
Abstract
Crassostrea ariakensis naturally distributes in the intertidal and estuary region with relative low salinity ranging from 10 to 25‰. To understand the adaptive capacity of oysters to salinity stress, we conducted transcriptome analysis to investigate the metabolic pathways of salinity stress effectors in oysters from two different geographical sites, namely at salinities of 16, 23, and 30‰. We completed transcriptome sequencing of 18 samples and a total of 52,392 unigenes were obtained after assembly. Differentially expressed gene (DEG) analysis and weighted gene correlation network analysis (WGCNA) were performed using RNA-Seq transcriptomic data from eye-spot larvae at different salinities and from different populations. The results showed that at moderately high salinities (23 and 30‰), genes related to osmotic agents, oxidation-reduction processes, and related regulatory networks of complex transcriptional regulation and signal transduction pathways dominated to counteract the salinity stress. Moreover, there were adaptive differences in salinity response mechanisms, especially at high salinity, in oyster larvae from different populations. These results provide a framework for understanding the interactions of multiple pathways at the system level and for elucidating the complex cellular processes involved in responding to osmotic stress and maintaining growth. Furthermore, the results facilitate further research into the biological processes underlying physiological adaptations to hypertonic stress in marine invertebrates and provide a molecular basis for our subsequent search for high salinity–tolerant populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The living environment of marine organisms is changing rapidly worldwide due to global warming and human activity (Philippart et al. 2011; Anderson et al. 2015). Salinity differences in sea areas limit the survival and distribution of marine organisms (Gunter 1961). The blood osmolarities of most marine invertebrates are close to the osmolarities of seawater. They can live in the ocean as osmoconformers, but lack the ability to regulate the osmotic pressure of the internal medium (Berger and Kharazova 1997). Salinity stress is a part of the environmental characteristics of intertidal zone (Macinnes and Calabrese 1979). In such areas, the environment varies from being nearly freshwater when there is rain to being highly saline or exhibiting dry salt conditions when there is drying between tidal inundations (Barnes 1999). Thus, oysters often have to experience large and sometimes rapid fluctuations in salinity, and they thrive within a wide range of salinities, from below 10‰ to over 35‰ (Pauley et al. 1988). Unlike other intertidal animals, oysters are attached to rocks and the lack of mobility forces them to evolve many regulatory mechanisms to adapt to highly dynamic and stressful environments (Barnes 1999). The tolerance of the oyster to extreme stress conditions makes it an excellent study module for examining stress responses and adaptation (Heilmayer et al. 2008; Abdelrahman et al. 2017).
Crassostrea ariakensis is one of the most common and most well-known oyster species in China (Ang et al. 1993). It has been bred for centuries in China and is widely distributed from the Lizijiang (the Oyster River) on the northern border with Korea to Beihai near the Vietnamese border (Zhou and Allen 2003). This disconnected distribution makes this species an ideal model to study adaptive divergence in marine invertebrates.
Unlike other oysters, C. ariakensis is mainly distributed in rivers and estuaries with salinities ranging from 10 to 25‰ (Wang et al. 2004). However, conditions of high water temperature and high salinity accompanying long droughts cause shellfish immunity to decline rapidly, leaving them prone to outbreaks of various diseases and large-scale deaths (Lacoste et al. 2002). In recent years, because of the scarcity of rainwater, the runoff of rivers into the sea has dropped sharply, and large-scale oyster deaths have occurred frequently. Therefore, the mechanisms of high salinity tolerance and the breeding of high salinity–tolerant varieties of C. ariakensis have attracted increasing attention. Previous transcriptomic, proteomic, and alternative splicing studies on C. gigas under salinity stress have shown that osmotic tolerance is a complex physiological characteristic that supports cell survival (Zhao et al. 2012; Zhou and Allen 2003; Yan et al. 2017; Huang et al. 2016). Salinity stress effectors and free amino acid (FAA) metabolism pathways have been suggested to be critical to adaptation to fluctuating salinities (Hosoi et al. 2007; Meng et al. 2013). A systematic study of cellular responses to salinity could provide new insights into the mechanisms of osmotic tolerance. Meng et al. (2013) performed transcriptomic analysis of the Pacific oyster at seven different salinities using RNA-Seq technology, and Zhao et al. (2016) performed weighted gene correlation network analysis (WGCNA) and further analyzed the related gene expression modules. Compared with those on C. gigas, only a few physiological studies have been investigated on C. ariakensis with regard to salinity responses, and there is a lack of relevant information at the molecular level on the adaptation of this species to high salinity.
Li et al. (2018) revealed that two geographical populations of C. gigas exhibited adaptive divergence of overall transcriptomic expression in response to acute heat stress, suggesting the potential for adaptive divergence in addition to plasticity in C. ariakensis with disconnected distributions. We selected oysters from different geographical sites of C. ariakensis, including Binzhou, Shandong Province China, and the western coast of North Korea. F1 oysters of the two populations cultured in the same environment showed significantly differential growth and survival, confirming the adaptive divergence of the geographic populations of the C. ariakensis (unpublished data). Compared with adults, oyster larvae are more sensitive to high salinity (Ko et al. 2014). And salt tolerance of larvae may be the main factor affecting the natural distribution of oysters. Thus, the eye-spot larvae were selected to study the salinity adaptive mechanism. A salinity of 16‰ is typical in an estuarine area, a salinity of 30‰ is normal for seawater in most regions of China, and a salinity of 23‰ is considered transitional. For C. ariakensis, a previous report has suggested that oyster larval growth and survival is significantly higher in 16‰ salinity than in 30‰ salinity (Xue et al. 2007).
