
Abstract
The aim of this study was to assess factors that can affect the in vivo and in vitro assays of the nitrate reductase activity (NRA) in sweet orange trees and then standardize conditions for tissue sampling and analysis. Seven-month-old plants grown in pots were used, of which, mature and healthy leaves between the third and the fifth position from the branch apex, stems, and roots were assessed. One half of each leaf was used for in vivo tests, and the other half was used for in vitro tests. In addition to varied incubation time and temperature, in vivo KNO3 and n-propanol concentrations and in vitro KNO3 and NADH concentrations were evaluated. The optimum conditions for the in vivo NRA assay in leaves were: 200 mmol L−1 KNO3 and 1 % n-propanol at 40 °C for 20 min. The highest leaf NRA occurred at 11:00 h for the in vivo assay and at 13:00 h for the in vitro assay, with both analysis showing similar results. Overestimation of the in vitro NRA occurred as compared to the in vivo analysis when accessed early morning and late afternoon. Branches bearing fruits show reduced NRA in young mature leaves, whereas sprouting significantly increases NRA in correspondent leaves. For the root assays, the optimized conditions for the NRA estimation were the same as for leaves. Although roots and stems (bark) have shown some NRA, it was six times lower than leaf NRA. Our data indicate that NO3 − reduction occurs mostly in leaves and there is a significant effect of daytime and leaf position in relation to fruit or sprouts on NRA of citrus trees.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nitrogen assimilation is vital for plant growth, a process related to the reduction of nitrate (NO3 −) absorbed by roots in all higher plants (Srivastava 1980; Kaiser et al. 1999) and mediated by the cytosolic enzyme nitrate reductase (NR, EC 1.6.6.1). NR activity (NRA) in situ is highly regulated by transcription factors and reversible phosphorylation, which determine its level of activation (Solomonson and Barber 1990; Lillo et al. 2004; Lea et al. 2006), which also depends on environmental factors such as light, CO2, oxygen, ATP, pH, and respiration inhibitors (Kaiser et al. 1999; Lea at al. 2006; Sánchez et al. 2009). The availability of NO3 − and the intracellular ammonium (NH4 +) concentration regulate the synthesis and activation of NR (Solomonson and Barber 1990; Chen et al. 2004; Correia et al. 2005; Miller et al. 2007; Nicodemus et al. 2008; Dias et al. 2011). Moreover, photosynthetic activity is also able to activate 60–80 % of the enzyme (Kaiser and Brendle-Behnisch 1991).
The NR is found in all plant organs (Bar-Akiva and Sagiv 1967; Carelli et al. 1990), with NO3 − reduction preferentially occurring in leaves. Nevertheless, higher NRA can be found in roots, which depends on specie, tissue age and type, environmental conditions, NO3 − availability, and plant ontogenetic state (Bar-Akiva and Sternbaum 1965; Bar-Akiva and Sagiv 1967; Chen et al. 2004; Correia et al. 2005; Oliveira et al. 2005). Therefore, it is necessary to clearly define the plant ontogenetic state and the sampled tissue to avoid bias when making comparisons.
While the in vitro method to estimate NRA involves cell rupture, enzyme extraction, and stabilization, and a subsequent enzyme activity test, without NO3 − and NADH limitations. The in vivo method, more fast and easy, requires previous absorption of NO3 − by tissues, a certain internal concentration of NADH, and the subsequent liberation of NO2 − produced in the medium. An important aspect is that the in vivo assay performed with NO3 − represents the in situ activity of the enzyme with the current endogenous level of cellular NADH (Chen et al. 2004). Therefore, nitrogen accumulation can be overestimated by the in vitro assay in a nonlinear manner (Lillo 1983; Kaiser et al. 2000; Tucker et al. 2004). Nonphosphorylated NR is evaluated and represents the active enzyme (NRactive) when in vitro tests are performed in the presence of Mg2+. If EDTA is added in medium assay, cations are chelated and NR becomes fully active (NRmaximum), being possible the evaluation of NR activation state (Kaiser et al. 2000). As there are many changes caused by extraction procedure in vitro assay, some phosphorylated NR would be activated non-deliberately (Chen et al. 2004).
