhance immunotherapeutic effects against melanoma. Int J Nanomed 15:1677–1691
Marzagalli M, Ebelt ND, Manuel ER (2019) Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin Cancer Biol 59:236–250. https://doi.org/10.1016/j.semcancer.2019.08.002
Bence C, Hofman V, Chamorey E, Long-Mira E, Lassalle S, Albertini AF, Liolios I, Zahaf K, Picard A, Montaudié H, Lacour JP (2020) Association of combined PD-L1 expression and tumour-infiltrating lymphocyte features with survival and treatment outcomes in patients with metastatic melanoma. J Eur Acad Dermatol Venereol 34:984–994. https://doi.org/10.1111/jdv.16016
Demaria O, Cornen S, Daëron M, Morel Y, Medzhitov R, Vivier E (2019) Harnessing innate immunity in cancer therapy. Nature 574:45–56. https://doi.org/10.1038/s41586-019-1593-5
Guo S, Deng CX (2018) Effect of stromal cells in tumor microenvironment on metastasis initiation. Int J Biol Sci 14:2083. https://doi.org/10.7150/ijbs.25720
Pach E, Brinckmann J, Rübsam M, Kümper M, Mauch C, Zigrino P (2021) Fibroblast mmp14-dependent collagen processing is necessary for melanoma growth. Cancers 13:1984. https://doi.org/10.3390/cancers13081984
Denton AE, Roberts EW, Fearon DT (2018) Stromal cells in the Tumor Microenvironment. Adv Exp Med Biol 1060:99–114. https://doi.org/10.1007/978-3-319-78127-3_6
Romano V, Belviso I, Venuta A, Ruocco MR, Masone S, Aliotta F, Fiume G, Montagnani S, Avagliano A, Arcucci A (2021) Influence of tumor microenvironment and fibroblast population plasticity on melanoma growth, therapy resistance and immunoescape. Int J Mol Sci 22:5283. https://doi.org/10.3390/ijms22105283
Tan Z, Chen X, Zuo J, Fu S, Wang H, Wang J (2023) Comprehensive analysis of scRNA-Seq and bulk RNA-Seq reveals dynamic changes in the tumor immune microenvironment of bladder cancer and establishes a prognostic model. J Transl Med 21:223. https://doi.org/10.1186/s12967-023-04056-z
Nip KM, Chiu R, Yang C, Chu J, Mohamadi H, Warren RL, Birol I (2020) RNA-bloom enables reference-free and reference-guided sequence assembly for single-cell transcriptomes. Genome Res 30:1191–1200. http://www.genome.org/cgi/doi/https://doi.org/10.1101/gr.260174.119
Noé A, Cargill TN, Nielsen CM, Russell AJ, Barnes E (2020) The application of single-cell RNA sequencing in vaccinology. J Immunol Res 2020:8624963. https://doi.org/10.1155/2020/8624963
Becht E, McInnes L, Healy J, Dutertre CA, Kwok IW, Ng LG, Ginhoux F, Newell EW (2019) Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 37:38–44. https://doi.org/10.1038/nbt.4314
Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ (2019) Reference-based analysis of lung single-cell sequencing reveals a transitional profbrotic macrophage. Nat Immunol 20:163–172. https://doi.org/10.1038/s41590-018-0276-y
He Y, Jiang Y, Chen Z, Wang C X (2018) Classification of triple-negative breast cancers based on immunogenomic profiling. J Exp Clin Cancer Res 37:1–13. https://doi.org/10.1186/s13046-018-1002-1
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K (2017) KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45:D353–D361. https://doi.org/10.1093/nar/gkw1092
Simon N, Friedman J, Hastie T, Tibshirani R (2011) Regularization paths for Cox’s proportional hazards model via coordinate descent. J Stat Softw 39:1. https://doi.org/10.18637/jss.v039.i05
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10:1523. https://doi.org/10.1038/s41467-019-09234-6
Liang D, Yu C, Ma Z, Yang X, Li Z, Dong X, Qin X, Du L, Li M (2022) Identification of anthelmintic parbendazole as a therapeutic molecule for HNSCC through connectivity map-based drug repositioning. Acta Pharm Sin B 12:2429–2442. https://doi.org/10.1016/j.apsb.2021.12.005
Mei S, Meyer CA, Zheng R, Qin Q, Wu Q, Jiang P, Li B, Shi X, Wang B, Fan J, Shih C (2017) Cistrome cancer: a web resource for integrative gene regulation modeling in cancer. Cancer Res 77:e19–e22. https://doi.org/10.1158/0008-5472.CAN-17-0327