In this study, we conducted detailed RNA-Seq analysis of the gene expression of eye-spot larvae from two populations under three different salinity treatments. Using pairwise comparisons and WGCNA, we identified differentially expressed genes (DEGs), modules of coexpressed genes, and candidate hub genes for each salinity level and for each population. In particular, we used these transcriptome results to discuss the adaptive divergence in salinity of the two geographical populations. This systematic work provides insights into the mechanism of adaptation of C. ariakensis to moderately high salinity at the molecular level.
Materials and Methods
Oyster Samples
Parental oysters collected from Binzhou (BZ), Shandong Province, China, and Yalu River Estuary of North Korea (CX) were maintained in a nursery in Rushan City, Shandong Province. Ripening took place indoors at a salinity of 23‰. Thirty males and 30 females were hybridized at two sampling sites. Thirty female eggs were mixed together and then divided into 30 beakers on average. The eggs in each beaker were fertilized with the sperms from each of the 30 mature male oysters. All samples were placed in large ponds and cultured at different salinities (16, 23, or 30‰; Sal16, Sal23, and Sal30, respectively). The population developed at the similar rate and eye-stage larvae were randomly collected at the same time and stored in liquid nitrogen. Sea water and aerated fresh water were used to produce the experimental water with required salinity gradient. The salinities were measured by a Knauer semimicro osmometer.
RNA Extraction
We completed transcriptome sequencing of 18 samples, which included three biological replicates for each of the three different salinities for each of the two geographical sites. Total RNA was extracted from approximately 20 mg of eye-spot larvae with an RNAprep Pure Tissue Kit (Tiangen, Beijing, China) according to the manufacturer’s instructions. RNA quality was assessed via 1.2% gel electrophoresis. RNA concentrations were measured at 260 nm using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, MA, USA).
cDNA Library Construction and Sequencing
The total RNA for each sample was sent to BioMarker (Beijing, China) for library construction and sequencing. Then, an Agilent 2100 Bioanalyzer and ABI StepOne Plus Real-Time PCR System were used for quantification and qualification of the sample library, after which the library was sequenced using an Illumina HiSeq X Ten platform. Raw data generated from Illumina sequencing were preprocessed to remove nonsense sequences containing adapter, poly-N, and low-quality reads. A minimum length of 50 bp after trimming was applied. After high-quality sequencing data were obtained, the transcriptome was assembled with Trinity (Grabherr et al. 2011).
Differential Expression, Cluster Analysis, and Enrichment Analysis
Differential expression analysis was performed using the DESeq2 (Anders and Huber 2010) R package. DEGs were filtered based on a false discovery rate (FDR) < 0.01 and a fold change (FC) ≥ 2 in each pairwise comparison. Then the collected DEGs were aligned with the database using BLAST algorithm with an E value of ≤ 1e−5. TopGO (Franceschini et al. 2013) software was used to perform enrichment analysis of the DEGs between samples with annotations from the Gene Ontology (GO) database. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database is the main public database of pathway information (Kanehisa et al. 2010) and was used to perform pathway analysis. Principal component analysis (PCA) was used based on the average fragments per kb of transcript per million mapped reads (FPKM) values for all the expressed genes in each sample. To help remove scale issues in PCA, all data were logarithmically transformed.
Gene Correlation Network Construction
The gene network was constructed using the R package WGCNA following the procedure described by Langfelder and Horvath (2008). After the low expression genes (FPKM < 1) had been filtered out from all gene expression libraries, correlation networks were constructed on the basis of pairwise correlations of gene expression across all samples. Modules are defined as highly interconnected clusters, and genes within the same modules have high correlation. In the weighted gene correlation network, any two genes were connected, and the edge weight was determined by the topology overlap measure calculated in the WGCNA package. The higher the weight is, the stronger the interaction between the two genes is. The sum of the weights of all edges of a node is defined as connectivity, and the top 1% (or 5%) of the genes with the highest connectivity in the network were called the hub gene (Yang et al. 2014).
Hub Gene Selection and Visualization
Hub genes with high connectivity may have more important biological significance in modules (Zhou et al. 2014). For each module, we selected the top ten genes with the highest connectivity as the hub genes of the module to analyze the different molecular mechanisms associated with the different wild populations and with adaptation to the different salinities. In addition, DEGs with high connectivity were selected as genes of interest. Correlation patterns and the interactions of the hub genes were visualized using Cytoscape (Shannon et al. 2003).
Result
Identification of the Expressed Transcripts of Eye-Spot Larvae
Eighteen cDNA libraries were sequenced from eye-spot larvae that had been subjected to three different salinities. Approximately 18.91–26.44 million 125-bp paired-end reads were generated for the 18 samples through RNA-Seq (Table 1). Among the clean reads, the number of sequences that could be mapped onto the reference transcriptome ranged from 14.9 to 19.8 million, and the percentage of clean reads ranged from 74.20 to 79.90% in the different libraries. The raw sequence reads have been submitted to the Sequence Read Archive (SRA) in the NCBI database and have been assigned SRA accession numbers (PRJNA513213).
Global Transcriptome Comparison Between the Two Wild Populations
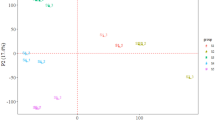
To investigate the global transcriptomic differences between BZ and CX oysters at different salinities, we performed PCA in each sample (Fig. 1). Under the 16‰ and 23‰ salinity treatments, there were clear differences in transcript expression between the two populations, although there was some overlap between the populations (Fig. S1, S2). Interestingly, the BZ and CX samples from the Sal30 treatment showed significant differences and could be clearly assigned to two groups.