Regardless of the chosen method, the NRA is affected by extraction and assay conditions and type of plant tissue (Lillo 1983, 1984a, b, Queiroz et al. 1991; Nievola and Mercier 2001). As NRA has a diurnal rhythm (Lillo and Henriksen 1984; Nievola and Mercier 2001; Oliveira et al. 2005), sampling time is also critically important for activity estimation.
Because the limiting step in NO3 − assimilation involves NR, its activity can be used to evaluate nitrogen uptake and metabolism, especially when NO3 − is the main source of nitrogen (Bar-Akiva and Sternbaum 1965; Nicodemus et al. 2008; Reis et al. 2009). Due to several differences observed for in vivo and in vitro assay in other species, the NR assay optimization is necessary. The aim of this study was to assess some factors that can affect the in vivo and in vitro assays of nitrate reductase activity in sweet orange trees and then standardize conditions for sampling and analysis.
Materials and methods
Seven-month-old sweet orange [Citrus sinensis (L.) Osbeck cv. Valencia] plants grafted on Rangpur lime (Citrus limonia Osbeck) were used for the optimization assays at varying incubation time and temperature and reagent concentrations. Plants were grown in 7-L plastic bags filled with organic substrate composed of 80 % pine bark, 5 % carbonized materials, and 15 % vermiculite (Multicitrus, Terra do Paraíso 9, Ltd., Holambra, SP, Brazil). The plants were grown under greenhouse conditions, irrigated daily, and fertilized every two days with 0.75 mL of a nutrient solution (Brennfeed®, Brenntag, Brazil) with the following concentration: 94.8 g N L−1 (85 % N–NO3); 24 g P L−1 (P2O5); 80 g K L−1 (K2O); 81.5 g Ca L−1 (CaO); 9.2 g Mg L−1 (MgO); 0.28 g Mn L−1; 0.2 g Zn L−1; 1.5 g Cu L−1; 0.72 g Fe L−1; and 0.12 g B L−1.
The protocols used by Bar-Akiva and Sagiv (1967), Queiroz et al. (1991), and Nievola & Mercier (2001) in different species were used as guidance in setting the protocol to in vivo NRA assay. Assays were designed to identify the optimum conditions for measuring in vivo NRA by testing the enzyme activity in samples of fully expanded, mature leaves located between the third and fifth position from the branch apex. Root samples consisted of fine roots less than 2 mm in diameter. Samples were collected between 09:00 and 11:00 h, washed with tap water, rinsed with deionized water and dried. The analyses were performed on leaves and roots sampled simultaneously during the summer season, with air temperature varying between 20 and 32 °C. At the same time, leaves were sampled and stored at −80 °C for in vitro assay.
The plant material (200 mg of fresh leaves and 1,000 mg of fresh roots) was cut with scissors into small fragments, which were placed in 10-mL syringes. The incubation medium was added to the syringes: 5 mL of sodium phosphate buffer solution (100 mmol L−1) at pH of 7.5 with KNO3 (0, 25, 50, 100, 150, 200, and 250 mmol L−1), and n-propanol (0, 1, 2, 3, 4, and 5 %). The samples inside the syringes were vacuum infiltrated six times, and incubated in the absence of light for 30 min at 30 °C. After evaluating the optimum time of incubation (0–90 min) and temperature (25–45 °C), n-propanol and KNO3 concentrations were also varied.
For in vitro assay of NRA, the method reported by Kato and Kubota (1982) was used, with some modifications. The plant material was ground in an extraction buffer composed of 100 mmol L−1 HEPES–NaOH (pH 7.5), 5 mmol L−1 MgCl2, 5 mmol L−1 of dithiothreitol, 10 mmol L−1 of ascorbic acid, 5 mmol L−1 of EDTA, 10 % glycerol (v:v), and 2 % of polyvinylpolypyrrolidone (v:v). This solution was centrifuged and desalted in PD10 columns (Sephadex G-25). The assay optimization was done as described by Kato and Kubota (1982), varying KNO3 (0, 10, 20, 30, 40 and 50 mmol L−1) and NADH (0, 25, 50, 100 and 200 µmol L−1) concentrations in the reaction medium, temperature (25–45 °C), and incubation time (0–90 min).
