
Abstract
The Pacific oyster Crassostrea gigas is a dominant aquaculture species in many intertidal zones throughout the Pacific and Atlantic Oceans and can tolerate a wide range of salinity. Studying the gene expression profiles of oyster gills had found differentially expressed genes (DEGs) involved in salinity tolerance. A systematic study of cellular response to salinity stress may provide insights into the mechanism of acquired salinity tolerance. Here, weighted gene co-expression network analysis (WGCNA) was carried out using RNA-seq data from gill transcriptome in response to different salinity. A total of 25,463 genes were parsed into 22 gene modules, of which 5 gene modules were identified as salinity-related modules. Brown module was the only one significantly correlated with salinity and free amino acids (FAAs) contents, which was associated with cellular metabolism, biosynthesis of amino acids, oxidation reduction, electron transport, nitrogen compound metabolism, and others. The enriched pathways in brown module were mainly about FAAs metabolism. The other four modules were significantly correlated with certain FAAs, and were over-represented in certain salinity. These results indicated that C. gigas triggered different FAAs in different salinity stress. This study represents the first RNA-seq gene network analysis in oysters responding to different salinity stresses. These results provide a systems-level framework to help understand the complexity of cellular process in response to osmotic stress and show the function and regulated genes of different FAAs at the molecular level.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Oysters are a major group of marine bivalves, which usually inhabit intertidal zones with fluctuating environmental factors, and they can tolerant a wide range of natural and anthropogenic stressors such as the changes of salinity and temperature, anoxia, acidification, and a variety of toxicants. The Pacific oyster Crassostrea gigas is a dominant species in many intertidal zones throughout the Pacific and Atlantic Oceans (Guo 2009). Because of their sessile lifestyle, they have evolved to acquire powerful defense mechanisms to withstand the highly dynamic and stressful environments. Meanwhile, with the genome of C. gigas released (Zhang et al. 2012a), they become an excellent model for studying stress adaption on the molecular levels.
Osmotic stress is one of common stresses that oysters usually face when there is rain or drying between tidal inundations (Barnes 1999). C. gigas can experience rapid and dramatic salinity fluctuations, from below salt 10 to in excess of salt 35 (Pauley et al. 1988). In previous studies, it has been shown that hypo-osmotic tolerance is rather a complex physiological trait incurring the coordination of cellular responses from transcriptome (Zhao et al. 2012) and proteome (Zhang et al. 2014). Salt stress effectors and free amino acids metabolism pathways were suggested to be a powerful ability to adapt to fluctuating salinities (Hosoi et al. 2007; Meng et al. 2013). A systematic study of cellular response to salinity would provide new insights into the mechanism of osmotic tolerance. Meng et al. (2013) have performed the transcriptome data of the Pacific oyster in seven different salinity gradients using RNA-seq technology. Although they uncovered several hundred marker genes for monitoring physiology status of oysters and the osmotic conditions, their analysis just focused on individual genes with high statistical significance and ignored gene interactions. Such analysis might lack scientifically sound hypotheses.
Gene co-expression network analysis is a system biology method for describing the correlation patterns among genes across a large-scale gene expression data, which is the merging of network theory with gene expression data analysis techniques (Fuller et al. 2011). Genes with similar expression patterns may participate in pathways and in regulatory and signaling circuits (Eisen et al. 1998). Weighted gene correlation network analysis (WGCNA) is an approach to network modeling that relies on easily understood statistical methods and improves on simple correlation networks (Zhang and Horvath 2005). This approach can identify modules of co-expressed genes, and relate these modules to phenotypic traits. Although, it has been widely applied in gene expression studies of humans and model organisms (Iancu et al. 2015; Lee et al. 2004; Liu and Ye 2014; Malki et al. 2013), the related studies on non-model organisms are still in its infancy (Fu et al. 2014; Zhang et al. 2012b).
Here, we constructed the co-expressed gene network of C. gigas transcriptome by reanalyzing RNA-seq datasets (Zhang et al. 2012a) to identify salinity stress-related modules and candidate key genes.
Materials and Methods
Transcriptome Data Acquisition
The transcriptome data from Zhang et al. (2012a) were downloaded from the Gene Expression Omnibus website, under the GEO accession number GSE31012 (GSM748453 ~ GSM768459).
Data Preprocessing
All the seven different gene expressed libraries were mapped to the genome of C. gigas (Zhang et al. 2012a) using tophat2 (Kim et al. 2013). Of the 28,027 coding sequences in the genome, 25,463 sequences could be mapped in at least two libraries. The transcript expression levels (FPKM) were estimated by RSEM software (Li and Dewey 2011), which were retained in subsequent analysis. Considering salt 30 as optimal salinity, differential expression analysis was performed using the R package DESeq (Anders 2010). Here, transcripts with nominal P value <0.05 and |log2(foldchange)| >1 were considered as differentially expressed genes (DEGs) to avoid omitting the positives.
Gene Co-Expression Network Construction
Gene network was constructed using R package WGCNA following the procedure described in Langfelder and Horvath (2008). Firstly, we clustered the samples to see if there were any obvious outliers. As a result, the group of salt 5 was removed as an outlier. Secondly, a power of nine was chosen so that the resulting networks exhibited approximate scale-free topology (model fitting index R 2 = 0.8). Thirdly, all the 25,463 genes in the other six libraries were hierarchically clustered using the topological overlap-based dissimilarity measure (Zhang and Horvath 2005). Finally, the resulting gene dendrogram was used for module detection using the dynamic tree cut method (minModuleSize = 100 and mergeCutHeight = 0.25). In a weighted gene co-expression network, any two genes were connected, and the edge weight was determined by the topology overlap measure provided in WGCNA. The weights ranged from 0 to 1, and reflected the strength of the communication between the two genes. Connectivity was defined as the sum of the weights across all the edges of a node, and the top 1 % (or 5 %) of the genes with the highest connectivity in the network were defined as hub genes (Yang et al. 2014). We defined the hub genes in a given module by the intramodular connectivity (Kwithin), which measures a gene’s connectivity in the specific module.
Identification of Salinity Stress-Related Modules
To identify modules that are significantly associated with the salinity, we first calculated the module eigengene using all genes in each module, and then correlate eigengenes with salinity treatments and free amino acids (FAAs) contents. The only modules with P values <0.05 were considered as salinity stress-related modules. To characterize those modules, GO and KEGG pathway enrichment analysis of the annotated genes in each salinity stress-related module using GSEABase package (Morgan et al. 2008). GO terms and KEGG pathways with P values <0.05 were considered to be significantly more enriched.
Hub Genes Selection and Visualization
Highly connected intramodular hub genes may be more biologically significant than hub genes in global network (Zhou et al. 2014). Based on the module sizes, we chose the top 2 % of the genes with the highest connectivity in the module as hub genes. In addition, the differential expressed genes with high connectivity would be selected as genes of interest. Co-expression patterns and interactions of hub genes were visualized using Cytoscape (Shannon et al. 2003).
Results
Construction of Gene Co-Expression Network
In the study, the transcriptome profiling of Zhang et al. (2012a) was performed using the high-throughput sequencing technologies for Pacific oysters that were sampled at different salinity (5, 10, 15, 20, 25, 30, and 40 ‰). Firstly, we clustered these samples and removed the salt 5 as an outlier (Fig. S1) and the left samples used the blockwiseConsensusModules R function to carry out network construction. To ensure that the network was biologically relevant, the scale-free topology model fit and the mean connectivity of the network was evaluated over a range of the soft threshold power β (β = 9, Fig. S2). All 25,463 coding sequences were parsed into 22 gene modules, with module size ranging from 128 to 12,942 (Table 1).
Identification of Salinity Stress-Related Modules
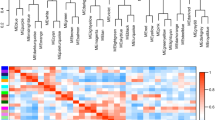
The eigengene-trait correlation analysis showed that brown module was significantly positively correlated with salinity stress and almost all the FAAs contents (except glutamate) after multiple testing correction (Fig. 1). In addition, pink module was correlated with taurine; lightyellow correlated with glycine, alanine, and proline; and cyan and royalblue correlated with glutamate. These five modules were conducted by over-representation analysis of DEGs using a hypergeometric test (Table 2) (Fig. 2). Among them, all the five modules showed differential expression at different salinity: brown module responded at salt 10 and salt 40; pink module responded at hypo-salinity (salt 10 and salt 15); cyan module responded at salt 20 and salt 40; lightyellow module responded at salt 40; and royalblue module responded at salt 25 and salt 40. Combining the results that four modules correlated with different FAAs contents, it may imply that different FAAs play a dominant role at different salinity.