Principal component analysis (PCA) plot showed the clustering of the transcriptomes of the two populations at a salinity of 30‰ (Sal30)
Identification of DEGs Between the Two Populations
In organisms, different gene products coordinate with each other to perform biological functions; pathway annotation analysis of DEGs is therefore helpful to further understand the functions of DEGs. To identify the genes that were associated with the adaptive divergence at different salinities, we conducted pairwise comparisons between populations BZ and CX at each salinity. At Sal16, pairwise comparisons of the BZSal16 and CXSal16 groups showed that 83 genes were significantly differentially expressed (Fig. 2(a)). Among these, 51 genes were significantly upregulated and 32 genes were significantly downregulated in BZ relative to CX samples. Representative upregulated and downregulated DEGs are listed in Tables 2 and 3, respectively, according to their functional categories. The data listed have been tested for significance and filtered based on a false discovery rate (FDR) < 0.01. The FPKMs in Table 2, 4, and 5 are average. Oxidation-reduction process genes were enriched and upregulated in the BZSal16 group, including dihydropyrimidine dehydrogenase [NADP(+)] and eosinophil peroxidase. The expression of calcium ion binding–related and aminopeptidase activity–related genes in the amino acid transport and metabolism category and of the carboxypeptidase B and eosinophil peroxidase genes in the chitin metabolic process category was approximately twofold higher in the BZSal16 group than in the CXSal16 group.

Number of DEGs for each population and salinity.(a) Number of DEGs between Sal16, Sal23, and Sal30 in each population. (b) Number of DEGs in BZ larvae at Sal16, Sal23, and Sal30. (c) Number of DEGs in CX larvae at Sal16, Sal23, and Sal30. The DEGs were filtered according to an FDR < 0.01 and an FC ≥ 2. The numbers in red and black indicate the number of upregulated and downregulated genes, respectively, in the sample at the top of the column compared with the sample at the beginning of the row
At Sal23, only 5 genes were downregulated in BZ samples compared with CX samples (Fig. 2(a)). Representative downregulated DEGs at Sal23 are listed in Table 3. More specifically, a DNA topoisomerase activity–related gene showed 5.55-fold greater expression in CX samples than in BZ samples. Similarly, a chitin metabolic process–related gene showed 22.03-fold greater expression in CX samples than in BZ samples.
Among eye-spot larvae subjected to Sal30, 65 and 13 genes were found to be significantly upregulated and downregulated, respectively, in BZ larvae compared with CX larvae (Fig. 2(a)). The upregulated and downregulated DEGs are shown in Tables 2 and 3, respectively. In our study, four candidate genes associated with stimulus responses were highly expressed in the BZ group. Eight metabolic process candidate genes were found to be significantly enriched, including genes associated with carbohydrate metabolic processes, lactate metabolic processes, and inorganic ion transport and metabolism. The representative genes included glutamine-fructose-6-phosphate aminotransferase [isomerizing] 2 isoform X7, glyoxalase 3–like, and glycosyltransferase-like family 2. A gene involved in oxidation-reduction processes (sushi, von Willebrand factor type A) showed over fivefold greater expression in the BZSal30 group than in the CXSal30 group. Notably, a large number of genes annotated as stimulus response genes, such as the slit-like protein, tumor necrosis factor family, and ATP-binding cassette subfamily A member 3-like genes, were upregulated in the BZSal30 group compared to the CXSal30 group.
The upregulated genes in the CXSal30 group compared to the BZSal30 group included DNA topoisomerase 1, which is associated with replication, recombination, and repair processes (KOG class annotation), and N-acetylneuraminate lyase B, which participates in amino acid transport and metabolism (eggNOG class annotation).
Identification of DEGs Among the Different Salinity Treatments
Pairwise comparisons were also performed to identify the DEGs among the three salinity treatments at both locations. The numbers of DEGs among the BZSal16, BZSal23, and BZSal30 groups are shown in Fig. 2(b). In the BZ population, compared with the BZSal16 group, the BZSal30 group demonstrated 200 upregulated genes, while the BZSal23 group showed 16 upregulated and 5 downregulated genes. No DEGs were evident in comparisons between the BZSal23 and BZSal30 groups.
A total of 40 genes associated with stimulus responses, amino acid transport, and metabolism and oxidation-reduction processes were annotated among the 200 genes upregulated in the BZSal30 group compared to the BZSal16 group. Some representative genes are shown in Table 4. Twelve genes involved in stimulus responses were enriched at high salinity. Multiple amino acid transport and metabolism genes were also highly enriched in the BZSal30 group. The analyses further revealed these genes to include diamine acetyltransferase 2, glucose dehydrogenase (acceptor), and glutamine-fructose-6-phosphate aminotransferase. In addition, 3 carbohydrate metabolic process–related genes were highly enriched in the BZSal30 group. Interestingly, the enrichment results showed that there were two apoptotic process–related genes among the DEGs, one of which was caspase-3 isoform X1.
Pairwise comparisons of the BZSal23 and BZSal16 groups showed that 21 genes were significantly differentially expressed (Fig. 2(b)). Among them were genes involved in amino acid transport and metabolism, oxidation-reduction processes, stimulus responses, and cell cycle control, and these genes were highly upregulated in the BZSal23 group compared to the BZSal16 group. Ion transmembrane transport–related and biological regulation–related genes were downregulated in the BZSal23 group compared to the BZSal16 group.
The numbers of DEGs among the CXSal16, CXSal23, and CXSal30 groups are shown in Fig. 2(c). The CX population had fewer DEGs than the BZ population. Analyses similar to those used for the BZ population were used to identify enriched DEGs in the CX population, and the results are shown in Table 5. Of the only 3 upregulated genes identified in the CXSal23 group compared to the CXSal16 group, 2 were enriched for signal transduction and are involved in stimulus responses. The other gene, cytochrome-c oxidase subunit III, was annotated as being involved in a variety of pathways, including oxidative phosphorylation (ko00190), cardiac muscle contraction (ko04260), and nonalcoholic fatty liver disease (NAFLD) (ko04932) in the KEGG database.