To quantify NO2 − produced, aliquots of 200–500 µL of extract (completing to 500 µL with buffer solution) were added to 500 µL of 1 % sulfanilamide solution in 2.4 N HCl + 0.02 % of N-[1-naphthyl] ethylenediamine dihydrochloride (NED; m:v). After 2 min, the samples were centrifuged at 3,000×g for 2 min and the absorbance evaluated in a spectrophotometer at 540 nm. Two aliquots per sample were read in spectrophotometer. The enzymatic activity was calculated in µmol NO2 − g−1 FM h−1 from a straight line of NaNO2 levels between 0.8 and 10 µmol L−1. The leaf assays were replicated four times while root assays three times (one plant represents one replication). Each test was conducted twice in consecutive days, at the same time of day (between 09:00 and 11:00 h).
Once the protocol was optimized, the diurnal course of leaf NRA was evaluated. Healthy leaves were sampled during the diurnal period, every 2 h from 07:00 to 17:00 h in mature leaves located between the third and fifth position from the branch apex, in four replications (plants). The samples were washed, dried, and divided by half. One portion was used in in vivo assay and the other portion was stored for in vitro analysis. Later, in another group of plants, net photosynthesis (A net) and NRA were evaluated every 2 h from 7:30 to 17:30 h in mature leaves located between the third and fifth position from the branch apex, in four replications. Photosynthesis was measured under natural conditions of light, air temperature, relative humidity, and CO2 concentration using a portable infrared gas analyzer (LI-6400 Li-Cor Inc., Lincoln, NE, USA), following the same procedure described by Ribeiro et al. (2009).
In a second experiment, a group of fifteen-year-old trees and another of seven-month-old plants of sweet orange cv. Valencia grafted on Rangpur lime, were used to evaluate the effects of leaf aging and branch phenological status on NRA. Leaves on old and new branches (different ages) and on branches with or without reproductive and vegetative structures (different phenological status) were evaluated for in vivo NRA. Trees were annually fertilized with N [300 g N (60 % of N-NO3)], P (100 g of P2O5), and K (70 g of K2O) and sprayed four times a year with a solution containing Zn (700 mg L−1), Mn (650 mg L−1), B (170 mg L−1), and N (2.25 g L−1; as urea). Healthy leaves were sampled between 10:00 and 11:00 h, the samples were immediately carried to laboratory, washed, dried, and use to in vivo NRA assay.
The experiments were arranged in a completely randomized design with three or four replications per treatment. Effects of each variable on NRA were tested with one-way ANOVA, followed by Tukey test when significance was detected. Normality and homogeneity of variances was tested with graphical methods (Q–Q plot and Res vs. Pred). When assumptions have not been fulfilled (NO3 concentration and photosynthesis diurnal cycle) the data were transformed using square root (\( \sqrt y \)). Data related to incubation time were analyzed through linear regression.
Results
Optimizing the in vivo and in vitro assays
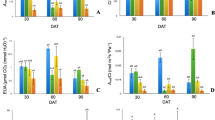
Leaf NRA exhibited a linear trend from 0 to 90 min, and no latent phase or downward slope was observed at the end of this period; a slope of 16.2 ± 0.6 was observed in the linear regression (P < 0.01) (Fig. 1). Root NO2 − production was linear from 0 to 30 min (P < 0.01) with a slope of 3.7 ± 0.2. After 30 min, a reduction in root NRA was observed and the slope decreased to 1.6 ± 0.2 (Fig. 1). NRA in leaves was more than six times higher than in roots (NRAleaf = 6.1*NRAroot; R 2 = 0.97; P < 0.01), indicating that citrus leaves are the primary organs responsible for the reduction of absorbed NO3 −.

