
Abstract
Coffea arabica L. is an important agricultural commodity, accounting for 60% of traded coffee worldwide. Nitrogen (N) is a macronutrient that is usually limiting to plant yield; however, molecular mechanisms of plant acclimation to N limitation remain largely unknown in tropical woody crops. In this study, we investigated the transcriptome of coffee roots under N starvation, analyzing poly-A+ libraries and small RNAs. We also evaluated the concentration of selected amino acids and N-source preferences in roots. Ammonium was preferentially taken up over nitrate, and asparagine and glutamate were the most abundant amino acids observed in coffee roots. We obtained 34,654 assembled contigs by mRNA sequencing, and validated the transcriptional profile of 12 genes by RT-qPCR. Illumina small RNA sequencing yielded 8,524,332 non-redundant reads, resulting in the identification of 86 microRNA families targeting 253 genes. The transcriptional pattern of eight miRNA families was also validated. To our knowledge, this is the first catalog of differentially regulated amino acids, N sources, mRNAs, and sRNAs in Arabica coffee roots.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Coffee is one of the most important agricultural commodities in the world, representing a significant source of income for several tropical developing countries (van der Vossen et al. 2015). Two species are economically important: Coffea arabica L. (Arabica coffee) and Coffea canephora Pierre ex Froehner (Conilon or Robusta coffee). C. arabica represents nearly 60% of traded coffee worldwide (van der Vossen et al. 2015).
With the development of functional genomics, large-scale transcriptome analysis based on expressed sequence tag (EST) sequencing, microarray hybridization, and RT-qPCR analysis of key genes have been extensively used to investigate the molecular basis of several features in Arabica coffee during fruit development stages (Joët et al. 2009, 2014; Ivamoto et al. 2017a; Ivamoto et al. 2017b), responses to abiotic (Marraccini et al. 2012), and biotic (Cardoso et al. 2014) stresses. More recently, next-generation sequencing technologies for quantitative determination of RNA levels (RNA-seq) have been incorporated into studies on the coffee transcriptome. mRNA sequencing (RNA-seq) allowed significant progress to improve our knowledge of the transcriptional profile of coffee genes under biotic and environmental challenges (Fernandez et al. 2012; Bertrand et al. 2015; Mofatto et al. 2016). However, most of these studies have focused on leaf or fruit development, while the transcriptional profile of below ground organs remains poorly understood. For instance, less than 2% of all Arabica coffee publicly available ESTs have originated from root cDNA libraries (Vieira et al. 2006; Mondego et al. 2011). Thus, there is a need for large-scale studies that unravel the repertoire of transcriptionally active genes in C. arabica roots.
Nitrate and ammonium are the main sources of N for agricultural systems. Both substances can be absorbed by the roots through specific transporters for ammonium (AMT1 and AMT2 families), and nitrate (NRT1 and NRT2 families) coded by multigenic families (Rennenberg et al. 2010; Xu et al. 2012). Léran et al. (2014) reviewed and proposed a new nomenclature for nitrate transporters, where NRT1/PTR family were denomined as NPF members. On average, angiosperms have five members of each AMT family, four NRT2, and more than 50 NRT1 genes (von Wittgenstein et al. 2014). Several studies have shown that these transporters are transcriptionally modulated by N availability (Scheible et al. 2004; Cai et al. 2012; Wei et al. 2013), and can also affect N net fluxes in roots (Sorgona et al. 2011; Luo et al. 2013). After uptake, nitrate is reduced to nitrite in the cytoplasm by nitrate reductase (NR) enzyme. Nitrite is then reduced to ammonium in the plastid by nitrite reductase (ammonium can enter directly at this step) for assimilation by glutamine synthetase (GS) enzyme (Coleman et al. 2012; Castro-Rodríguez et al. 2015). Additionally, N assimilation processes can also be transcriptionally regulated under N starvation (Ruffel et al. 2008; Krapp et al. 2011).
Gutiérrez et al. (2007) were the first to suggest that N metabolism could also be regulated by non-coding short RNAs, like microRNAs (miRNAs). For example, in Arabidopsis thaliana, miR156, miR169, and miR398 responded to N limitation (Pant et al. 2009; Zhao et al. 2011), whereas Liang et al. (2012) identified five miRNA families (miR160, miR780, miR826, miR842, and miR846) that are induced by N deficiency in Arabidopsis using small RNA deep sequencing analysis. Additionally, miR393 and one of its targets, the auxin receptor AFB3, act as molecular players mediating NO3− regulation of primary and lateral root growth (Vidal et al. 2010). Thus, it is now well accepted that miRNAs play important roles as regulators of nutrient metabolism in plant responses to low N stress (Kulcheski et al. 2015; Nath and Tuteja 2016). RNA-seq can also be used to analyze the small RNA (sRNA) component of the transcriptome when libraries are prepared from low-molecular weight RNA fractions. To date, large-scale analyses of C. arabica miRNAs have been based only on Sanger ESTs, and genome survey sequences (GSSs) (Akter et al. 2014; Devi et al. 2016), but with a clear bias imposed by the scarcity of available data from roots. Based on these informations, and knowing that integrated mRNA-seq and sRNA-seq analysis can make important contributions to understand root molecular responses to N starvation (Vidal et al. 2013), our study used high-throughput sequencing technologies to characterize a C. arabica root transcriptome in control conditions, with and without N supply. To support our analysis, we conducted experiments to address N uptake, assimilation, and amino acid profiles in coffee roots. This is the first study to simultaneously profile the mRNAs and miRNAs transcriptome in Arabica coffee and to perform a molecular characterization of Arabica coffee roots using bioinformatics and biochemical and molecular approaches.
Material and methods
Plant material and experimental conditions
Basic procedures for obtaining N-starved roots followed previous studies from our research group (de Carvalho et al. 2013; dos Santos et al. 2017). Arabica coffee seeds (Coffea arabica L. cv. IAPAR59) were germinated in greenhouse using boxes containing sterilized sand (Fig. S1). Seedlings were irrigated twice weekly with distilled water in the first 30 days, and with Clark nutritive solution (Clark et al. 1975) with pH adjusted to 5.5–6.0 for the remainder of the study. After 4 months, plants were selected by size uniformity (plants with three leaf pairs). Roots were washed thoroughly with distilled water and the plantlets were transferred to 4 L pots (three plants per pot) with nutrient solution, constant aeration, and cultivated in a growth chamber (12/12 h light/dark cycle, 25/23 °C day/night temperature, 45% humidity, and photosynthetic photon flux density of ~ 250 μmol/m2/s). Plants were grown in nutrient solution containing 800 μM K2SO4, 250 μM, MgSO4, 200 μM KH2PO4, 500 μM CaCl2, 4000 μM NH4NO3, 100 μM NaFeEDTA, 5 μM H3BO3, 3 μM MnSO4, 2.5 μM ZnSO4, 0.1 μM CuSO4, and 0.7 μM NaMoO with pH 5.8. The pH of the nutrient solution was adjusted to 5.5–6.0 every 2 days using sodium hydroxide or hydrochloric acid, and renewed weekly. After 4 weeks, lateral roots were harvested with a minimum of three biological replicates per sampling at 0, 1 day (24 h), and 10 days after transfer into an N-free nutrient solution. Sampling was always done between 9:30 and 10:30 h. Biological replicates were represented by pools of lateral roots of at least nine plants at the same developmental stage. After harvesting, all samples were immediately frozen in liquid nitrogen and stored at − 80 °C until use. These samples were used for all analyses, except for the 15N influx experiments.
Total N concentration
Total nitrogen content in roots was analyzed by spectrophotometry according to Miyazawa et al. (1999).
Glutamine synthetase activity
GS activity was measured using procedures similar to those used in previous studies of coffee plants (Pompelli et al. 2010). Plant material was homogenized in buffer containing 25 mM Tris-HCl (pH 7.6), 1 mM MgCl, 1 mM EDTA, 14 mM β-mercaptoethanol, and 1% (w/v) polyvinylpyrrolidone (PVP). The homogenate was centrifuged at 25,000 g for 30 min at 4 °C. Glutamine synthetase activity was determined using hydroxylamine as a substrate and the formation of γ-glutamyl hydroxamate (γ-GHM) was determined using acidified ferric chloride (Wallsgrove et al. 1979; Cruz et al. 2004). The GHM γ-glutamine was quantified as standard and the absorbance was measured at 540 nm. For each time point, GS activity was measured in three biological replicates.
Nitrate reductase activity
In vitro nitrate reductase (NR) activity was measured according to Rossa (1999) with modifications made by Dovis et al. (2014). Approximately 70 mg of tissue were ground in liquid N in a mortar, suspended in an extraction buffer (0.2 M sodium phosphate buffer, pH 8.0 [2.8% (w/v) and NaH2PO4.H2O and 97.2% (w/v) Na2HPO4. 7 H2O]; 5 mM EDTA; 1 mM DTT; 0.3% (w/v) BSA), and centrifuged at 12,000 rpm and 4 °C for 15 min. The supernatant was collected and incubated in the reaction buffer (0.2 M sodium phosphate buffer, pH 8.0; 6 mM KNO3, 0.5 mM MgSO4) at 25 °C, and 0.04 mM NADH was then added to start enzymatic reaction. Controls without the addition of NADH were included for each treatment. The reaction was stopped with 1.4 mM ZnSO and 43% (v/v) ethanol and the material centrifuged at 12,000 rpm and 4 °C for 10 min. The nitrite produced by NR activity was analyzed spectrophotometrically at 543 nm using a 1-cm polypropylene optical path cuvette after adding 9.6 mM sulfanilamide and 0.7 mM N-(1-naphthyl) ethylenediamine dihydrochloride (NED) to the extract. A 0.1 mM NaNO solution was used for the preparation of the standard curve. The in vitro activity of NR (NRA) was calculated according to the formula:
Where EBt = total volume of crude extract, EBi = volume of crude extract incubated, t = incubation time, and FW = fresh weight. The result is expressed as U g−1, assuming that the activity of one NR unit (U) corresponds to 1 μmol nitrite produced per minute (Chapman and Harrison 1988) at a constant temperature of 25 °C. Three biological replicates were analyzed per time point.
Total protein content
To determine total protein concentration, frozen samples were homogenized in 2 ml of 50 mM potassium phosphate buffer (pH 7.3), 1% PVP (w/v), and 0.1 mM EDTA previously cooled to 4 °C. After centrifugation for 10 min at 4 °C and 7500 rpm, the supernatant was transferred to a different tube and kept on ice until the time of analysis. The extracts were quantified by the Bradford method (1976) using bovine serum albumin (BSA) as standard.