Heagerty PJ, Zheng Y (2005) Survival model predictive accuracy and ROC curves. Biometrics 61:92–105. https://doi.org/10.1111/j.0006-341X.2005.030814.x
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, Diehn M (2015) The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21:938–945. https://doi.org/10.1038/nm.3909
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA (2017) IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Investig 127:2930–2940. https://doi.org/10.1172/JCI91190
Wan Q, Liu C, Liu C, Liu W, Wang X, Wang Z (2020) Discovery and validation of a metastasis-related prognostic and diagnostic biomarker for melanoma based on single cell and gene expression datasets. Front Oncol 10:585980. https://doi.org/10.3389/fonc.2020.585980
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, Wang S (2018) A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175:984–997. https://doi.org/10.1016/j.cell.2018.09.006
Reichenbach G, Starzinski-Powitz A, Doll M, Hrgovic I, Maria Valesky E, Kippenberger S, Bernd A, Kaufmann R, Meissner M (2012) Ligand activation of peroxisome proliferator-activated receptor delta suppresses cathepsin B expression in human endothelial cells in a posttranslational manner. Exp Dermatol 21:751–757. https://doi.org/10.1111/exd.12002
Zhang H, Xu X, Xu R, Ye T (2022) Drug repurposing of ivermectin abrogates neutrophil extracellular traps and prevents melanoma metastasis. Front Oncol 12:989167. https://doi.org/10.3389/fonc.2022.989167
Aschacher T, Sampl S, Käser L, Bernhard D, Spittler A, Holzmann K, Bergmann M (2012) The combined use of known antiviral reverse transcriptase inhibitors AZT and DDI induce anticancer effects at low concentrations. Neoplasia 14:44–53. https://doi.org/10.1593/neo.11426
Li Y, Li Y, Liu Q, Wang A (2015) Tyrphostin AG1296, a platelet-derived growth factor receptor inhibitor, induces apoptosis, and reduces viability and migration of PLX4032-resistant melanoma cells. OncoTargets Ther 2015:1043–1051. https://doi.org/10.2147/OTT.S70691
Barzegar-Fallah A, Alimoradi H, Dunlop JL, Torbati E, Baird SK (2021) Serotonin type-3 receptor antagonists selectively kill melanoma cells through classical apoptosis, microtubule depolymerisation, ERK activation, and NF-κ B downregulation. Cell Biol Toxicol 2021:1–17. https://doi.org/10.1007/s10565-021-09667-0
Isola AL, Eddy K, Zembrzuski K, Goydos JS, Chen S (2018) Exosomes released by metabotropic glutamate receptor 1 (GRM1) expressing melanoma cells increase cell migration and invasiveness. Oncotarget 9:1187. https://doi.org/10.18632/oncotarget.23455
Brzostek J, Gascoigne NR, Rybakin V (2016) Cell type-specific regulation of immunological synapse dynamics by B7 ligand recognition. Front Immunol 7:24. https://doi.org/10.3389/fimmu.2016.00024
Kennedy A, Waters E, Rowshanravan B, Hinze C, Williams C, Janman D, Fox TA, Booth C, Pesenacker AM, Halliday N, Soskic B (2022) Differences in CD80 and CD86 transendocytosis reveal CD86 as a key target for CTLA-4 immune regulation. Nat Immunol 23:1365–1378. https://doi.org/10.1038/s41590-022-01289-w
Payandeh Z, Yarahmadi M, Nariman-Saleh‐Fam Z, Tarhriz V, Islami M, Aghdam AM, Eyvazi S (2019) Immune therapy of melanoma: overview of therapeutic vaccines. J Cell Physiol 234:14612–14621. https://doi.org/10.1002/jcp.28181
Qiu H, Tian W, He Y, Li J, He C, Li Y, Liu N, Li J (2021) Integrated analysis reveals prognostic value and immune correlates of CD86 expression in lower grade glioma. Front Oncol 11:654350. https://doi.org/10.3389/fonc.2021.654350
Ch’ng ES, Kumanogoh A (2010) Roles of Sema4D and Plexin-B1 in tumor progression. Mol Cancer 9:1–9. https://doi.org/10.1186/1476-4598-9-251