Incubation time for in vivo nitrate reductase activity (NRA) in leaves and roots of seven-month-old plants of ‘Valencia’ sweet orange grafted onto ‘Rangpur’ lime. Values shown are the mean ± SD of four replications for leaves and three replications for roots
The highest NRA in leaves was observed at 40 and 45 °C (P < 0.01), being 2.7-times higher than the activity at 35 °C (Fig. 2a). In roots, no significant differences were observed in NRA when temperature increased from 25 to 40 °C (Table 1). Increases in NRA activity of 12.4 % were observed in leaves when 1 % n-propanol was added (P < 0.05), with higher concentrations causing reduction in NRA (Fig. 2b). In roots no significant difference was observed (Table 1).

In vivo nitrate reductase activity (NRA) in leaves of seven-month-old plants of ‘Valencia’ sweet orange grafted onto ‘Rangpur’ lime, considering different: a temperature of incubation, b n-propanol in the incubation medium, and c KNO3 concentration in the incubation medium. Values shown are the means of four replications ± SD. Letters indicate significant differences (Tukey P < 0.1)
In leaves, considerable NRA was detected when no substrate was added in the reaction medium, indicating that a pool of readily available NO3 − existed inside cell vacuoles (Fig. 2c). However, NRA increases in 2.5-times (P < 0.01) with 200 mmol L−1 of KNO3 in the incubation medium. In roots, adding 100 mmol L−1 of KNO3 in the incubation medium increased NRA (Table 1).
The optimization of in vitro assay conditions indicated that NRA increased (P < 0.05) when NO3 − and NADH were both added to the incubation medium (Fig. 3a, b). No difference was found between NO3 concentrations above 10 µmol L−1 of KNO3. The addition of 200 µmol L−1 of NADH was adequate for the maximum NRA. Under these conditions, the optimum temperature was 35 °C (P < 0.13) and NO2 − production was linear from 0 to 75 min (Fig. 3c, d).

In vitro nitrate reductase activity (NRA) in leaves of ‘Valencia’ sweet orange plants grafted onto ‘Rangpur’ lime, considering different: a KNO3 concentration in the incubation medium; b NADH concentration in the incubation medium; c temperature of incubation; and d incubation time. Values shown are the means of four replications ± SD. Letters indicate significant differences (Tukey P < 0.1)
Diurnal course and differences among plant organs
The diurnal course of in vivo NRA differed from the course of in vitro NRA (Fig. 4). Most of the in vivo NRA occurred during the morning, varying from 0.25 to 0.86 µmol NO2 g−1 FM h−1 between 7:00 and 11:00 h. In vitro, the highest NRA was recorded at 13:00 h, reaching 1.0 µmol NO2 g−1 FM h−1. The capacity for NO3 − reduction was very similar for both in vitro and in vivo assays only between 09:00 and 11:00 h (Fig. 4). In the second experiment, we observed that the diurnal course of the in vivo NRA followed the diurnal course of CO2 assimilation, with maximum NRA and photosynthesis found around 10:00 h (Fig. 5). A difference in maxima between both NRA diurnal courses was attributed to phenological stage and environmental condition which plants were grown. The highest temperature in the morning and the presence of developing shoots in plants could be responsible for both the highest NRA observed early in the morning and the abrupt fall at midday. Diurnal course of root NRA was similar to leaf NRA, with maximum values around 9:00 h (Fig. 5).

Diurnal course of nitrate reductase activity (NRA) in leaves of seven-month-old plants of ‘Valencia’ sweet orange grafted onto ‘Rangpur’ lime. a In vitro and b in vivo assays. Values shown are the means of four replications ± SD. Letters indicate significant differences (Tukey P < 0.1)

a Diurnal course of in vivo nitrate reductase activity (NRA) in leaves b roots, and c diurnal course of CO2 assimilation (A net) in leaves of seven-month-old plants of ‘Valencia’ sweet orange grafted onto ‘Rangpur’ lime. Values shown are the means of four replications ± SD. Letters indicate significant differences (Tukey P < 0.1)
In young plants, NRA was detected in leaves, stem, and roots; however, leaves are the most important organs for reducing NO3 − (Table 2). In bearing trees, there were no significant differences in NRA due to leaf aging, with >6-months and 2-weeks old leaves showing similar activity. However, the presence of fruiting and sprouting caused a large difference in NRA, with leaves close to sprouting buds showing higher NRA (Table 2).