Total free amino acids content
The stored roots (− 80 °C) were macerated in liquid nitrogen using a mortar and pestle and added 80% ethanol. Next, the mixture was homogenized, incubated at 80 °C for 20 min, and centrifuged at 10,000 rpm for 10 min. Total amino acid concentrations were determined based on the methodology described by Praxedes et al. (2006). The reaction mixture contained 50 μl of 1 kmol m−3 citrate-NaOH buffer (pH 5.2) with 0.2% (w/v) ascorbic acid, 100 ml of ethanolic extract, and 70 ml of 1% (v/v) ninhydrin in 70% (v/v) ethanol. The free amino acids concentration was estimated using a standard curve with an equimolecular mixture of glycine, glutamic acid, phenylalanine, and arginine in 70% (v/v) ethanol. The absorbance of the mixture was measured spectrophotometrically at 570 nm.
Individual amino acids analysis by UPLC-MS
For amino acids analysis, ground samples (100 mg) were extracted with 1 ml of methanol/water (50:50 v/v) solution for 30 min in ultrasonic bath. Next, the mixture was centrifuged at 7000 rpm for 10 min. The supernatant was collected into a vial and directly analyzed by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS). The samples were analyzed in an Acquity UPLC-MS system (Waters, Milford, MA USA) and data were acquired by injection of 7 μl of supernatant. Chromatographic separation was performed in a Waters Acquity C18 BEH analytical column (150 mm × 2.1 mm i.d., 1.7 μm) at 30 °C. Methanol (A) and 0.1% formic acid in water (B) were used as the mobile phase at a flow rate of 0.2 ml/min. The initial gradient condition was kept at 1% A and 99% B for 2.5 min, followed by a change to 50% A and 50% B for 2.5 min and back to the initial condition at 8 min. ESI(+)-MS and tandem ESI(+)-MS/MS were obtained under the following conditions: capillary, 3.5 KV; cone, 30 V; source and desolvation temperature, 150 and 300 °C, respectively. For ESI(+)MS/MS, the energy for the collision-induced dissociations (CID) was 15 eV. Data were acquired in the 50–300 m/z range. Ions were identified by the comparison of their m/z, retention time, and ESI(+)MS/MS dissociation patterns with pure standards.
15N influx experiment
15N uptake in coffee roots was measured using 15N-labeled ammonium nitrate as N source with either nitrate (K15NO) or ammonium (15NH4SO4). To measure high and low-affinity N transport, 15N-labeled ammonium nitrate was added to the N-free nutrient solution at two concentrations (0.2 mM and 2.0 mM) (Kojima et al. 2007; Camañes et al. 2009). During the acclimation period of the C. arabica L. cv. IAPAR59, seedlings were grown hydroponically under non-sterile conditions in a greenhouse with the following regime: 14/10 h light/dark and 28/18 °C temperature. Plants were grown in nutrient solution containing 1 mM KH2PO4, 1 mM MgSO4, 250 μM K2SO4, 250 μM CaCl2, 100 μM Na-Fe-EDTA, 50 μM KCl, 50 μM H3BO3, 5 μM MnSO4, 1 μM ZnSO4, 1 μM CuSO4, and 1 μM NaMoO4 (pH adjusted using 2 mM MES, pH 5.8; Sigma-Aldrich, St. Louis, MO, USA). The nutrient solution was replaced every 2 days. After a 1-week acclimation period, plants were supplied with N-sufficient (+ N, 2 mM NH4NO3) or N-starvation (− N, without N supply) nutrient solution for 3 days. Influx measurements of 15N-labeled N in plant roots were conducted after rinsing the roots in 1 mM CaSO4 solution for 1 min followed by incubation for 10 min in nutrient solution containing 0.2 mM of 15N-labeled with the equimolar concentration of either K15NO3 (98.18 atom% 15N) or 15(NH4)2SO4 (60 atom% 15N) as the sole nitrogen source, and washing in 1 mM CaSO4 solution. Roots were harvested and stored at − 70 °C until use. Each sample was ground in liquid N and dried at 55 °C for 5 days. 15N and % N determination was performed by isotope ratio mass spectrometry (IRMS) (ANCA-SL, Sercon, Crewe, CHS, UK) with 5 mg of dried samples. The results are presented as the mean and standard error (SE) of three biological replicates (n = 3).
RNA extraction
Total RNA from C. arabica L. cv. IAPAR59 roots was extracted using an adapted CTAB method for plants (Chang et al. 1993; Tong et al. 2012). RNA integrity was examined by 1% agarose gel electrophoresis and RNA preparations were treated with DNase (RNase-free; Invitrogen, Carlsbad, CA, USA). Absence of DNA contamination was confirmed by PCR using GAPDH primers and approximately 100 ng of RNA (data not shown). All samples were purified using the PureLink™ Micro-to-Midi Total RNA Purification System (Invitrogen, Carlsbad, CA, USA) to have an A260/A280 > 2.0. RNA concentration and absorbance ratios were determined using a NanoDrop™ 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
High-throughput sequencing: mRNA and sRNA libraries
mRNA was isolated from 20 μg of total RNA using DynaBeads mRNA purification kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. RNA was quantified using the Quant-iT RiboGreen RNA Reagent (Invitrogen) and assessed for integrity by electrophoresis using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA).
Five hundred nanograms of mRNA were used for cDNA library construction and sequencing. First, the mRNA was fragmented using ZnCl2 solution at 70 °C for 30 s. The first-strand cDNA was synthesized applying random primers according to the standard cDNA Synthesis System (Roche Diagnostics, Basel, Switzerland). The cDNA was then subjected to end-repair followed by adaptor ligation using the cDNA Rapid Library Prep kit (Roche Diagnostics). cDNA libraries were purified with Agencourt AMPure XP beads (Beckman Coulter Inc., Brea, CA, USA) and subjected to emulsion PCR amplification by applying two molecules of cDNA per bead (Roche Diagnostics) according to the manufacturer’s instructions. Beads with clonally amplified cDNA library were selected and deposited in a picotiter plate for pyrosequencing using Titanium Sequencing Chemistry (Roche Diagnostics) with 200 flow cycles on a GS Junior 454 Sequencing System (Roche Diagnostics) following the manufacturer’s protocols. In total, six libraries were constructed and sequenced, two for each time point (0, 1, and 10 days without N supply).
sRNA libraries were constructed using 1 μg of total RNA. The sRNAs were ligated with 3′ and 5′ adaptors, and RT-PCR was performed using the TruSeq Small RNA kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Library quality and quantity were tested using an Agilent 2100 Bioanalyzer (Agilent). Following purification, the cDNA from sRNA libraries was sequenced (50 bp [base pairs], single-end) on an Illumina Genome Analyzer II at LC Sciences (Houston, USA). In total, nine sRNA libraries (three for each time point) were sequenced. Sequencing data containing data from mRNA-seq and sRNA-seq libraries have been deposited at the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/ena) under accession number ERP017352.
mRNA-seq: data processing, assembly, annotation, and differential expression analyses
Fastq-mcf version 1.04.662 (Andrews 2010) was used to process and trim reads from the 454 platform. We applied a quality filter to trim reads with mean Phred quality scores < 25, reads covered by more than 90% of homopolymers and low complexity sequences, reads with at least 5 Ns or 5 poly-A/Ts and adapters at the ends, and removing reads with minimum length of 40 after trimming.
De novo transcriptome assembly was initially performed using three assemblers: Newbler, CLC Genome Suite, and Trinity (data not shown). We opted to use Trinity assembly for further analyses because it yielded in the highest mean, median, and N50 contig length values and the lowest singleton number. Assembly with Trinity was performed using default parameters (Grabherr et al. 2011), except for min_contig_length = 200, and included the CuffFly parameter. We further checked the quality of our assembled transcripts by comparing their sequences to a core set of eukaryotic genes using BUSCO (Simão et al. 2015). The comparison revealed 71.1% of BUSCO genes, indicating that assembly quality was similar to that of high-quality plant transcriptome assemblies (Xu et al. 2015).
Differential expression analysis mostly followed the methods of Ranjan et al. (2014). We used RNA-seq by expectation maximization (RSEM), which allows for an assessment of transcript abundances based on the mapping of trimmed RNA-seq reads to the assembled transcriptome, for transcript abundance estimation of the de novo assembled transcripts (Li and Dewey 2011). Reads from individual libraries were mapped to final transcripts using default RSEM parameters using the script un_RSEM_align_n_estimate.pl, followed by joining RSEM-estimated abundance values for each sample using merge_R-SEM_frag_counts_single_table.pl. Lastly, differential expression analysis was performed using run_DE_analysis.pl as implemented in the Bioconductor package EdgeR in the R statistical environment (Robinson and Oshlack 2010; R Development Core Team 2011). Differentially expressed genes (available at https://doi.org/10.7910/DVN/OIN1UW) were selected using two criteria: fold change > 2 and < − 2 and p value < 0.05.
For annotation, Blast2GO (Conesa and Gotz 2008) was first used to obtain Gene Ontology (GO) attribution of the assembled transcripts. Next, the GO functional classifications of unigenes were performed with Web Gene Ontology Annotation Plot (WEGO) (Ye et al. 2006). Assembled contigs were also annotated using TRAPID (Van Bel et al. 2013) to obtain overall functional statistics, protein domains, closest species homologs, and comparative analyses.
Transposable elements identification
All assembled contigs were compared to the RepBase (Jurka et al. 2005) plant nucleotide database (version 21.08; http://www.girinst.org/repbase/) and to reference transposable elements (TEs) from the C. canephora genome (Denoeud et al. 2014; Guyot et al. 2014) using tBLASTx. Contigs were considered related to TEs when there was a minimum alignment of 100 bp, a score > 150, 75% of identity, and a 1e-05 e value in tBLASTx. Intersections between the two databases were highlighted using InteractiVenn (Heberle et al. 2015).