The enrichment results for the comparison of the CXSal23 and CXSal30 groups showed that the expression of phosphoethanolamine N-methyltransferase 3 isoform X2 was threefold higher in the CXSal30 group than in the CXSal23 group; this gene is involved in glycerophospholipid metabolism and methylation. In addition, the expression of three genes encoding signaling molecules, including early growth response protein 3 and a gene involved in chitin metabolism, was significantly reduced in the CXSal30 group compared to the CXSal23 group. Of the 18 upregulated DEGs identified in the CXSal30 group compared to the CXSal16 group, two were associated with cell cycle control, cell division, and chromosome partitioning, and one was identified as carbohydrate sulfotransferase 15. Of the 11 downregulated DEGs in the CXS30 group compared to the CXS16 group, three interesting genes were enriched: cytochrome P450 3A24, carnosine synthase 1, and pyridoxal 5′-phosphate synthase subunit SNZERR. KEGG pathway analysis showed that these genes are involved in linoleic acid metabolism (ko00591) and retinol metabolism (ko00830); arginine and proline metabolism (ko00330), histidine metabolism (ko00340), and beta-alanine metabolism (ko00410); and vitamin B6 metabolism (ko00750), respectively.
Construction of Gene Correlation Networks
To obtain a comprehensive understanding of the genes expressed in the gradually rising salinities at the two sites and to identify the specific genes that are highly associated with salinity adaptation, WGCNA was performed. After the low expression genes (FPKM < 1) had been filtered out from all gene expression libraries, 5664 genes were retained. Correlation networks were constructed on the basis of pairwise correlations of gene expression across all samples. Modules were defined as clusters of highly interconnected genes; genes within the same cluster had high correlation coefficients. This analysis identified 11 distinct modules (labeled with different colors), which are shown in the dendrogram in Fig. 3a. The major tree branches define the modules.

WGCNA of genes in BZ and CX at each salinity. a Hierarchical cluster tree showed correlation modules identified by WGCNA. Each leaf in the tree represents one gene. The major tree branches constitute 11 modules, labeled with different color. b Module–sample association. Each row corresponds to a module, labeled with ME (module eigengene) and a color as in (a). The number of genes in each module is indicated on the left. The color in the boxes represents –log(P), the Fisher exact test P value, according to the color legend on the right
The green module, with 430 identified genes, was highly associated with Sal30. In addition, the 2145 genes identified as being in the turquoise module were also highly correlated with Sal30. The royal blue module (52 genes) was associated with Sal23. Many modules were highly associated with Sal16, including the red module (1027 genes), the magenta module (744 genes), the light cyan module (138 genes), the midnight blue module (169 genes), the royal blue module (52 genes), and the tan module (111 genes). Regarding the different locations, the turquoise module, representing 2145 genes, was highly associated with BZ. The dark red module (50 genes), the midnight blue module (169 genes), and the tan module (111 genes) were highly related to CX.
WGCNA can also be employed to construct gene networks, in which each node represents a gene and the connecting lines (edges) between genes represent correlations (Yang et al. 2014). Hub genes are the genes that show the most connections in the network, as indicated by their high KME (eigengene connectivity) values. The top ten genes with the highest KME values in each of the specific modules are shown in Table 6. Notably, hub genes in highly connected modules may be more biologically significant than hub genes in global networks (Zhou et al. 2014). We chose the genes with the highest connectivity in each module as the hub genes to discuss the molecular mechanisms associated with the different wild populations and with adaptation to the different salinities.
The correlation patterns and the interactions of the hub genes were visualized with Cytoscape (Shannon et al. 2003). The correlation network of the dark red module (CX-specific) is shown in Fig. 4a. Genes encoding ETS-related transcription factor Elf-3, a toll-like protein, a slit-like protein, two myeloid differentiation primary response MyD88 proteins, proline-rich receptor-like protein kinase PERK6, and myeloid differentiation factor 88-1 were identified as candidate hub genes for this module (Fig. 4a, Table 6). Most of the genes were annotated with the signal transduction function, and the rest were annotated as being involved in negative regulation of peptidase activity, transcription, and regulation of transcription.

Correlation network analysis of specific modules. The correlation networks in each module that were highly associated with CX, BZ, Sal16, Sal23, and Sal30 are shown in a, b, c, d, and e, respectively. The candidate hub genes are shown in yellow
The correlation network of the turquoise module (BZ-specific) is shown in Fig. 4b. Genes encoding or associated with protein TANC1 isoform X1, phospholipid-transporting ATPase VA, titin isoform X10, posttranslational modification, protein broad–minded isoform X3, protein argonaute-2 isoform X5, protein DENND6A isoform X1, leucine-rich repeats, and immunoglobulin-like domains protein 2 and capicua-like protein were identified as candidate hub genes for this module (Fig. 4b, Table 6). These genes are related to zinc ion binding, magnesium ion binding, nucleic acid binding, transcription, and posttranslational modification.
The correlation network of the red module (Sal16-specific) is shown in Fig. 4c. Scavenger receptor cysteine–rich domain protein, sarcoplasmic calcium–binding protein, collagen alpha-1(XII) chain-like isoform X1, chitin-binding peritrophin-A domain protein, ATP-dependent RNA helicase DHX58, and perlucin were identified as candidate hub genes for the red module. These candidate genes are involved in calcium binding, signal transduction, chitin metabolic processes, and apoptotic processes.
The correlation network of the royal blue module (Sal23-specific) is shown in Fig. 4d. The candidate hub genes for this module included an expansin family protein and a 31-kDa antigen that bind oxygen during oxygen transport processes.
The correlation network of the green module (Sal30-specific) is shown in Fig. 4e. Genes encoding or related to protein-coupled receptor activity, the universal stress protein family, mucin-2-like protein, animal heme peroxidase, and phospholipid scramblase 1–like isoform X6 were identified as candidate hub genes for this module. In addition, these genes were enriched for the G protein–coupled receptor signaling pathway, stress responses, proteolysis, transmembrane transport, phospholipid scrambling, and chitin metabolic processes.