Neufeld G, Mumblat Y, Smolkin T, Toledano S, Nir-Zvi I, Ziv K, Kessler O (2016) The semaphorins and their receptors as modulators of tumor progression. Drug Resist Updat 29:1–12. https://doi.org/10.1016/j.drup.2016.08.001
Roumenina LT, Daugan MV, Petitprez F, Sautès-Fridman C, Fridman WH (2019) Context-dependent roles of complement in cancer. Nat Rev Cancer 19:698–715. https://doi.org/10.1038/s41568-019-0210-0
Feng H, Zhang YB, Gui JF, Lemon SM, Yamane D (2021) Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PLOS Pathog 17:e1009220. https://doi.org/10.1371/journal.ppat.1009220
Jiang F, Wu C, Wang M, Wei K, Zhou G, Wang J (2020) Multi-omics analysis of tumor mutation burden combined with immune infiltrates in melanoma. Clin Chim Acta 511:306–318. https://doi.org/10.1016/j.cca.2020.10.030
Wang Z, Hou H, Zhang H, Duan X, Li L, Meng L (2022) Effect of MUC16 mutations on tumor mutation burden and its potential prognostic significance for cutaneous melanoma. Am J Transl Res 14:849
Wang Q, Yang Y, Yang M, Li X, Chen K (2020) High mutation load, immune-activated microenvironment, favorable outcome, and better immunotherapeutic efficacy in melanoma patients harboring MUC16/CA125 mutations. Aging 12:10827. https://doi.org/10.18632/aging.103296
Femel J, Van Hooren L, Herre M, Cedervall J, Saupe F, Huijbers EJ, Verboogen DR, Reichel M, Thijssen VL, Griffioen AW, Hellman L (2022) Vaccination against galectin-1 promotes cytotoxic T-cell infiltration in melanoma and reduces tumor burden. Cancer Immunol Immunother 71:2029–2040. https://doi.org/10.1007/s00262-021-03139-4
Wang H, Yang L, Wang D, Zhang Q, Zhang L (2017) Pro-tumor activities of macrophages in the progression of melanoma. Hum Vaccin Immunother 13:1556–1562. https://doi.org/10.1080/21645515.2017.1312043
Weber JS, Poretta T, Stwalley BD, Sakkal LA, Du EX, Wang T, Chen Y, Wang Y, Betts KA, Shoushtari AN (2022) Nivolumab versus placebo as adjuvant therapy for resected stage III melanoma: a propensity weighted indirect treatment comparison and number needed to treat analysis for recurrence-free survival and overall survival. Cancer Immunol Immunother 2022:1–10. https://doi.org/10.1007/s00262-022-03302-5
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, Liu J (2018) Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24:1550–1558. https://doi.org/10.1038/s41591-018-0136-1
Acknowledgements
We thank TCGA and GEO databases for the availability of the data.
Funding
This research was funded by National Natural Science Foundation of China (Nos. 82070288, 11772198), the Science & Technology Department of Sichuan Province (No. 2022YFS0627), the young and middle-aged talent development fund of Shanghai Institute of Technology (No. ZQ2021-23).
Author information
Authors and Affiliations
Contributions
Conceptualization: Shanshan Li, Junjie Zhao, and Wei Wang; Methodology: Shanshan Li, Junjie Zhao and Wei Wang; Software: Junjie Zhao and Wei Wang; Validation: Shanshan Li, Junjie Zhao and Wei Wang; Formal Analysis: Shanshan Li, Junjie Zhao, Guangyu Wang, Zhe Leng, Qinglei Liu and Wei Wang; Investigation: Junjie Zhao, Guangyu Wang and Shanshan Li; Resources: Wei Wang, Qingping Yao and Jun Jiang; Data curation: Junjie Zhao and Shanshan Li; Writing – original draft: Junjie Zhao and Shanshan Li; Writing – review & editing: Shanshan Li, Junjie Zhao, and Wei Wang; Visualization: Shanshan Li, Junjie Zhao, Guangyu Wang and Zhe Leng; Supervision: Junjie Zhao and Wei Wang; Project administration: Wei Wang; Funding acquisition: Shanshan Li, Qingping Yao and Jun Jiang. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, S., Zhao, J., Wang, G. et al. Based on scRNA-seq and bulk RNA-seq to establish tumor immune microenvironment-associated signature of skin melanoma and predict immunotherapy response. Arch Dermatol Res 316, 262 (2024). https://doi.org/10.1007/s00403-024-03080-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00403-024-03080-3