Identification of non-coding mRNA transcripts
The coding potential of assembled contigs was estimated using coding potential calculator (CPC) to discriminate coding and non-coding RNAs (Kong et al. 2007). Contigs were considered as non-coding sequences if they had a CPC score < 0 in both senses and returned “no hit” when submitted to tBLASTx analysis against Uniref90 protein database.
sRNA-seq: data analysis, miRNA prediction, and target prediction
Basic analyses were performed based on previous studies that aimed to identify microRNAs in plants (Guzman et al. 2012; Katiyar et al. 2015; da Silva et al. 2016). All procedures for miRNA prediction used the UEA sRNA workbench V3.2-Plant version (http://srna-tools.cmp.uea.ac.uk/) (Stocks et al. 2012). Low-quality reads (FASTq values < 13), low complexity sequences (sequences with < 3 distinct nucleotides), and 5′ and 3′ adapter sequences were all trimmed using the “sequence file pre-processing tool.” Plant t/rRNAs from Rfam (excepted miRNAs), Arabidopsis tRNAs from “The Genomic tRNA Database,” and plant t/rRNA sequences from “EMBL” release 95 were removed using the “filter pipeline” from UEA sRNA workbench. Sequences shorter than 18 nt and larger than 25 nt were also excluded from further analysis. For the identification of miRNAs, all unique reads obtained after filtering were submitted to the miRCat pipeline from UEA sRNA toolkit-Plant version, and default criteria were used to predict specific and conserved miRNAs. Reads were first mapped against the C. canephora genome (Denoeud et al. 2014). The remaining reads were re-mapped against the de novo assembled transcriptome. Potential precursors were identified following the criteria for plant microRNAs annotation (Meyers et al. 2008) to recover the potential miRNA candidates. To identify conserved miRNAs, all the predicted miRNA sequences were mapped to known mature plant miRNAs deposited in miRBase v21 (Kozomara and Griffiths-Jones 2014) using an in-house Pearl script. A maximum of three mismatches were allowed during the alignment. Sequences with more than three mismatches or no homology to any previously known plant miRNAs were considered as novel miRNAs. We used the assembled root transcriptome as input reference for the identification of putative miRNAs targets in psRNATarget (http://plantgrn.noble.org/psRNATarget/) (Dai and Zhao 2011) with default settings: maximum expectation: 3; length for complementarity scoring: 20; maximum energy to unpair the target site (UPE): 25; flanking length around target site for target accessibility analysis: 17 bp; and range of central mismatch leading to translation inhibition: 9–11 nt.
Clean reads fully matching other RNAs, including repeat RNA, rRNA, snRNA, snoRNA, and tRNA, were excluded from the analysis. For newly discovered miRNAs, we included only those with their miRNA’s co-existing in at least one library. To further validate the existence of miRNA precursors, we aligned miRNAs against the C. canephora genome (Denoeud et al. 2014) and C. arabica transcriptome datasets (Mondego et al. 2011).
RT-qPCR analysis
Total RNA extraction of root cDNA was performed as described in the “RNA isolation” section. Complementary DNA (cDNA) was synthesized using the SuperScript III Reverse Transcriptase kit (Invitrogen) following the manufacturer’s instructions and 5 μg of total RNA for a final volume of 20 μl. For each 1 μg of total RNA extracted, 500 ng of oligo (dT) 12–18 and 1 mM dNTP mix were used. Removal of the original RNA was performed by incubating this reaction at 37 °C for 20 min with two units of RNase H (Invitrogen). The final cDNA products were diluted in 50 ng for use in RT-qPCR.
The transcriptional levels of six differentially expressed genes and six N transporters were evaluated by quantitative PCR (RT-qPCR) on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Primers specific for each gene were designed using Primer Express (Tables S1 and S2). Thermal conditions were 95 °C for 10 min, followed by 40 cycles at 95 °C for 30 s, and 60 °C for 60 s. All reactions were performed in triplicate for each of the three biological replicates, following the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines (Bustin et al. 2009). Dissociation curves were constructed to determine the presence of nonspecific products and primers were used only in the case of a single peak, with amplicons checked in an agarose gel. The mean amplification efficiency of each primer pair was checked using the LinRegPCR program (Ramakers et al. 2003). Data were analyzed using the (1 + E)−ΔΔct method (Livak and Schmittgen 2001; de Carvalho et al. 2014), where E represents the efficiency, with day 0 used as calibrator. RT-qPCR reactions were normalized using the CaEF1α gene (de Carvalho et al. 2013).
cDNA synthesis for miRNAs
Basic procedures for cDNA synthesis followed those used in a recent transcriptional evaluation of miRNAs in coffee plants (Chaves et al. 2015). Stem-loop reverse transcription primers were designed according to Chen et al. (2005) and Varkonyi-Gasic et al. (2007). For cDNA synthesis reaction, we used 1 μg of total RNA, 1 μl of 10 mM dNTP, 2 μl of the specific primer for each miRNA, and 1 μl of oligo (dT) (500 ng/μl). The samples were incubated at 70 °C for 10 min for denaturation of the secondary structures and later incubated at 4 °C for 10 min. Next, we added 5 μl of 5X First-Strand Buffer (Invitrogen), 1.2 μl of 25 mM MgCl, 0.6 μl of RNase out (Invitrogen), and 1 μl of SuperScript III Reverse Transcriptase (Invitrogen). Reactions were run in a thermocycler under the following conditions: 16 °C for 30 min followed by 60 cycles at 30 °C for 30 s, 42 °C for 30 s and 50 °C for 1 s and 70 °C for 5 min. cDNA products were diluted in ~ 750 ng/μl for use in RT-qPCR analyses.
miRNAs transcriptional profile by RT-qPCR
We determined the transcriptional profile of eight miRNAs (miR159, miR166, miR167, miR169, miR171, miR393, miR858, and miRcar1; Table S3) in coffee roots by pulsed stem-loop RT-qPCR (Varkonyi-Gasic et al. 2007; Chaves et al. 2015). All reactions were run on a 7500 Fast Real-Time PCR System (Applied Biosystems). Each PCR reaction (10 μl) included 1 μl of cDNA, 5 μl of SYBR Green Master Mix (Life Technologies), 0.5 μl of sequence-specific forward primer (10 μM), 0.5 μl of universal reverse primer (10 μM), and 3 μl of water. The reaction conditions were 5 min at 94 °C followed by 15 s at 95 °C and 40 cycles of amplification at 60 °C for 10 s and 72 °C for 15 s. For all reactions, dissociation curve analysis was performed to determine the presence of nonspecific products. CaEF1α was used as the reference gene (de Carvalho et al. 2013). Dissociation curves were constructed for biological and technical triplicates. Amplification efficiency was evaluated using LinRegPCR (Ramakers et al. 2003). The relative expression levels of miRNAs were calculated according to the cycle threshold (Ct) method ([1 + E]-ΔΔct) (Livak and Schmittgen 2001; de Carvalho et al. 2014) using day 0 as calibrator.
Statistical analysis
Statistical tests were performed using SISVAR software (Ferreira 2011) and means were compared by the Tukey’s test at 5% significance. The Duncan, Dunnett’s, and Student’s t tests were used for amino acid analysis by UPLC-MS in SAS software (SAS Institute 2004). Data for differentially expressed gene candidates, genes regulated by N starvation, and miRNAs were analyzed by analysis of variance (ANOVA) and the means were compared by the Student’s t test (p < 0.05) using ASSISTAT software (Silva and Azevedo 2013).
Results
Amino acid levels and N influx increase in coffee roots in response to N starvation
To characterize the root-specific response to N starvation, coffee plants grown in nutrient solution under a long-day cycle and high N supply were transferred to N-free nutrient solution for 10 days. After short-term (1 day) and long-term (10 days) N starvation, levels of N assimilation-related metabolites, total amino acids, total protein, and the activity of key enzymes involved in N metabolism, namely nitrate reductase (NR) and and glutamine synthetase (GS), were measured to characterize root responses to N starvation.
Total N content in coffee roots during N starvation showed a slight increase of ~ 8% after 1 day followed by a reduction of ~ 2% at 10 days after N restriction (Fig. S2a). Although NR and GS activity in roots increased slightly, no significant differences were detected after 1 and 10 days of N restriction, indicating that N assimilation in the roots of N-limited coffee plants was stable (Figs. S2b and S2c).
High-affinity transport systems (HATS) regulate N influx to the roots as an adaptive response to N starvation (Forde 2000; Loqué and von Wirén 2004; Glass et al. 2002). To measure the temporal regulation of HATS during N starvation, nitrate and ammonium influxes were measured at a concentration of 0.2 mM 15N-labeled in plants grown in N-free nutrient solution for 1, 3, and 10 days. Short-term influx analysis after 1 and 3 days of N deprivation showed that 15NO3 uptake increased 1.8- and 2-fold in coffee roots, respectively, and was below half that of N-sufficient roots after prolonged N-deficiency (10 days) (Fig. 1). HATS activity for ammonium showed a transient, 5-fold 15NH4 influx increase in the roots after 1 and 3 days of N starvation. In contrast, extended N starvation reduced 15NH4 influx to roughly the levels of N-sufficient roots, with only a 1.4-fold increase in 15NH4 influx relative to the onset of N starvation (Fig. 1). These results indicate that for both inorganic N forms, HATS activity is transiently induced under N deficiency, resulting in a temporal increase in N uptake capacity in coffee roots. In addition, at the beginning of N starvation, 15NH4 influx exceeded that of 15NO3 by 65%, and at 10 days of N deficiency, HATS activity for ammonium represented approximately 70% of the total 15NO3 uptake (Fig. 1), suggesting a relative preference of coffee roots for uptake of NH4+ over NO3 when N availability is limited.

Measurement of labeled nitrogen uptake in coffee roots along nitrogen starvation period. The coffee plants were pre-cultivated hydroponically in 2 mM NH4NO3 for 3 days before influx measurements of labeled 15N. Values are means ± SE of three replicates
Total amino acid levels increased by ~ 5% after 1 day of starvation and remained constant after long-term starvation (Fig. 2b). The initial increase in total amino acids (TAA) concentration in roots may have been caused either by a remobilization of amino acids from the shoots to the roots or by the degradation of proteins in the roots. Indeed, total protein levels in roots increased by ~ 10% only 10 days after N deprivation (Fig. 2a), indicating that amino acids transported from the shoots may support the synthesis of essential proteins.