Functional Annotation of Salinity Stress–Related Modules
To identify the characteristics of the salinity stress–related modules and the biological responses to salinity stress, the modules were functionally annotated according to the included genes (Table 7). The results of GO annotation showed that the metabolic process, cellular process, and single-organism process terms were enriched in the red module, royal blue module, and green module. These three modules had distinct characteristics.
In addition to the oxidation-reduction process, regulation of transcription (DNA-templated), and proteolysis terms, which were also enriched in the other two modules, the metabolic process and carbohydrate metabolic process terms were highly enriched in the red module. In addition, compared with the other modules, the red module (Sal16-specific) had greater enrichment of developmental process and biological adhesion genes.
For the royal blue module (Sal23-specific), the GO enrichment results showed that besides oxidation-reduction processes and single-organism metabolic processes, neuropeptide signaling pathways, oxygen transport, and lipid homeostasis also play important roles in this module. The genes related to sensory perception of pain were also enriched.
The green module was highly associated with Sal30. More biological process and molecular function GO terms, e.g., protein phosphorylation, ion transmembrane transport, carbohydrate metabolic process, and antioxidant activity, were enriched in the green module than in the other two modules. The top-ranked biological process functional annotations enriched in the Sal30-responsive green module included the oxidation-reduction process, translation, regulation of transcription (DNA-templated), proteolysis, and G protein–coupled receptor signaling pathway terms.
To further explore the characteristics of relevant modules under high-salinity stress, the green module was selected, and pathway analysis was performed (Fig. 5). Pathway enrichment analysis (Fig. 5) showed that the phosphatidylinositol signaling system, metabolism of xenobiotics by cytochrome P450, drug metabolism by cytochrome P450, glutathione metabolism, arachidonic acid metabolism, and amino sugar and nucleotide sugar metabolism were the most enriched pathways in the green module.

KEGG pathway analysis of the green module
Discussion
In this study, we performed transcriptomic comparisons of eye-spot larvae from two sites subjected to different salinity treatments. WGCNA was performed to identify sample-specific modules and candidate hub genes. In data analysis, we focused not only on highly significant individual DEGs but also on interactions among genes. Such analysis might contribute to scientifically sound hypotheses. The results provide comprehensive information on genes involved in salinity adaptation mechanisms.
Osmolytes
As previous studies have reported, osmolytes, such as large amounts of nitrogenous solutes, amino acids, and inorganic ions, play primary roles in the osmoregulatory activities of osmoconformers (Berger and Kharazova 1997; Yuan et al. 2017; Jiang et al. 2019). In our study, the identified DEG- and WGCNA-enriched genes may be related to the metabolism and biosynthesis of osmotic regulators.
Ion Channels
In this study, enrichment analysis of DEGs and the genes identified in WGCNA revealed some binding-related molecular functions, many of which were associated with ions. Our study highlighted significantly upregulated ion-binding genes such as neuronal acetylcholine receptor subunit beta-2, sodium-coupled monocarboxylate transporter 1–like, and protein TANC1 isoform X1. Previous studies have indicated that ion channels help maintain the homeostasis of potassium and calcium ions under osmotic stress (Hasegawa et al. 2000). Calcium ions could regulate cell volume by controlling membrane activity during low osmotic stress in mollusks (Hirschi 1999; Mccarty and Oneil 1992). In addition, studies on C. gigas have shown that increased expression of calcium channels may activate calcium-activated potassium ion channels to maintain potassium ion concentration. Our results indicated that increased Ca2+ channel gene expression may have altered Ca2+ currents, increasing the activity of calcium-gated K+ channels to maintain ionic balance under hyperosmotic conditions (Sal23 or Sal30).
FAAs
In the BZ population, many amino acid regulatory genes, including glucose dehydrogenase, were differentially expressed at high salinity compared with low salinity. In addition, hydrolysis-related serine-type endopeptidase activity–related genes and N-acetylneuraminate lyase B and aminopeptidase activity–related genes were enriched among the DEGs in other groups. These findings suggest that amino acids may be involved in osmotic stress regulation in high-salinity environments. Previous studies have shown that intracellular FAAs regulation in one of the main mechanisms to regulate cell volume and respond to osmotic stress in bivalves (Pierce and Amende 1981; Robertson et al. 2017). In addition, Meng et al. (2013) has been studied the molecular mechanism of FAA osmotic adjustment in oysters. They also explained that one of the mechanisms respond to low osmotic pressure is volume reduction, such as increased metabolic rate, which releases ammonia from organs into hemolymph or bathing medium in oysters.
Our conclusions not only supplement the existing information on responses to hyperosmotic conditions but also confirm previous research. An important component of the oyster response to salinity stress is osmotic adaptation. In order to adapt to the rapid and large fluctuation of seawater salinity in the intertidal environment, oysters and other marine organisms adapt to the environment by adjusting the intracellular osmotic concentration to change the cell volume. The osmotic regulation of FAAs is one of these important mechanisms. Amino acids tend to be hydrolyzed to release ammonia under both hypertonic and hypotonic stress. Previous studies have indicated that glycine, proline, and taurine are the main osmotic pressure–responsive FAA in C. gigas (Hosoi et al. 2003).
Compared with that in the BZSal16 group, the expression of the diamine acetyltransferase 2 gene, which is involved in arginine and proline metabolism, was 1384-fold greater in the BZSal30 group (Table 4). In addition, some DEGs between CXSal23 and CXSal30 were enriched in the arginine and proline metabolism (ko00330) pathway in KEGG analysis. The expression of genes related to this coding pathway increases under hyperosmotic conditions. In animal cell, intracellular proline concentration is mainly controlled via biosynthetic and catabolic (Szabados and Savoure 2010). Under osmotic stress conditions, the regulation mechanism is the same (Szabados and Savoure 2010). Many previous studies have consistently shown that proline accumulates under high osmotic stress (Bishop et al. 1994; Meng et al. 2013; Yang et al. 2014). Such accumulation of proline, a compatible solute, leads to an increase in cell osmotic pressure, which can drive water inflow or reduce water outflow.