Discussion
In vivo and in vitro NRA assay
The in vivo method is relatively simple and fast, allowing NO3 − reduction capacity to be estimated based on energy availability and the activation state of the enzyme in situ (Solomonson and Barber 1990; Aslam et al. 2001). The linearity of leaf NRA from 0 to 90 min suggests that NO3 − was rapidly absorbed and subsequently diffused into the reaction medium (Srivastava 1980), most likely due to the vacuum infiltration or/and the use of n-propanol (Lillo 1983). In roots, a linear relationship was maintained until 30 min and a reduction in slope was observed later, which was probably caused by the limiting availability of NADH (Queiroz et al. 1991). In general, 20 min is considered more appropriate to avoid metabolic disturbances in leaf and root tissues (Nievola and Mercier 2001). The highest NRA in citrus leaves was observed at 40 °C (Fig. 2a), which is higher than the optimum temperatures reported in other species (Lillo 1983; Lillo and Henriksen 1984; Queiroz et al. 1991; Nievola and Mercier 2001). Chopra (1983) observed that the optimum temperature for NRA was higher in C4 species than in C3 species, and it was correlated with thermal conditions during the growing season. Therefore, a possible explanation to the high optimum temperature found herein is the environmental condition. The Citrus plants were grown under greenhouse conditions, where air temperature reached values higher than 35 °C during the warmest days.
N-propanol alters the permeability of membranes and improves NO3 − and NO2 − diffusion into and out of the tissue (Lillo 1983). Nevertheless, the use of propanol at high concentrations may cause diffusion of chlorophyll into the incubation medium and affect colorimetric estimation of NRA (Streeter and Bosler 1972). The addition of 1 % n-propanol to the incubation medium for leaf NRA was lower as compared to the recommendation for other species (Queiroz et al. 1991; Nievola and Mercier 2001).
Nitrate concentration in the incubation medium is the most variable component in protocols. Concentration between 25 and 100 mmol L−1 of NO3 − were required to maximize NRA in species from different growing habitats (Carelli et al. 1990; Lee and Titus 1992; Machado et al. 1992; Nievola and Mercier 2001; Oliveira et al. 2005). Those differences may be caused by varying metabolic pools of NO3 − (Aslam et el. 2001), leaf segment sizes, capacities of diffusion into the buffer (Lillo 1983) and K m of NRA among species (Chen et al. 2004). As found herein, reduction in NRA has been observed at NO3 − concentrations above the optimum and the underlying mechanism of this response remains unknown (Carelli et al. 1990; Nievola and Mercier 2001; Oliveira et al. 2005).
In vitro NRA assay was performed without EDTA in assay medium and represents the active nitrate reductase enzyme (Kaiser et al. 1999, 2000). The concentration of 10 mmol L−1 of KNO3 observed in our experiment (Fig. 3a) is the same of that used for several species (Lillo 1983; Kaiser et al. 2000; Tucker et al. 2004). The optimum temperature of 35 °C for the in vitro assay was lower than the optimum temperature of 40 °C for the in vivo assay (Fig. 3b). When enzymatic activity is determined by in vivo assay, NRA is also affected by the input rate of substrate (NO3 −) in the cytoplasm of intact cells. This is an active process that consumes energy and depends on the activity of membrane transporters, being affected by temperature and NO3 − concentration inside and outside of cell (Crawford and Glass 1998). NADH concentration needed to saturate in vitro NRA (200 µmol L−1) was similar to one used in other species (Lillo 1983; Kaiser et al. 2000; Tucker et al. 2004). Fifteen minutes of reaction time to assay NRA was considered appropriate in citrus tissues (Fig. 3d).