Amino acid quantification results during nitrogen starvation experiment. a Total protein content and b total free amino acid concentration in coffee roots under nitrogen suppression. c Amino acid levels during nitrogen starvation. Values are means ± SE. Different letters above the bars indicate statistically significant differences (p < 0.05; t test). FW fresh weight
UPLC-MS analysis revealed that amino acid depletion was affected in response to N starvation (Fig. 2c). The most abundant amino acids in coffee roots were asparagine (Asn) and glutamate (Glu), whose concentrations increased by ~ 5 and ~ 10%, respectively, after 10 days of N starvation (Fig. 2c). Rapid and transient increases were observed for glutamine (Gln) and aspartate (Asp) levels after 1 day, but their concentration was reduced to ~ 60% of the initial levels after 10 days (Fig. 2c). Alanine (Ala), proline (Pro), and serine (Ser) showed lower absolute levels compared to other amino acids, but their concentration increased during short-term starvation (1 day) (Fig. 2c). Other minor amino acids are not shown. These changes in amino acid levels likely reflect the metabolic changes of coffee roots to adjust N supply in response to N limitation. The changes in TAA and protein levels described above indicate that N starvation in coffee roots leads to a temporal adaptation that relies on inducing HATS activity and amino acid transport to cope with N-limited conditions.
mRNA-seq of Arabica coffee roots under N starvation
In total, 809,517 raw reads were generated from roots of control plants and plants grown for 1 and 10 days without N supply (Table 1). To perform the de novo assembly, we used 774,529 high-quality reads, generating 34,654 contigs. Our dataset (34,654 contigs, Table 1) is comparable to most coffee transcriptomic resources available: previous studies reported 35,113 contigs for C. arabica (Mondego et al. 2011) and 36,935 contigs for Coffea eugenioides (Yuyama et al. 2016).
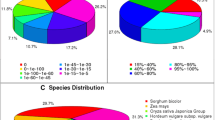
Contigs ranged from 200 to 8514 bp, with a mean contig length of 966 bp, an average open reading frame (ORF) length of 569 bp, a N50 of 1314 bp, and a contig GC content of 43.4% (Tables 1 and 2). Most contigs had BlastX hits in the nr database with an e value cut-off of 1 e−5 (Fig. 3a). Almost 85% of all assembled genes have one or two isoforms (Fig. 3b).

Coffee roots transcripts general information. a Length distribution of assembled contigs. Histogram of assembled transcripts and transcripts showing BlastX hits. b Isoform abundance in the assembled transcriptome. c The best 12 top-hit species distribution observed during C. arabica roots transcriptome annotation process. d Functional annotation of coffee root transcriptome based on WEGO classification. Classes were divided into three main categories: “cellular component,” “molecular function,” and “biological process.” The x-axis indicates different categories described, the y-axis on the left indicates the percentages of genes involved, and the y-axis on the right indicates the number of genes
We used BlastX tool to compare, annotate, and study the conservation of our transcriptome contigs from coffee roots to other plant species genome sequences. A total of 25,026 contigs had correspondents in sequenced plant genomes. The top three species with first hits to Arabica coffee sequences were Vitis vinifera (23.1%, 5774 contigs), Theobroma cacao (16.3%, 4080 contigs), and Manihot esculenta (11.8%, 2947 contigs) (Fig. 3c). Only 1.1% of the total contigs had their first hit in A. thaliana.
We mapped contigs of C. arabica roots against Gene Ontology using BlastX analysis. We were able to classify 20,963 contigs in GO terms and 22,501 transcripts had a protein domain. A summary of the contigs annotated according to the GO terms classification is shown in Fig. 3d.
Validation of differentially expressed genes
To validate the differential expression of genes among libraries, we selected six differentially expressed genes (Table 3). Contigs comp10027_seq3 (TE-related transcript) and comp11089_seq8 (expansin) were selected in the 0 × 1 day combination; comp5715_seq2 (glycosyltransferase) and comp10285_seq3 (peroxidase) were differentially expressed at 0 and 10 days; and comp10246_seq1 (cytochrome P450) and comp8633_seq2 (plant kinase) were chosen based on their differential expression between days 1 and 10. The transcriptional validation of these genes using RT-qPCR revealed that this expression pattern is in agreement with the in silico expression analysis (Fig. 4).

Transcriptional analysis of differentially expressed genes in coffee roots under nitrogen starvation. Transcript levels were normalized using CaEF1α gene and day 0 as calibrator (relative gene expression = 1). Values are means ± standard errors (bars). Statistically significant differences are indicated by different letters (Student’s t test; p < 0.05)
Transcriptional analysis of N transporter genes
To investigate the transcriptional patterns of N transporter genes by RT-qPCR, we selected three ammonium transporters (AMTs) and three nitrate transporters (NRTs) based on a recent study by our research group (Table 4; dos Santos et al. 2017). Most N transporters analyzed showed higher transcript abundance under long-term N starvation when compared to the N-sufficient condition (day 0) (Fig. 5). In particular, expression of CaAMTa was more than eight times higher in roots without N supply for 10 days than in control roots. We also observed similar upregulation for the CaNRTc gene (Fig. 5).

Transcriptional analysis of N transporters. Transcript levels were normalized using CaEF1α gene and day 0 as calibrator (relative gene expression = 1). Values are means ± standard errors (bars). Statistically significant differences are indicated by different letters (Student’s t test; p < 0.05)
Transposable elements and non-coding transcripts
We identified 862 contigs in our analysis that contained TE fragments (Table S4). Most hits (806) were related to C. canephora transposable elements, whereas 35 contigs were related to TEs uniquely based on RepBase reference TEs. In addition, 21 contigs were identified by both approaches (Fig. S3). Most annotated contigs (86%, 744 contigs) were identified as belonging to class I elements (retrotransposons), which is I agreement with most analyses of transcriptionally active TEs in plants (Vicient 2010; Santos et al. 2015). In addition, 6192 transcripts had a CPC score < 0 and no hits in the Uniref database (Online Resource 1) and can be considered putative long ncRNAs.
Identification of miRNAs in N-deficient coffee roots by small RNA library sequencing
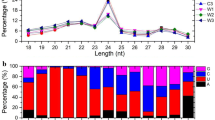
Nine small RNA libraries were prepared from roots maintained in N-sufficient and N-free nutrient solution. Small RNA libraries were sequenced using Illumina Hiseq2000, yielding a total of 86,826,830 redundant reads and 25,498,103 unique reads. After cleaning and trimming, 8,524,332 reads were retained for miRNA analysis. For redundant reads and unique reads, 21 nt was the most frequent length followed by 24 nt, similarly to what was reported for Arabidopsis roots under N starvation (Fig. 6; Vidal et al. 2013).

The total amount and reads length of small RNAs identified in our coffee root transcriptome analysis: a day 0, b day 1, and c day 10. Numbers in the x-axis indicate read size in nucleotides
We identified 86 miRNA families when considering only sequences in the same library with miRNA and miRNA* strands and with precursors in the C. canephora genome or in our transcriptome. Among these families, 30 correspond to previously known miRNA families, whereas 29 families were found in the C. canephora genome (Table S5) and one family was found in the assembled contigs (Table S6). Fifty-six novel miRNA candidates were identified based on the presence of mature miRNA and miRNA* sequences in the same library using the miRCat pipeline; only one new family was identified in transcripts (Table S7) and 55 families were identified in the genome (Table S7). Putative hairpins are shown in Figs. S4, S5, S6, and S7.
Predicting miRNA targets
Subsequently, we identified targets for miRNAs using psRNATarget (Dai and Zhao 2011). For all miRNA families, we identified 253 target genes. These genes were functionally annotated using WEGO classification and according to their respective cellular component, molecular function, and biological process results (Fig. S8).
Transcriptional profile of miRNAs in response to N starvation
We selected eight miRNA families for RT-qPCR transcriptional analyses (Table 5). Seven families were selected because they represent conserved miRNA families in plants with implications in N homeostasis (miR159, miR166, miR167, miR169, miR171, miR393, and miR858), and we validated the transcriptional profile of one new miRNA family (miRcar1) by stem-loop RT-qPCR (Table S8). The relative miRNAs expression profiles were transiently altered in coffee roots subjected to N deficiency (Fig. 7). Five miRNA families (miR169, miR171, miR167, miR393, and miR858) were upregulated after short-term N starvation (1 day). In contrast, miR159 was significantly overexpressed in roots after 10 days of N starvation, whereas miRcar1 was downregulated after prolonged N restriction (Fig. 7). These results reveal distinct expression patterns of N-responsive miRNAs modulating the adaptive response to N fluctuations in coffee roots.

Validation of the miRNAs transcriptional profile modulated by N starvation using RT-qPCR. miRNAs transcriptional levels were normalized with respect to the mean transcript level of the CaEF1α gene. Values are means ± standard errors (bars). Statistically significant differences are indicated by different letters (Student’s t test; p < 0.05)
Discussion
Adaptive responses to N starvation in coffee roots
Nitrate and ammonium are taken up by roots and assimilated according to the plants demand for N. When N becomes limiting, the synthesis and degradation of individual amino acids are essential to provide N to sustain C metabolism to support plant growth and development (Stitt and Krapp 1999; Stitt et al. 2002). In the current study, TAA levels in coffee roots increased in the first day of starvation and remained high until 10 days of N restriction, and a similar response was observed for total protein levels in N-starved root at day 10 (Fig. 2a). The increase in total protein content likely reflects protein synthesis in roots as a result of the amino acids being exported from the shoots to the roots (Krapp 2011). Alternatively, the initial increase in TAA levels in coffee roots may be related to the induction of ammonium and nitrate HATS activity in N-starved roots after 1 day (Fig. 1). Induction of HATS activity for ammonium and nitrate production is a common response in plant roots to N limitation (Crawford and Glass 1998; Yuan et al. 2009); however, an intimate relationship between N uptake rates and N-assimilation was not observed in coffee roots, because NR and GS activity were not correlated with transient increases in N influxes (Fig. S2b and S2c). In addition, analysis of individual amino acids in coffee roots after 1 day of N starvation revealed that Gln remained at levels similar to those of N-sufficient plant roots, whereas Glu levels increased rapidly at 1 day of N restriction and remained high after 10 days (Fig. 2c), which supports the lack of GS activity at 1 day (Fig. S2c). Other nitrogen-containing metabolites such as Asp and Asn exhibited a different accumulation pattern in response to N starvation (Fig. 2c), confirming the alteration of the plants N status and suggesting a role for Asp, Asn, and Glu as N sources in the biosynthesis of other amino acids when N availability is limited in coffee roots (Forde and Lea 2007; Nunes-Nesi et al. 2010). Interestingly, the early accumulation of amino acids and N remobilization occurred before N was depleted in roots (Fig. 2b), clearly revealing a metabolic adaptation in this organ. A similar adaptive mechanism was previously shown to support root growth in Arabidopsis roots under long-term N deficiency (Krapp et al. 2011).