ROS Signal Transduction Pathways
Among the WGCNA results, the most interesting results were found for the red, royal blue, and green modules, which were highly correlated with salinities of 16, 23, and 30‰, respectively. Among the top 5 hub genes in these three modules, the first-ranked genes were found to be involved in oxidation-reduction, and several hub genes in these modules participated in this process. In addition, a large number of DEGs in the different populations and salinities were enriched for oxidation-reduction processes, and the expression of these genes differed by tens of times (Tables 2, 4, and 5). Changes in temperature, oxygen levels, and salinity in natural and artificial conditions can cause stress by disrupting the balance between the production and elimination of reactive oxygen species (ROS) (Lushchak 2011). Previous studies have shown that reduced expression of oxidation-reduction process genes in hypoosmotic conditions represents a protective strategy to reduce cell stress and ROS, but the strategy is different in hyperosmotic conditions (Meng et al. 2013; Zhao et al. 2016). In the study of osmotic stress in crabs and Japanese eels, the expression of oxidation-reduction-associated genes was also observed (Paital and Chainy 2010). Our results support these findings and suggest that oysters also need elevated oxidation-reduction gene expression to cope with high salinity. This increased expression helps to eliminate the imbalance between ROS production and elimination caused by changes in temperature, oxygen content, and salinity under natural or artificial conditions (Lushchak 2011). ROS signal transduction pathways have been identified as salinity stress signal transduction pathways.
Transcriptional Regulation and Signal Transduction
The DEGs in the oysters from the different populations subjected to the different salinity treatments were mainly enriched for transcriptional regulation and signal transduction, and analysis of biological processes related to developmental regulation revealed enrichment for chitin metabolism. Previous studies have shown that transcriptional regulation and signal transduction are widely involved in the salinity stress responses of organisms (Brewster et al. 1993; Causton et al. 2001). In addition, these mechanisms also exist in C. gigas (Ko et al. 2014). Enrichment of transcriptional regulation–related, G protein–coupled receptor signaling pathway–related, and transmembrane transport–related genes was identified in biological processes related to larval growth and development, and these genes were selected as candidate hub genes at Sal16 (Table 6). Our results are in line with the findings of other studies on oysters and other organisms. We found that the positive regulation of developmental process–related genes was abundant at the mRNA level. This enrichment of a large number of developmental regulation–related genes may be due to our selection of eye-stage larvae as experimental materials, as this stage is very important for oyster development. In addition, moderately high salinity did not completely destroy the growth and development of the oysters from the BZ or CX populations. Therefore, salinity stimulation has an important impact on the development process, leading to the expression of relevant genes that support appropriate responses to help organisms adapt to adverse environments.
Different Salinity Stress Responses in Different Populations
We selected two specific wild populations to investigate adaptive divergence of oysters under high-salinity stress. In this study, there was somewhat distinct transcriptional activity between the BZ and CX populations at Sal16 and Sal23 (Fig. S1, S2), but the two populations showed greater transcriptomic differences at Sal30 (Fig. 1). Adaptive divergence for oysters is more common than previously thought due to environmental variation in the intertidal zone. Li et al. (2017) found that temperature drives the divergence of subspecies C. gigas and C. angulata. In addition, environmental stress also contributes to the divergence of organisms between the intertidal and subtidal zones and between connected distributions of the same species (Weng and Wang 2014). Our study complements these findings on divergence with information on a disconnected distribution of C. ariakensis.
DEG analysis between the two populations showed that the DEGs at Sal30 were annotated with metabolic process, oxidation-reduction process, amino acid transport, and metabolism terms; in addition, the expression levels of these genes in the BZ population were higher than those in the CX population. Moreover, at high salinity, more DEGs were identified in the BZ population than in the CX population, which corresponds to the relative higher environmental salinity level of the BZ population than CX. The enrichment results for hub genes in WGCNA revealed many transcription-related genes that were selected as candidate hub genes for CX (Table 6). For BZ, most of the candidate genes are related to ion channels. In this study, differences in salinity responses between different populations existed at the gene expression level. Kim et al. (2014) assessed the levels and patterns of genetic variation of five concatenated mtDNA regions in C. ariakensis in Korea via DNA sequence analysis. The two populations we used are considered to be in the same clades as those in Kim et al.’s study. Clearly, our method has higher resolution, and transcriptome analysis can further identify divergence between populations. Furthermore, the F1 oysters of the two populations cultured in the same environment exhibited clear differences in larval growth at salinities of 16‰, 23‰, and 30‰, and in a high-salinity stress experiment for the juvenile oysters, the BZ population had a higher survival rate than the CX population (unpublished data). The overall expression differences, the differences in adaptation mechanisms, and the preliminary growth and survival data (unpublished data) in this study all indicate that there is divergence in tolerance to high salinity between the two populations. We can safely infer that there is significant genetic adaptation between these wild populations of C. ariakensis.
Temporal and spatial variations in salinity and other environmental factors are characteristic of the intertidal zone and place evolutionary pressure on intertidal organisms through negative impacts or through selection for fitness (Barranger et al. 2014; Sanford and Kelly 2011; Weng and Wang 2014). These two effects can be passed on to offspring, leading to genetic diversity and species formation. The discontinuous distribution of C. ariakensis and environmental variation may lead to the development of different response mechanisms to high salinity among wild populations under different salinities. In addition to salinity, many other evolutionary forces have acted on oysters. Profiling of genes responsive to high temperature in C. gigas and C. angulata in China has suggested that temperature may be the driving force of the divergence of the two subspecies (Li et al. 2017). In addition to temperature, metals, pathogens, and aquaculture may be evolutionary forces affecting the genetic structure of oyster populations (Li et al. 2013; Weng and Wang 2014).