Plant material and sampling time standardization
During the diurnal cycle, in vivo NRA and leaf CO2 assimilation presented similar variation, which was different to the diurnal cycle of in vitro NRA (Figs. 4, 5). In vivo and in vitro NRA in leaves was similar only by 11:00 h (Fig. 4). This can be explained because of diurnal variations of the in vitro NRA reflects changes in soluble proteins (Kaiset et al. 1999; Tucker et al. 2004), in gene expression (Lillo et al. 2004; Wickert et al. 2007) and in activation state (Kaiser et al. 2000; Barreto et al. 2007). Moreover, the diurnal cycle of in vivo NRA is regulated by energy availability, transpiration rate, and other organic compounds (Lillo 1984a; Aslam et al. 2001; Kaiser et al. 2001; Yang and Midmore 2005) that affect nitrate uptake and transport in plants (Aslam et al. 2001). In this situation, cytosolic NADH appears as the major limiting factor for in situ nitrate reduction, and rates are only similar to in vitro assay under optimal conditions for CO2 assimilation (Kaiser et al. 2000). With that, in vivo NRA assay reflects more accurately plant metabolism and the rate of nitrogen uptake in situ. Finally, NRA in vivo could be reflecting more accurately the response of applied treatments regarding to physiological condition of evaluated plant material.
Although NRA has been detected in leaves, roots, and stem, the capacity of reducing nitrate in roots and stem was between 17 and 10 % of that observed in leaves, respectively (Table 2). This was highlighted by Bar-Akiva and Sagiv (1967) in seedlings and bearing trees of some Citrus species. Accordingly, various genes codifying NR synthesis are well conserved in Citrus species and were preferentially expressed in leaves (Wickert et al. 2007). Plants differ in tissue localization of nitrate reduction and assimilation. Generally, substantial proportion of nitrate assimilation occurs in shoots of tropical and subtropical species (Andrews 1986), as found in peach palm (Oliveira et al. 2005) and sunflower (Correia et al. 2005). Nevertheless, the relative contribution of roots and shoots to NO3 − reduction is changed with environmental conditions, nutritional status, and plant age (Andrews et al. 1986; DaMatta et al. 1999; Dias et al. 2011). While leaf aging did not affect NRA in citrus trees, the phenological condition of citrus branches (e.g., sprouting buds vs. fruiting) has a significant effect on NRA (Table 2). Therefore, it is important standardize not only sampled time but also the type of plant material in order to reduce the variability between replicates and compare treatments and experiments.
Conclusion
The optimum conditions for the in vivo NRA are: 100 mmol L−1 of phosphate buffer with 200 mmol L−1 of KNO3 and 1 % of n-propanol, and incubation for 20 min at 40 °C for leaves and 35 °C for roots. For the in vitro NRA assay, the concentration of KNO3 is 10 mmol L−1 and 200 µmol L−1 of NADH, and incubation for 15 min at 35 °C. In citrus leaves, in vivo NRA reflects CO2 assimilation and physiological condition of plant more accurately than in vitro NRA.
The citrus leaves are responsible for most of NO3 − reduction, as NRA activity is higher in leaves than in roots and stem. Leaf and root sampling procedure should consider daytime, branch phenological state, and injuries for reducing undesirable variability among replications and comparing treatments. In vivo and in vitro assays provided comparable results only when leaves were collected around 11:00 h, occurring overestimation of NRA by the in vitro as compared to the in vivo method.