The lack of regulation of NR activity in coffee roots under N-limited conditions (Fig. S2b) indicates that nitrate assimilation occurs in the shoots (Carelli and Fahl 1991; Queiroz et al. 1991, 1993; Carelli et al. 2006). Moreover, accumulation of N metabolites such as Gln and Glu, as observed in N-starved coffee roots (Fig. 2c), has a negative feedback regulatory effect on NR activity in different plant species (Stitt et al. 2002; Miller et al. 2007; Krapp et al. 2011). These results suggest that the nitrate taken up via HATS activity by coffee roots under N deficiency (Fig. 1) may be transported to the shoots to be assimilated or may act as signal to modulate N metabolism in the roots (Wang et al. 2000; Scheible et al. 2004; Krouk et al. 2010).
Altogether, these results indicate that N starvation induces a distinct temporal adaptation pattern in coffee roots through induction of nitrate and ammonium HATS activity and a rapid response for accumulation of amino acids via N remobilization before N is depleted in roots.
Differentially expressed genes
This is the first study to use mRNA-seq to understand the transcriptional patterns of genes under N starvation in coffee. We analyzed six differentially expressed genes that were modulated in Arabica coffee roots in response to N starvation (Table 3). The transcriptional pattern of comp10027, comp8633, and comp5715 revealed by RT-qPCR showed that the three contigs were upregulated in roots after 10 days of N deprivation (Fig. 4). Contig comp5715_seq2, an UDP-glycosyltransferase, has a homolog with higher expression in roots (At1g70570), whereas comp8633 is a zinc-finger kinase that is reported to have diverse functions in the regulation of specific genes (Noguero et al. 2013).
Expression of contig comp10246_seq, a P450 gene, was induced only in the first day of N deprivation. Cytochromes P450 is one of the largest gene super families of enzyme proteins and is found in the genome of all plants (Nelson 2011). P450 genes are often recruited in the biosynthesis of secondary metabolites and thus play important roles in several metabolic processes (Bak et al. 2012). Because At4g31940, the closest homolog of this gene in Arabidopsis, is preferentially expressed in roots (Ensembl Baseline Plant Expression Atlas, https://www.ebi.ac.uk/gxa/home; data not shown), we can speculate that this P450 is possibly involved in the production of secondary metabolites during short-term N starvation. However, the transcriptional profile of comp11089 and comp10285 revealed that both contigs were specifically expressed during the first day of N starvation (1 day). Thus, we believe that Arabica coffee may also have different adaptations to short-term N stress.
AMTs and NRTs transcriptional patterns in coffee roots
We transcriptionally validated three AMT genes that were found in our mRNA-seq analysis of Arabica coffee roots under N starvation (Fig. 5). The AMT genes have been characterized in several plant species including, among others, Citrus spp. (Camañes et al. 2009), Solanum lycopersicum L. (Graff et al. 2011), Puccinellia tenuiflora (Bu et al. 2013), Medicago truncatula (Straub et al. 2014), Pyrus betulaefolia Bunge (Li et al. 2016), and Brassica alboglabra L. (Song et al. 2017).
The relative gene expression levels of all AMT and NRT family members analyzed in our study increased at 10 days, indicating that in short-term N deprivation, uptake is regulated at protein level, although both low- and high-affinity systems were induced by N deprivation. Interestingly, the only gene repressed at 1 day (Fig. 5) was the low-affinity transporter CaNRTa (dos Santos et al. 2017).
Even though this study was not aimed at assessing the role of ammonium and nitrate in root development, our results indicate that these ions transporters may play different roles in N assimilation (Fig. S2). Of the six genes that we transcriptionally analyzed (Fig. 5), AMT1 family members CaAMTa and CaAMTb were identified as candidate high-affinity NH4+ uptake transporters, whereas CaAMTc was classified as a low-affinity transporter based on dos Santos et al. (2017). According to Loqué et al. (2006) and Yuan et al. (2007), AMT1 family members account for approximately 70–80% of the high-affinity ammonium uptake capacity in roots. HATS activity is transiently induced under N deficiency (Fig. 1), resulting in a temporal increase in N uptake capacity in coffee roots, probably due to changes in the phosphorilation status of AMT transporters. Phosphorylated AMT transporters have restricted ammonium uptake capacity (Lanquar et al. 2009); in this sense, the dephosphorylation triggered by kinases is probably the first molecular transition in response to N deprivation. Nevertheless, a more precise characterization of the number and expression pattern of other AMT homologs in other C. arabica tissues is required.
We also analyzed the relative expression of three NRTs in coffee roots during N starvation. Based on the transcriptional pattern observed in our study, the CaNRTs examined are likely to be classified as members of the NRT1/PTR family in coffee (dos Santos et al. 2017). However, further research is needed to determine the role of CaNRTs in coffee plants and measure the expression of this spatiotemporally regulated gene family. Hence, with all the presented results, we suggested a model for the ammonium and nitrate transporters in coffee roots under N starvation summarized in Fig. S9.
N starvation altered the expression pattern of miRNAs in coffee roots
The families of miRNA studied in our work, as already described in dicots and monocots plants, might play an important process role in response to the uptake and assimilation according to the availability of nitrogen in coffee roots. We observed an increase in the expression of miR159 after 10 days (Fig. 7), suggesting that this miRNA may play an important role in coffee roots under N starvation. miR159 has been shown to play an important role in regulating MYB and TCP transcription factors that are active in roots (Zhao et al. 2012; Wang et al. 2013). In potato (Solanum tuberosum), a reduction in miR159 expression resulted in increased expression of GAMyb-like genes possibly in response to drought stress (Yang et al. 2014).
In the analysis of novel and conserved miRNAs in C. arabica and C. canephora, Loss-Morais et al. (2014) identified eight miRNA sequences belonging to the miR166 family. This family of miRNAs is suggested to play vital roles in numerous biotic and abiotic stress responses. For example, miR166 was upregulated by cold treatment in S. lycopersicum (Valiollahi et al. 2014) and was involved in soybean defense against phytophthora root resistance (Wong et al. 2014). Here, we showed that miR166 is downregulated in coffee roots after long-term N starvation (10 days) (Fig. 7).
Similarly, Boualem et al. (2008) also reported that miR166 is significantly downregulated in M. truncatula roots grown under N stress. miR167 was significantly upregulated in coffee roots at the beginning (1 day) of the N starvation period (Fig. 7). In Arabidopsis and maize, this miRNA was reported to be responsive to N deficiency, indicating a conserved role for miR167 in regulating plant adaptation to N (Liang et al. 2012; Xu et al. 2011; Zhao et al. 2012). miR167 has two transcription factors regulated by auxin as targets (ARF6 and ARF8), which are involved in lateral root development (Gutierrez et al. 2009). Chaves et al. (2015) also identified and characterized miR167 in coffee plants and reported a high complementarity to the ARF8 sequence in Arabidopsis and to the putative ARF8 gene in coffee.
Several studies have shown that miR169 is downregulated by N deficiency, triggering the expression of its target genes NFYA2, NFYA3, NFYA5, and NFYA8 (Hsieh et al. 2009; Liang et al. 2012; Pant et al. 2009; Zhao et al. 2011; Sorin et al. 2014). In other plant species such as maize and soybean, miR169 has been implicated in the response to N deficiency (Trevisan et al. 2012; Xu et al. 2013; Zhao et al. 2013). In coffee roots, this miRNA was upregulated after 1 day, returning to baseline expression levels after 10 days of N starvation (Fig. 7).
The role of miR858 in plants is still poorly understood. miR858 expression has been reported in Arabidopsis (Rajagopalan et al. 2006), apple (Xia et al. 2012), cotton (Guan et al. 2014), and, more recently, tomato (Jia et al. 2015). The current study is the first to provide information about the transcriptional changes of miR858 under N starvation (Fig. 7), and the first to present a transcriptome study of miRNAs in coffee roots. It has been shown that this microRNA participates in the regulation of MYB transcription factors involved in the biosynthesis of anthocyanins (Sharma et al. 2016), which can be affected by nitrogen supply (Soubeyrand et al. 2014). Thus, the modulation of miR858 observed in our study indicates that, in coffee plants under N starvation, the regulation of transcription factors in the MYB family may be dependent on miR858 regulation.
Conclusions
This study provides novel data on expressed sequences of coffee roots under N starvation, which may contribute to improve genome-wide research and analysis in this perennial species. In addition, we identified microRNAs and their putative targets, providing relevant information on gene transcriptional regulation of N metabolism in C. arabica. Finally, the information generated here provides a biotechnological basis for the characterization of genes and gene expression regulators that will guide future research aimed at developing coffee plants that are more adept at growing under suboptimal nutritional conditions.