Comparing adaptive differences between populations and related species is a promising method for identifying biological processes. Such analysis is important for establishing environmental tolerance ranges and revealing the molecular basis of stress adaptation (Tomanek 2014). In general, the wide and disconnected geographical distribution of C. ariakensis makes it an excellent model with which to study stress responses and the mechanisms of divergence of these responses. The results of this study provide a molecular basis for subsequent research on the development of high salinity–tolerant populations that can be cultured at normal seawater salinity.
Overall, we found that oyster larvae may activate several cellular processes to adapt to osmotic stress and that amino acids and ammonia are the major regulators of osmotic balance through ion transport. Genes involved in oxidation-reduction may reduce the production of ROS that can damage all cellular components. At moderately low salinity (16‰), cellular macromolecule biosynthesis and carbohydrate metabolism are abundant. Such activation of energy supply and cell differentiation processes indicates that 16‰ is a relatively suitable salinity. At moderately high salinity (23 and 30‰), osmotic agents, oxidation-reduction processes, and related regulatory networks of complex transcriptional regulation and signal transduction pathways dominate to counteract the salinity stress. Moreover, there are differences in the mechanisms of salinity responses in oysters from different geographical sites. Our findings provide a molecular basis for our subsequent research on the culture of high salinity–tolerant populations. Overall, our systematic study provides a framework for understanding the interactions of multiple pathways at the system level by which oyster larvae respond to moderate levels of high-salinity stress.
Change history
05 August 2019
The original version of this article unfortunately contained a mistake in the authorgroup section. Letters A, B, C, D, E and F were inadvertently added before each author’s name. The original article has been corrected.
References
Abdelrahman H et al (2017) Aquaculture genomics, genetics and breeding in the United States: current status, challenges, and priorities for future research. BMC Genomics 18:191
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106
Anderson K, Taylor DA, Thompson EL, Melwani AR, Nair SV, Raftos DA (2015) Meta-analysis of studies using suppression subtractive hybridization and microarrays to investigate the effects of environmental stress on gene transcription in oysters. PLoS One 10:e0118839
Ang R, Wang Z, Zhang J (1993) Marine Molluscan Culture. Qingdao Ocean University Press, Qingdao (in Chinese)
Barnes DKA (1999) High diversity of tropical intertidal zone sponges in temperature, salinity and current extremes. Afr J Ecol 37:424–434
Barranger A, Akcha F, Rouxel J, Brizard R, Maurouard E, Pallud M, Menard D, Tapie N, Budzinski H, Burgeot T, Benabdelmouna A (2014) Study of genetic damage in the Japanese oyster induced by an environmentally-relevant exposure to diuron: evidence of vertical transmission of DNA damage. Aquat Toxicol 146:93–104
Berger VJ, Kharazova AD (1997) Mechanisms of salinity adaptations in marine molluscs. Hydrobiologia 355:115–126
Bishop SH, Greenwalt DE, Kapper MA, Paynter KT, Ellis LL (1994) Metabolic-regulation of proline, glycine, and alanine accumulation as intracellular osmolytes in ribbed mussel gill tissue. J Exp Zool 268:151–161
Brewster JL, Devaloir T, Dwyer ND, Winter E, Gustin MC (1993) An Osmosensing signal transduction pathway in yeast. Science 259:1760–1763
Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA (2001) Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell 12:323–337
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41:D808–D815
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–U130
Gunter G (1961) Some relations of estuarine organisms to salinity. Limnol Oceanogr 6(2):182–190
Hasegawa PM, Bressan RA, Zhu JK, Bohnert HJ (2000) Plant cellular and molecular responses to high salinity. Annu Rev Plant Physiol 51:463–499
Heilmayer O, Digialleonardo J, Qian LF, Roesijadi G (2008) Stress tolerance of a subtropical Crassostrea virginica population to the combined effects of temperature and salinity. Estuar Coast Shelf Sci 79:179–185
Hirschi KD (1999) Expression of Arabidopsis CAX1 in tobacco: altered calcium homeostasis and increased stress sensitivity. Plant Cell 11:2113–2122
Hosoi M, Kubota S, Toyohara M, Toyohara H, Hayashi I (2003) Effect of salinity change on free amino acid content in Pacific oyster. Fish Sci 69:395–400
Hosoi M, Shinzato C, Takagi M, Hosoi-Tanabe S, Sawada H, Terasawa E, Toyohara H (2007) Taurine transporter from the giant Pacific oyster Crassostrea gigas: function and expression in response to hyper- and hypo-osmotic stress. Fish Sci 73:385–394
Huang B, Zhang L, Tang X, Zhang G, Li L (2016) Genome-wide analysis of alternative splicing provides insights into stress adaptation of the Pacific oyster. Mar Biotechnol (NY) 18(5):598–609
Jiang DL, Gu XH, Li BJ, Zhu ZX, Qin H, Meng ZN, Lin HR, Xia JH (2019) Identifying a long QTL cluster across chrLG18 associated with salt tolerance in tilapia using GWAS and QTL-seq. Mar Biotechnol (NY) 21:250–261
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M (2010) KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38:D355–D360
Kim WJ, Dammannagoda ST, Jung H, Baek IS, Yoon HS, Choi SD (2014) Mitochondrial DNA sequence analysis from multiple gene fragments reveals genetic heterogeneity of Crassostrea ariakensis in East Asia. Genes Genom 36:611–624
Ko GWK, Dineshram R, Campanati C, Chan VBS, Havenhand J, Thiyagarajan V (2014) Interactive effects of ocean acidification, elevated temperature, and reduced salinity on early-life stages of the Pacific oyster. Environ Sci Technol 48:10079–10088
Lacoste A, Malham SK, Gélébart F, Cueff A, Poulet SA (2002) Stress-induced immune changes in the oyster Crassostrea gigas. Dev Comp Immunol 26(1):1–9
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559
Li L, Wu XY, Yu ZN (2013) Genetic diversity and substantial population differentiation in Crassostrea hongkongensis revealed by mitochondrial DNA. Mar Genomics 11:31–37
Li A, L, L, Song K, et al. (2017) Temperature, energy metabolism, and adaptive divergence in two oyster subspecies. Ecol Evol 7:6151–6162.