References
Andrews M (1986) The partitioning of nitrate assimilation between root and shoot of higher plants. Plant Cell Environ 9:511–519
Aslam M, Travis RL, Rains DW (2001) Diurnal fluctuations of nitrate uptake and in vivo nitrate reductase activity in Pima and Acala cotton. Crop Sci 41:372–378
Bar-Akiva A, Sagiv J (1967) Nitrate reductase in the citrus plant: properties, assay conditions and distribution within the plant. Physiol Plant 20:500–506
Bar-Akiva A, Sternbaum J (1965) Possible use of nitrate reductase activity of leaves as a measure on the nitrogen requirement of citrus trees. Plant Cell Physiol 6:575–577
Barreto DCS, Gonçalves JFC, Santos Júnior UM, Fernandes AV, Bariani A, Sampaio PTB (2007) Biomass accumulation, photochemical efficiency of photosystem II, nutrient contents and nitrate reductase activity in young rosewood plants (Aniba rosaeodora Ducke) submitted to different NO3 −:NH4 + ratios. Acta Amazônica 37:533–542
Carelli MLC, Fahl JI, Magalhães AC (1990) Atividade da reductase de nitrato em folhas e raízes de plantas de café (Coffea arabica L.). Rev Bras Bot 13:119–123
Chen B, Wang Z, Li S, Wang G, Song H, Wang X (2004) Effects of nitrate supply on plant growth, nitrate accumulation, metabolic nitrate concentration and nitrate reductase activity in three leafy vegetables. Plant Sci 167:635–643
Chopra RK (1983) Effects of temperature on the in vivo assay of nitrate reductase in some C3 and C4 species. Ann Bot 51:617–620
Correia MJ, Fonseca F, Azevedo-Silva J, Dias C, David MM, Barrote I, Osório ML, Osório J (2005) Effects of water deficit on the activity of nitrate reductase and content of sugars, nitrate and free amino acids in the leaves and roots of sunflower and white lupin plants growing under two nutrient supply regimes. Physiol Plant 124:61–70
Crawford NM, Glass ADM (1998) Molecular and physiological aspects of nitrate uptake in plants. Trends Plant Sci 3:389–395
DaMatta FM, Amaral JAT, Rena AB (1999) Growth periodicity in trees of Coffea arabica L. in relation to nitrogen supply and nitrate reductase activity. Field Crops Res 60:223–229
Dias DN, Martins-Loução MA, Sheppard L, Cruz C (2011) Patterns of nitrate reductase activity vary according to the plant functional group in a Mediterranean maquis. Plant Soil 347:363–376
Kaiser WM, Brendle-Behnisch E (1991) Rapid modulation of spinach leaf nitrate reductase activity by photosynthesis. Plant Physiol 96:363–367
Kaiser WM, Huber SC (2001) Post-translational regulation of nitrate reductase: mechanism, physiological relevance and environmental triggers. J Exp Bot 52:1981–1989
Kaiser WM, Weiner H, Huber SC (1999) Nitrate reductase in higher plants: a case study for transduction of environmental stimuli into control of catalytic activity. Physiol Plant 105:385–390
Kaiser WM, Kandlbinder A, Stoimenova M, Glaab J (2000) Discrepancy between nitrate reductase activity in leaf extracts: what limits nitrate reduction in situ? Planta 210:801–807
Kato T, Kubota S (1982) Reduction and assimilation of 15N-nitrate by citrus trees in cold season in comparison with summer. J Jpn Soc Hortic Sci 50:413–420
Lea US, Leydecker MT, Quilleré I, Meyer C, Lillo C (2006) Posttranslational regulation of nitrate reductase strongly affects the levels of free amino acids and nitrate, whereas transcriptional regulation has only minor influence. Plant Physiol 140:1085–1094
Lee HJ, Titus JS (1992) Factors affecting the in vivo nitrate reductase assay from MM106 apple trees. Commun Soil Sci Plant Anal 23:981–991
Lillo C (1983) Studies of diurnal variations of nitrate reductase activity in barley leaves using various assay methods. Physiol Plant 57:357–362
Lillo C (1984a) Circadian rhythmicity of nitrate reductase activity in barley leaves. Physiol Plant 61:219–223