References
Akter A, Islam MM, Mondal SI, Mahmud Z, Jewel NA, Ferdous S, Amin MR, Rahman MM (2014) Computational identification of miRNA and targets from expressed sequence tags of coffee (Coffea arabica). Saudi J Biol Sci 21:3–12
Andrews S (2010) FASTQC. A quality control tool for high throughput sequence data. URL http://www.Bioinformatics.Babraham.Ac.Uk/projects/fastqc
Bak S, Beisson F, Bishop G, Hamberger B, Hofer R, Paquette S, Werck-Reichhart D (2012) Cytochromes p450. Arabidopsis Book 9:e0144
Bertrand B, Bardil A, Baraille H, Dussert S, Doulbeau S, Dubois E, Severac D, Dereeper A, Etienne H (2015) The greater phenotypic homeostasis of the allopolyploid Coffea arabica improved the transcriptional homeostasis over that of both diploid parents. Plant Cell Physiol 56:2035–2051
Boualem A, Laporte P, Jovanovic M, Laffont C, Plet J, Combier JP, Niebel A, Crespi M, Frugier F (2008) MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J 54:876–887
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Bu Y, Sun B, Zhou A, Zhang X, Lee I, Liu S (2013) Identification and characterization of a PutAMT1; 1 Gene from Puccinellia tenuiflora. PLoS One 8(12):e83111
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55:611–622
Cai H, Lu Y, Xie W, Zhu T, Lian X (2012) Transcriptome response to nitrogen starvation in rice. J Biosci 37:731–747
Camañes G, Cerezo M, Primo-Millo E, Gojon A, Garcia-Agustin P (2009) Ammonium transport and CitAMT1 expression are regulated by light and sucrose in Citrus plants. J Exp Bot 58:2811–2825
Cardoso DC, Martinati JC, Giachetto PF, Vidal RO, Carazzolle MF, Padilha L, Guerreiro-Filho O, Maluf MP (2014) Large-scale analysis of differential gene expression in coffee genotypes resistant and susceptible to leaf miner-toward the identification of candidate genes for marker assisted-selection. BMC Genomics 15:66
Carelli MLC, Fahl JI (1991) Distribuição da assimilação do nitrato e da matéria seca em plantas jovens de café cultivadas em diferentes níveis de nitrogênio. Bragantia 50:29–37
Carelli MLC, Fahl JI, Ramalho JDC (2006) Aspects of nitrogen metabolism in coffee plants. Braz J Plant Physiol 18:9–21
Castro-Rodríguez V, García-Gutiérrez A, Cañas RA, Pascual MB, Avila C, Cánovas FM (2015) Redundancy and metabolic function of the glutamine synthetase gene family in poplar. BMC Plant Biol 15:20
Chang S, Puryear J, Cairney J (1993) A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Report 11:113–116
Chapman DJ, Harrison PJ (1988) Nitrogen metabolism and measurement of nitrate reductase activity. In: Lobban CS, Chapman DJ, Kremer B (eds) Experimental phycology. A laboratory manual. Cambridge University Press, Cambridge, pp 196–202
Chaves SS, Fernandes-Brum CN, Silva GFF, Ferrara-Barbosa BC, Paiva LV, Nogueira FTS, Cardoso TCS, Amaral LR, de Souza GM, Chalfun-Junior A (2015) New insights on Coffea miRNAs: features and evolutionary conservation. Appl Biochem Biotech 177:879–908
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ (2005) Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33(20):e179–e179
Clark RB (1975) Characterization of phosphatase of intact maize roots. J Agr Food Chem 23:458–460
Coleman HD, Canovas FM, Man H, Kirby EG, Mansfield SD (2012) Enhanced expression of glutamine synthetase (GS1a) confers altered fibre and wood chemistry in field grown hybrid poplar (Populus tremula x alba) (717-1B4). Plant Biotech J 10:883–889
Conesa A, Gotz S (2008) Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008:1–13
Crawford N, Glass A (1998) Molecular and physiological aspects of nitrate uptake in plants. Trends Plant Sci 3:389–395
Cruz JL, Mosquim PR, Pelacani CR, Araújo WL, DaMatta FM (2004) Effects of nitrate nutrition on nitrogen metabolism in cassava. Biol Plantarum 48:67–72
da Silva AC, Grativol C, Thiebaut F, Hemerly AS, Ferreira PCG (2016) Computational identification and comparative analysis of miRNA precursors in three palm species. Planta 243:1265–1277
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159
de Carvalho K, Bespalhok Filho JC, dos Santos TB, de Souza SGH, Vieira LGE, Pereira LFP, Domingues DS (2013) Nitrogen starvation, salt and heat stress in coffee (Coffea arabica L.): identification and validation of new genes for qPCR normalization. Mol Biotechnol 53:315–325
de Carvalho K, Petkowicz CL, Nagashima GT, Bespalhok Filho JC, Vieira LG, Pereira LFP, Domingues DS (2014) Homeologous genes involved in mannitol synthesis reveal unequal contributions in response to abiotic stress in Coffea arabica. Mol Gen Genomics 289:951–963
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F, Aprea G, Aury JM, Bento P, Bernard M, Bocs S, Campa C, Cenci A, Combes MC, Crouzillat D, Da Silva C, Daddiego L, De Bellis F, Dussert S, Garsmeur O, Gayraud T, Guignon V, Jahn K, Jamilloux V, Joët T, Labadie K, Lan T, Leclercq J, Lepelley J, Leroy T, Li LT, Librado P, Lopez L, Muñoz A, Noel B, Pallavicini A, Perrotta G, Poncet V, Pot D, Priyono RM, Rouard M, Rozas J, Tranchant-Dubreui C, VanBuren R, Zhang Q, Andrade AC, Argout X, Bertrand B, Kochko A, Graziosi G, Henry RJ, Jayarama MR, Nagai C, Rounsley S, Sankof D, Giuliano G, Albert VA, Wincker P, Lashermes P (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181–1184
Devi KJ, Chakrabortya S, Debb B, Rajwanshia R (2016) Computational identification and functional annotation of microRNAs and their targets from expressed sequence tags (ESTs) and genome survey sequences (GSSs) of coffee (Coffea arabica L.). Plant Gene 6:30–42
dos Santos TB, Lima JE, Felicio MS, Soares JDM, Domingues DS (2017) Genome- wide identification, classification and transcriptional analysis of nitrate and ammonium transporters in Coffea. Genet Mol Biol 40:346–359
Dovis VL, Hippler FWR, Silva KI, Ribeiro RV, Machado EC, Mattos D Jr (2014) Optimization of the nitrate reductase activity assay for citrus trees. Braz J Bot 37:383–390
Fernandez D, Tisserant E, Talhinhas P, Azinheira H, Vieira A, Petitot AS, Loureiro A, Poulain J, da Silva C, Do Céu Silva M, Duplessis S (2012) 454-pyrosequencing of Coffea arabica leaves infected by the rust fungus Hemileia vastatrix reveals in planta-expressed pathogen-secreted proteins and plant functions in a late compatible plant–rust interaction. Mol Plant Pathol 13:17–37
Ferreira DF (2011) Sisvar: Sistema de análise de variância para dados balanceados. Versão 5.3, Build 75. Lavras: DEX/ UFLA
Forde BG (2000) Nitrate transporters in plants: structure, function and regulation. BBA-Biomembranes 1465:219–235
Forde BG, Lea PJ (2007) Glutamate in plants: metabolism, regulation, and signaling. J Exp Bot 58:2339–2358
Glass ADM, Britto DT, Kaiser BN, Kinghorn JR, Kronzucker HJ, Kumar A, Okamoto M, Rawat S, Siddiqi MY, Unkles SE, Vidmar JJ (2002) The regulation of nitrate and ammonium transport systems in plants. J Exp Bot 53:855–864
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma BBW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol 29:644–652
Graff L, Obrdlik P, Yuan L, Loqué D, Frommer WB, von Wirén N (2011) N-terminal cysteines affect oligomer stability of the allosterically regulated ammonium transporter LeAMT1;1. J Exp Bot 62:1361–1373
Guan X, Pang M, Nah G, Shi X, Ye W, Stelly DM, Chen ZJ (2014) miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat Commun 5:3050
Gutiérrez RA, Lejay LV, Dean A, Chiaromonte F, Shasha DE, Coruzzi GM (2007) Qualitative network models and genome-wide expression data define carbon/nitrogen-responsive molecular machines in Arabidopsis. Genome Biol 8:R7
Gutierrez L, Bussell JD, Pacurar DI, Schwambach J, Pacurar M, Bellini C (2009) Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 21:3119–3132
Guyot R, Chaparro C, Bocs S, Dereeper A, Jamilloux V, Domingues DS, Lashermes P (2014) The repetitive landscape of the Robusta coffee genome. In: 11th Solanaceae Conference: 52–52
Guzman F, Almerão MP, Körbes AP, Loss-Morais G, Margis R (2012) Identification of microRNAs from Eugenia uniflora by high-throughput sequencing and bioinformatics analysis. PLoS One 7(11):e49811
Heberle H, Vaz Meirelles G, da Silva FR, Telles GP, Minghim R (2015) InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics 16:169
Hsieh LC, Lin SI, Shih AC, Chen JW, Lin WY, Tseng CY, Li WH, Chiou TJ (2009) Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Phys 151:2120–2132
Ivamoto ST, Sakuray LM, Ferreira LP, Kitzberger CSG, Scholz MBS, Pot D, Leroy T, Vieira LGE, Domingues DS, Pereira LFP (2017a) Diterpenes biochemical profile and transcriptional analysis of cytochrome P450s genes in leaves, roots, flowers, and during Coffea arabica L. fruit development. Plant Physiol Bioch 111:340–347
Ivamoto ST, Reis Júnior O, Domingues DS, dos Santos TB, de Oliveira FF, Pot D, Leroy T, Vieira LGE, Carazzolle MF, Pereira GAG, LFP P (2017b) Transcriptome analysis of leaves, flowers and fruits perisperm of Coffea arabica L. reveals the differential expression of genes Involved in raffinose biosynthesis. PloS ONE 12(1):e0169595
Jia X, Jie S, Hui L, Fang L, Na D, Changgyong G, Sitakanta P, Barunava P, Runzhi L, Ling Y (2015) Small tandem target mimic-mediated blockage of microRNA858 induces anthocyanin accumulation in tomato. Planta 242:283–293
Joët T, Laffargue A, Salmona J, Doulbeau S, Descroix F, Bertrand B, Kochko A, Dussert S (2009) Metabolic pathways in tropical dicot albuminous seeds: Coffea arabica as a case study. New Phytol 182:146–162
Joët T, Laffargue A, Salmona J, Doulbeau S, Descroix F, Bertrand B, Lashermes F, Dussert S (2014) Regulation of galactomannan biosynthesis in coffee seeds. J Exp Bot 65:323–337
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005) Repbase update, a database of eukaryotic repetitive elements. Cytoge Gen Res 110:462–467