Li L, Li A, Song K, Meng J, Guo X, Li S, Li C, de Wit P, Que H, Wu F, Wang W, Qi H, Xu F, Cong R, Huang B, Li Y, Wang T, Tang X, Liu S, Li B, Shi R, Liu Y, Bu C, Zhang C, He W, Zhao S, Li H, Zhang S, Zhang L, Zhang G (2018) Divergence and plasticity shape adaptive potential of the Pacific oyster. Nat Ecol Evol 2:1751–1760
Lushchak VI (2011) Environmentally induced oxidative stress in aquatic animals. Aquat Toxicol 101:13–30
Macinnes JR, Calabrese A (1979) Combined effects of salinity, temperature, and copper on embryos and early larvae of the American oyster, Crassostrea virginica. Arch Environ Contam Toxicol 8:553–562
Mccarty NA, Oneil RG (1992) Calcium signaling in cell-volume regulation. Physiol Rev 72:1037–1061
Meng J, Zhu Q, Zhang L, Li C, Li L, She Z, Huang B, Zhang G (2013) Genome and transcriptome analyses provide insight into the euryhaline adaptation mechanism of Crassostrea gigas. PLoS One 8(3):e58563
Paital B, Chainy GBN (2010) Antioxidant defenses and oxidative stress parameters in tissues of mud crab (Scylla serrata) with reference to changing salinity. Comp Biochem Physiol C 151:142–151
Pauley GB, Bowers KL, Thomas GL (1988) Species profiles: life histories and environmental requirements of coastal fishes and invertebrates (Pacific northwest): chum salmon, U.S. Fish and Wildlife Service Biological Report 82(11.81)
Philippart CJM, Anadón R, Danovaro R, Dippner JW, Drinkwater KF, Hawkins SJ, Oguz T, O'Sullivan G, Reid PC (2011) Impacts of climate change on European marine ecosystems: observations, expectations and indicators. J Exp Mar Biol Ecol 400:52–69
Pierce SK, Amende LM (1981) Control mechanisms of amino acid-mediated cell-volume regulation in salinity stressed mollusks. J Exp Zool 215:247–257
Robertson LS, Galbraith HS, Iwanowicz D, Blakeslee CJ, Cornman RS (2017) RNA sequencing analysis of transcriptional change in the freshwater mussel Elliptio complanata after environmentally relevant sodium chloride exposure. Environ Toxicol Chem 36:2352–2366
Sanford E, Kelly MW (2011) Local adaptation in marine invertebrates. Annu Rev Mar Sci 3:509–535
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Szabados L, Savoure A (2010) Proline: a multifunctional amino acid. Trends Plant Sci 15:89–97
Tomanek L (2014) Proteomics to study adaptations in marine organisms to environmental stress. J Proteome 105:92–106
Wang HY, Guo XM, Zhang GF, Zhang FS (2004) Classification of Jinjiang oysters Crassostrea rivularis (Gould, 1861) from China, based on morphology and phylogenetic analysis. Aquaculture 242:137–155
Weng NY, Wang WX (2014) Improved tolerance of metals in contaminated oyster larvae. Aquat Toxicol 146:61–69
Xue L, Que H, Zhang G (2007) The effect of salinity on growth and survival of Crassostrea rivularis larvae. Mar Sci. https://doi.org/10.3969/j.issn.1000-3096.2007.09.014. (in Chinese)
Yan LL, Su J, Wang Z, Yan X, Yu R, Ma P, Li Y, du J (2017) Transcriptomic analysis of Crassostrea sikamea x Crassostrea angulata hybrids in response to low salinity stress. PLoS One 12:e0171483
Yang Y, Han L, Yuan Y, Li J, Hei NN, Liang H (2014) Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat Commun 5:3231
Yuan J, Zhang X, Liu C, Duan H, Li F, Xiang J (2017) Convergent evolution of the osmoregulation system in decapod shrimps. Mar Biotechnol (NY) 19:76–88
Zhao XL, Yu H, Kong LF, Li Q (2012) Transcriptomic responses to salinity stress in the Pacific oyster Crassostrea gigas. PLoS One 7:e46244
Zhao XL, Yu H, Kong LF, Li Q (2016) Gene co-expression network analysis reveals the correlation patterns among genes in euryhaline adaptation of Crassostrea gigas. Mar Biotechnol (NY) 18:535–544
Zhou MF, Allen SK (2003) A review of published work on Crassostrea ariakensis. J Shellfish Res 22:1–20
Zhou Y, Xu JC, Liu YQ, Li JT, Chang CF, Xu CS (2014) Rat hepatocytes weighted gene co-expression network analysis identifies specific modules and hub genes related to liver regeneration after partial hepatectomy. PLoS One 9:e94868
Acknowledgements
We thank Dr. J. Meng for suggestions on the article writing. We thank Dr. Zhaoxing Qiu and his team for the help in larval rearing.
Funding
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA23050402), National Key R&D Program of China (2018YFD0900304) and the Earmarked Fund for Modern Agro–industry Technology Research System (CARS–49).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: Letters A, B, C, D, E and F were inadvertently added before each author’s name.
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Liu, X., Li, L., Li, A. et al. Transcriptome and Gene Coexpression Network Analyses of Two Wild Populations Provides Insight into the High-Salinity Adaptation Mechanisms of Crassostrea ariakensis. Mar Biotechnol 21, 596–612 (2019). https://doi.org/10.1007/s10126-019-09896-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-019-09896-9