Lillo C (1984b) Diurnal variations of nitrite reductase, glutamine synthetase, glutamate synthase, lanine aminitransferase and aspartate aminotransferase in barley leaves. Physiol Plant 61:214–218
Lillo C, Henriksen A (1984) Comparative studies of diurnal variations of nitrate reductase activity in wheat, oat and barley. Physiol Plant 62:89–94
Lillo C, Meyer C, Lea US, Provan F, Oltedal S (2004) Mechanism and importance of post-translational regulation of nitrate reductase. J Exp Bot 55:1275–1282
Machado AT, Magalhães JR, Magnavaca R, Silva MR (1992) Determinação da atividade de enzimas envolvidas no metabolismo do N em diferentes genótipos de milho. Rev Bras Fisiol Veg 4:45–47
Miller AJ, Fan X, Shen Q, Smith SJ (2007) Amino acids and nitrate as signals for the regulation of nitrogen acquisition. J Exp Bot 59:111–119
Nicodemus MA, Francis Salifu K, Jacobs DF (2008) Nitrate reductase activity and nitrogen compounds in xylem exudate of Juglans nigra seedlings: relation to nitrogen source and supply. Trees 22:685–695
Nievola CC, Mercier H (2001) Variações diurnas da atividade in vivo da redutase do nitrato em abacaxizeiro (Ananas comosus (L.) Merr.-Bromeliaceae). Rev Bras Bot 24:295–301
Oliveira MAJ, Machado EC, Bovi MLA, Rodrigues JD (2005) Atividade da redutase de nitrato em mudas de pupunheira (Bactris gasipaes). Ciênc Rural 35:515–522
Queiroz CGS, Alves JD, Rena AB, Cordeiro AT (1991) Efeito do cloranfenicol, propanol, pH e temperatura sobre a atividade in vivo da redutase do nitrato em cafeeiros jovens. Rev Bras Bot 14:73–77
Reis AR, Favarin JL, Gallo LA, Malavolta E, Moraes MF, Lavres JJ (2009) Nitrate reductase and glutamine synthetase activity in coffee leaves during fruit development. Rev Bras Cienc Solo 33:315–324
Ribeiro RV, Machado EC, Santos MG, Oliveira RF (2009) Photosynthesis and water relations of well-watered orange plants as affected by winter and summer conditions. Photosynthetica 47:215–222
Sánchez E, Ávila-Quezada G, Gardea AA, Muñoz E, Ruiz JM, Romero L (2009) Nitrogen metabolism in roots and leaves of green bean plants exposed to different phosphorus doses. Phyton 78:11–16
Solomonson LP, Barber MJ (1990) Assimilatory nitrate reductase: functional properties and regulation. Ann Rev Plant Physiol Mol Biol 41:225–253
Srivastava HS (1980) Regulation of nitrate reductase activity in higher plants. Phytochemistry 19:725–733
Streeter JG, Bosler ME (1972) Comparison of in vitro and in vivo assays for nitrate reductase in soybean leaves. Plant Physiol 49:448–450
Tucker DE, Allen DJ, Ort DR (2004) Control of nitrate reductase by circadian and diurnal rhythms in tomato. Planta 219:277–285
Wickert E, Marcondes J, Lemos MV, Lemos EGM (2007) Nitrogen assimilation in Citrus based on CitEST data mining. Genet Mol Biol 30:810–818
Yang Z, Midmore DJ (2005) A model for the circadian oscillations in expression and activity of nitrate reductase in higher plants. Ann Bot 96:1019–1026
Acknowledgments
ECM, RVR, and DMJ acknowledge the fellowships and VLD the scholarship granted by the National Council for Scientific and Technological Development (CNPq, Brazil). FWRH and KIS received scholarships from the São Paulo Research Foundation (FAPESP, Brazil; Grant #2010/02981-0) and Coordination for the Improvement of Higher Level Personnel (CAPES, Brazil), respectively. This research was funded by FAPESP (Grant #2005/57862-8).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dovis, V.L., Hippler, F.W.R., Silva, K.I. et al. Optimization of the nitrate reductase activity assay for citrus trees. Braz. J. Bot 37, 383–390 (2014). https://doi.org/10.1007/s40415-014-0083-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40415-014-0083-0