Katiyar A, Smita S, Muthusamy SK, Chinnusamy V, Pandey DM, Bansal KC (2015) Identification of novel drought-responsive microRNAs and trans-acting siRNAs from Sorghum bicolor (L.) Moench by high-throughput sequencing analysis. Frontiers in. Plant Sci 6:506
Kojima S, Bohner A, Gassert B, Yuan L, von Wirén N (2007) AtDUR3 represents the major transporter for high-affinity urea transport across the plasma membrane of nitrogen-deficient Arabidopsis roots. Plant J 52:30–40
Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G (2007) CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35:W345–W349
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73
Krapp A, Berthomé R, Orsel M, Mercey-Boutet S, Yu A, Castaings L, Elftieh S, Major H, Renou JP, Daniel-Vedele F (2011) Arabidopsis roots and shoots show distinct temporal adaptation patterns toward nitrogen starvation. Plant Phys 157:1255–1258
Krouk G, Crawford NM, Coruzzi GM, Tsay YF (2010) Nitrate signalling: adaptation to fluctuating environments. Curr Opin Plant Biol 13:266–273
Kulcheski FR, Côrrea R, Gomes IA, de Lima JC, Margis R (2015) NPK macronutrients and microRNA homeostasis. Frontiers in Plant Sci 6:451
Lanquar V, Loqué D, Hörmann F, Yuan L, Bohner A, Engelsberger WR, Lalonde S, Schulze WX, von Wirén N, Frommer WB (2009) Feedback inhibition of ammonium uptake by a phospho-dependent allosteric mechanism in Arabidopsis. Plant Cell 21:3610–3622
Léran S, Varala K, Boyer JC, Chiurazzi M, Crawford N, Daniel-Vedele F, David L, Dickstein R, Fernandez E, Forde B, Gassmann W, Geiger D, Gojon A, Gong JM, Halkier BA, Harris JM, Hedrich R, Limami AM, Rentsch D, Seo M, Tsay YF, Zhang M, Coruzzi G, Lacombe B (2014) A unified nomenclature of nitrate transporter 1/peptide transporter family members in plants. Trends Plant Sci 19:5–9
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323
Li H, Han JL, Chang YH, Lin J, Yang QS (2016) Gene characterization and transcription analysis of two new ammonium transporters in pear rootstock (Pyrus betulaefolia). J Plant Res 129:737–748
Liang G, He H, Yu D (2012) Identification of nitrogen starvation-responsive microRNAs in Arabidopsis thaliana. PLoS One 7(11):e48951
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2− CT method. Methods 25:402–408
Loqué D, von Wirén N (2004) Regulatory levels for the transport of ammonium in plant roots. J Exp Bot 55:1293–1305
Loqué D, Yuan L, Kojima S, Gojon A, Wirth J, Gazzarrini S, Ishiyama K, Takahashi H, von Wiren N (2006) Additive contribution of AMT1;1 and AMT1;3 to high-affinity ammonium uptake across the plasma membrane of nitrogen-deficient Arabidopsis roots. The Plant J 48:522–534
Loss-Morais G, Ferreira DCR, Margis R, Alves-Ferreira M, Corrêa RL (2014) Identification of novel and conserved microRNAs in Coffea canephora and Coffea arabica. Genet Mol Biol 37:671–682
Luo J, Qin J, He F, Li H, Liu T, Polle A, Peng C, Luo ZB (2013) Net fluxes of ammonium and nitrate in association with H+ fluxes in fine roots of Populus popularis. Planta 237:919–931
Marraccini P, Vinecky F, Alves GS, Ramos HJ, Elbelt S, Vieira NG, Carneiro FA, Sujii PS, Alekcevetch JC, Silva VA, DaMatta FM, Ferrão MAG, Leroy T, Pot D, Vieira LGE, da Silva FR, Andrade AC (2012) Differentially expressed genes and proteins upon drought acclimation in tolerant and sensitive genotypes of Coffea canephora. J Exp Bot 63:4191–4212
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qio Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK (2008) Criteria for annotation of plant MicroRNAs. Plant Cell 20:3186–3190
Miller AJ, Fan X, Shen Q, Smith SJ (2007) Amino acids and nitrate as signals for the regulation of nitrogen acquisition. J Exp Bot 59:111–119
Miyazawa M et al (1999) Análise química de tecido vegetal. In: Silva, f. c. da (Org.). Manual de análises químicas de solos, plantas e fertilizantes. Embrapa, Brasília, pp 171–223
Mofatto LS, de Araújo CF, Vieira NG, Duarte KE, Vidal RO, Alekcevetch JC, Cotta MG, Verdeil JL, Lapeyre-Montes F, Lartaud M, Leroy T, De Bellis F, Pot D, Rodrigues GC, Carazzolle MF, Pereira GAG, Andrade AC, Marraccini P (2016) Identification of candidate genes for drought tolerance in coffee by high-throughput sequencing in the shoot apex of different Coffea arabica cultivars. BMC Plant Biol 16:94
Mondego JMC, Vidal RO, Carazzolle MF, Tokuda EK, Parizzi LP, Costa GGL (2011) An EST-based analysis identifies new genes and reveals distinctive gene expression features of Coffea arabica and Coffea canephora. BMC Plant Biol 11:30
Nath M, Tuteja N (2016) NPKS uptake, sensing, and signaling and miRNAs in plant nutrient stress. Protoplasma 253:767–786
Nelson DR (2011) Progress in tracing the evolutionary paths of cytochrome P450. BBA-Proteins and Proteomics 1814:14–18
Noguero M, Atif RM, Ochatt S, Thompson RD (2013) The role of the DNA-binding one zinc finger (DOF) transcription factor family in plants. Plant Sci 209:32–45
Nunes-Nesi A, Fernie AR, Stitt M (2010) Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol Plant 3:973–996
Pant BD, Musialak-Lange M, Nuc P, May P, Buhtz A, Kehr J, Walther D, Scheible WR (2009) Identification of nutrient-responsive Arabidopsis and rapeseed microRNAs by comprehensive real-time polymerase chain reaction profiling and small RNA sequencing. Plant Phys 150:1541–1555
Pompelli MF, Martins SCV, Antunes WC, Chaves ARM, DaMatta FM (2010) Photosynthesis and photoprotection in coffee leaves is affected by nitrogen and light availabilities in winter conditions. J Plant Physiol 13:1052–1060
Praxedes SC, DaMatta FM, Loureiro ME, Ferrão MAG, Cordeiro AT (2006) Effects of long-term soil drought on photosynthesis and carbohydrate metabolism in mature robusta coffee (Coffea canephora Pierre var. kouillou) leaves. Environ Exp Bot 56:263–273
Queiroz CGS, Alves JD, Rena AB, Cordeiro AT (1991) Efeito do cloranfenicol, n- propanol, pH e temperatura sobre a atividade in vivo da redutase do nitrato em cafeeiros jovens. Rev Bras Bot 14:73–77
Queiroz CGS, Rena AB, Cordeiro AT, Alves JD (1993) Ritmo diurno na atividade da redutase de nitrato em folhas e raízes de Coffea arabica L. Pesqui Agropecu Bras 28:787–795
R Development Core Team (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Available in: <http://www.r-project.org>
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425
Ramakers C, Ruijter JM, Deprez RHL, Moorman AF (2003) Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339:62–66
Ranjan A, Ichihashi Y, Farhi M, Zumstein K, Townsley B, David-Schwartz R, Sinha NR (2014) De novo assembly and characterization of the transcriptome of the parasitic weed dodder identifies genes associated with plant parasitism. Plant Phys 166:1186–1199
Rennenberg H, Wildhagen H, Ehlting B (2010) Nitrogen nutrition of poplar trees. Plant Biol 12:275–291
Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11:R25
Rossa MM (1999) A luz como fator de regulação da atividade enzimática da nitrato redutase (NR) e superóxido dismutase (SOD) em Gracilariopsis tenuifrons (Rhodophyta). Dissertação de mestrado, Universidade de São Paulo, São Paulo
Ruffel S, Freixes S, Balzergue S, Tillard P, Jeudy C, Martin-Magniette ML, van der Merwe MJ, Kakar K, Gouzy J, Fernie AR, Udvardi M, Salon C, Gojon A, Lepetit M (2008) Systemic signaling of the plant nitrogen status triggers specific transcriptome responses depending on the nitrogen source in Medicago truncatula. Plant Phys 146:2020–2035
Santos FC, Guyot R, Do Valle CB, Chiari L, Techio VH, Heslop-Harrison P, Vanzela ALL (2015) Chromosomal distribution and evolution of abundant retrotransposons in plants: gypsy elements in diploid and polyploid Brachiaria forage grasses. Chromosom Res 23:571–582
SAS Institute (2004) SAS/GRAPH 9.1 reference. SAS Institute
Scheible WR, Morcuende R, Czechowski T, Fritz C, Osuna D, Palacios-Rojas N, Schindelasch D, Thimm O, Udvardi MK, Stitt M (2004) Genome-wide reprogramming of primary and secondary metabolism, protein synthesis, cellular growth processes, and the regulatory infrastructure of Arabidopsis in response to nitrogen. Plant Phys 136:2483–2499
Sharma D, Tiwari M, Pandey A, Bhatia C, Sharma A, Trivedi PK (2016) MicroRNA858 is a potential regulator of phenylpropanoid pathway and plant development Plant Physiol 171:944–959
Silva FAZ, Azevedo CAV (2013) Versão atualizada do software estatístico Assistat. Versão 7.6 beta <http://www.assistat.com>
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212
Song S, He Z, Huang X, Zhong L, Liu H, Sun G, Chen R (2017) Cloning and characterization of the ammonium transporter genes BaAMT1; 1 and BaAMT1; 3 from Chinese kale. Hortic Environ Biotechnol 58:178–186
Sorgona A, Lupini A, Mercati F, Di Dio L, Sunseri F, Abenavoli MR (2011) Nitrate uptake along the maize primary root: an integrated physiological and molecular approach. Plant Cell Environ 34:1127–1140
Sorin C, Declerck M, Christ A, Blein T, Ma L, Lelandais-Brière C, Njo MF, Beeckman T, Crespi M, Hartmann C (2014) A miR169 isoform regulates specific NF-YA targets and root architecture in Arabidopsis. New Phytol 202:1197–1211
Soubeyrand E, Basteau C, Hilbert G, van Leeuwen C, Delrot S, Gomès E (2014) Nitrogen supply affects anthocyanin biosynthetic and regulatory genes in grapevine cv. Cabernet-Sauvignon berries. Phytochemistry 103:38–49
Stitt M, Krapp A (1999) The interaction between elevated carbon dioxide and nitrogen nutrition: the physiological and molecular background. Plant Cell Environ 22:583–621
Stitt M, Muller C, Matt P, Gibon Y, Carillo P, Morcuende R, Scheible WR, Krapp A (2002) Steps towards an integrated view of nitrogen metabolism. J Exp Bot 53:959–970
Stocks MB, Moxon S, Mapleson D, Woolfenden HC, Mohorianu I, Folkes L, Schwach F, Dalmay T, Moulton V (2012) The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 28:2059–2061
Straub D, Ludewig U, Neuhäuser B (2014) A nitrogen-dependent switch in the high affinity ammonium transport in Medicago truncatula. Plant Mol Biol 86:485–494
Tong Z, Qu S, Zhang J, Wang F, Tao J, Gao Z, Zhang Z (2012) A modified protocol for RNA extraction from different peach tissues suitable for gene isolation and real-time PCR analysis. Mol Biotechnol 50:229–236
Trevisan S, Nonis A, Begheldo M, Manoli A, Palme K, Caporale G, Ruperti B, Quaggiotti S (2012) Expression and tissue-specific localization of nitrate-responsive miRNAs in roots of maize seedlings. Plant Cell Environ 35:1137–1155
Valiollahi E, Farsi M, Kakhki AM (2014) Sly-miR166 and Sly-miR319 are components of the cold stress response in Solanum lycopersicum. Plant Biotech Rep 8:349–356
Van Bel M, Proost S, Van Neste C, Deforce D, Van de Peer Y, Vandepoele K (2013) TRAPID: an eficient online tool for the functional and comparative analysis of de novo RNA-Seq transcriptomes. Genome Biol 14(1):R134
van der Vossen H, Bertrand B, Charrier A (2015) Next generation variety development for sustainable production of arabica coffee (Coffea arabica L.): a review. Euphytica 204:243–256
Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP (2007) Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3:12
Vicient CM (2010) Transcriptional activity of transposable elements in maize. BMC Genomics 11:601
Vidal EA, Araus V, Lu C, Parry G, Green PJ, Coruzzi GM, Gutiérrez RA (2010) Nitrate-responsive miR393/AFB3 regulatory module controls root system architecture in Arabidopsis thaliana. P Natl Acad Sci 107:4477–4482
Vidal EA, Moyano TC, Krouk G, Katari MS, Tanurdzic M, McCombie WR, Coruzzi GM, Gutiérrez RA (2013) Integrated RNA-seq and sRNA-seq analysis identifies novel nitrate-responsive genes in Arabidopsis thaliana roots. BMC Genomics 14:701
Vieira LGE, Andrade AC, Colombo CA, Moraes AHDA, Metha Â, Oliveira ACD, Labate CA, Marino CL, Monteiro-Vitorello CB, Monte DC, Giglioti E, Kimura ET, Romano E, Kuramae EE, Lemos EGM, Almeira ERP, Jorge EC, Albuquerque EVS, Silva FR, Vinecky F, Sawazaki HE, Dorry HFA, Carrer H, Abreu IN, Batista JAN, Teixeira JB, Kitajima JP, Xavier KG, Lima LM, Camargo LEA, Pereira LFP, Coutinho LL, Lemos MVF, Romano MR, Machado MA, Costa MMC, Sá MFG, Goldman MHS, Ferro MIT, Tinoco MLP, Oliveira MC, Sluys MAV, Shimizu MM, Maluf MP, Eira MTS, Filho OG, Arruda P, Mazzafera P, Mariani PDSC, Oliveira RLBC, Harakava R, Balbao SF, Tsai SM, Mauro SMZ, Santos SN, Siqueira WJ, Costa GGL, Formighieri EF, Carazzolle MF, Pereira GAG (2006) Brazilian coffee genome project: an EST-based genomic resource. Braz J Plant Physiol 18:95–108
von Wittgenstein NJ, Le CH, Hawkins BJ, Ehlting J (2014) Evolutionary classification of ammonium, nitrate, and peptide transporters in land plants. BMC Evol Biol 14:11
Wallsgrove RM, Lea PJ, Miflin BJ (1979) Distribution of the enzymes of nitrogen assimilation within the pea leaf cell. Plant Phys 63:232–236
Wang R, Guegler K, LaBrie ST, Crawford NM (2000) Genomic analysis of a nutrient response in Arabidopsis reveals diverse expression patterns and novel metabolic and potential regulatory genes induced by nitrate. Plant Cell 12:1491–1509
Wang Y, Zhang C, Hao Q, Sha A, Zhou R, Zhou X, Yuan L (2013) Elucidation of miRNAs mediated responses to low nitrogen stress by deep sequencing of two soybean genotypes. PLoS One 8(7):e67423
Wei H, Yordanov YS, Georgieva T, Li X, Busov V (2013) Nitrogen deprivation promotes Populus root growth through global transcriptome reprogramming and activation of hierarchical genetic networks. New Phytol 200:483–497
Wong J, Gao L, Yang Y, Zhai J, Arikit S, Yu Y, Duan S, Chan V, Xiong Q, Yan J, Li S, Liu R, Wang Y, Tang G, Meyers BC, Chen X, Ma W (2014) Roles of small RNAs in soybean defense against Phytophthora sojae infection. Plant J 79:928–940
Xia R, Zhu H, An YQ, Beers EP, Liu Z (2012) Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol 13:R47
Xu Z, Zhong S, Li X, Li W, Rothstein SJ, Zhang S, Bi Y, Xie C (2011) Genome-wide identification of microRNAs in response to low nitrate availability in maize leaves and roots. PLoS One 6(11):e28009
Xu G, Fan X, Miller AJ (2012) Plant nitrogen assimilation and use efficiency. Annu Rev Plant Biol 63:153–182
Xu MY, Zhang L, Li WW, Hu XL, Wang MB, Fan YL, Zhang CY, Wang L (2013) Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J Exp Bot 65:89–101
Xu ZC, Peters RJ, Weirather J, Luo HM, Liao BS, Zhang X, Zhu Y, Ji A, Zhang B, Hu S, Au KF, Song J, Chen S (2015) Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J 82:951–961
Yang J, Zhang N, Mi X, Wu L, Ma R, Zhu X, Yao L, Jin X, Si H, Wang D (2014) Identification of miR159s and their target genes and expression analysis under drought stress in potato. Comput Biol Chem 53:204–213
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:W293–W297
Yuan L, Loqué D, Kojima S, Rauch S, Ishiyama K, Inoue E, Takahashi H, von Wiren N (2007) The organization of high-affinity ammonium uptake in Arabidopsis roots depends on the spatial arrangement and biochemical properties of AMT1-type transporters. Plant Cell 19:2636–2652
Yuan L, Graff L, Loque D, Kojima S, Tsuchiya YN, Takahashi H, von Wiren N (2009) AtAMT1;4, a affinity ammonium transporter of the plasma membrane in Arabidopsis. Plant Cell Physiol 50:13–25
Yuyama PM, Reis Junior O, Ivamoto ST, Domingues DS, Carazzole MF, Pereira GAG, Charmetant P, Leroy T, Pereira LFP (2016) Transcriptome analysis in Coffea eugenioides, an Arabica coffee ancestor, reveals differentially expressed genes in leaves and fruits. Mol Gen Genomics 291:323–336
Zhao M, Ding H, Zhu JK, Zhang F, Li WX (2011) Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol 190:906–915
Zhao M, Tai H, Sun S, Zhang F, Xu Y, Li WX (2012) Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS One 7(1):e29669
Zhao Y, Xu Z, Mo Q, Zou C, Li W, Xu Y, Xie C (2013) Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann Bot 112:633–642
Acknowledgments
This study received grants from Brazilian Coffee Research Consortium, Fundação Araucária, CAPES (Ciência sem Fronteiras PVE 084/2013) and CNPq. TBS, JDMS, JCS, VYB, and STI acknowledge CAPES, CNPq, and FAPESP for the student fellowships. DSD, LGEV, and LFPP are CNPq research productivity fellows.
Author information
Authors and Affiliations
Contributions
TBS, LGEV, and LFPP conceived and designed the research, performed transcriptional experiments, wrote and revised the final manuscript; ARM assisted in designing the experiment; JEL performed experiments using 15N and wrote the manuscript; STI RT-qPCR statistical analysis, revised the final manuscript; JDMS and VYB RT-qPCR and statistical analyses; SGHS performed GS and NR analyses; JBM and PM performed the amino acid analysis; MYNJ, JCS, ORJ, APRL, and ARP bioinformatic analysis; ILMJA and UCO library construction and mRNAseq sequencing; RG and DSD transposable element analysis, coordinated the research, and revised the final manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(XLS 280 kb)
ESM 2
(FA 2908 kb)
Fig. S1
N starvation experiment scheme. a Initially, seeds were germinated in sand boxes and transfered to pots with continuous N supply. Three-month-old plants were transferred to a nutrient solution based on Clark’s (1975). After acclimation, plants were harvested (experimental control, day 0) and then transferred to N-starvation solution for 10 days. b Lateral roots were collected for analysis at different sampling times (day 0, day 1, and day 10). (JPG 63 kb)
Fig. S2
Quantification of total N and related-enzymes in coffee root. a Total concentration of N in coffee roots. b Nitrate reductase (NR) enzyme activity in coffee roots. c Glutamine synthetase (GS) enzyme activity in coffee roots under N starvation. Different letters above the bars indicate statistically significant differences (Student’s t test; p < 0.05). FW fresh weight (JPG 50 kb)
Fig. S3
Number of assembled sequences with hits in the Coffea canephora reference transposable elements (TE) database (806), RepBase Plants transposable elements database (35), and both databases (21). (JPG 63 kb)
Fig. S4
miRNA hairpin sequences of conserved families and precursors found in the Coffea canephora genome. Expected miRNA and miRNA* sequences are represented in red and blue, respectively. (DOC 541 kb)
Fig. S5
miRNA hairpin sequences of conserved families and precursors found in Coffea arabica root transcriptome. Expected miRNA and miRNA* sequences are represented in red and blue, respectively. (DOC 40 kb)
Fig. S6
New miRNA families and precursors identified in Coffea canephora genome. Expected miRNA and miRNA* sequences are represented in red and blue, respectively. (DOC 903 kb)
Fig. S7
Eight new miRNA families and precursors identified in Coffea arabica root transcriptome. Expected miRNA and miRNA* sequences are represented in red and blue, respectively. (DOC 40 kb)
Fig. S8
Annotation of the 253 putative miRNAs target genes identified in our coffee root trancriptome database. The most represented classes were divided into three main categories: “cellular component,” “molecular function,” and “biological process.” (JPG 102 kb)
Fig. S9
Proposed model for the representation of ammonium and nitrate transporters in coffee roots under N starvation. (JPG 81 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
dos Santos, T.B., Soares, J.D.M., Lima, J.E. et al. An integrated analysis of mRNA and sRNA transcriptional profiles in Coffea arabica L. roots: insights on nitrogen starvation responses. Funct Integr Genomics 19, 151–169 (2019). https://doi.org/10.1007/s10142-018-0634-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-018-0634-8