Module-trait associations. Each row corresponds to a module eigengene, column to a trait. Each cell contains the corresponding correlation and p value. The table is color-coded by correlation according to the color legend

Network visualization of interactions between hub genes in five stress-responsive modules. Each node represents a gene, which is labeled with sequence ID. Green nodes represent DEGs. Genes in the same rectangle belong to the same module. Node size represents total connectivity (ktotal)
Functional Annotation of Salinity Stress-Related Modules
To identify modular features with their biological roles response to salinity, functional annotations of the salinity stress-related modules were performed on the basis of their gene composition using GSEABase software (Table 3). A detailed functional enrichment of GO annotations in these modules was provided in Table S1 with P value <0.05. According to functional annotations, brown module was significantly enriched with genes functioning in “cellular metabolic process,” “primary metabolic process,” “catalytic activity,” “oxidoreductase activity,” and so on. These biological activities were closely associated with metabolic process of C. gigas. Pink module was closely associated with “biological regulation,” and lightyellow module was involved in “ATP metabolic process,” “actin filament organization,” and “peptide metabolic process.” Royalblue and cyan modules were related to “glucide metabolism” and “macromolecule biosynthesis,” respectively.
The pathway annotations were assigned to KEGG database (http://www.genome.jp/kegg/). Pathways enrichment analysis (Table 4) showed that selenocompound metabolism, methane metabolism, biosynthesis of amino acids, and glycine, serine, and threonine metabolism were top enriched in brown module. “Cysteine and methionine metabolism” was enriched in both brown module and pink module. It was also found that “oxidative phosphorylation” and “protein export” were enriched in lightyellow module, “starch and sucrose metabolism” and “flavone and flavonol biosynthesis” were enriched in royalblue module, and “ribosome” was enriched in cyan module. To investigate the interactions between KEGG pathways and show the systematic osmoregulation pathways, we extracted the genes in brown module and in enriched KEGG pathways (Table S2), and constructed a sketch of the hypothetical pathways network that are significantly correlated to osmoregulation (Fig. 3). In this framework, C. gigas response to salinity was initiated upon the sensing of ion via cell membrane receptors. The signals subsequently transmitted down through the AMPK signaling pathways and triggered the production of a series of FAAs as the end results. In parallel, energy metabolism was activated to provide energy.

Overview of salinity-related enriched pathways of genes in brown modules during Crassostrea gigas response to salinity stress. Only pathway components interacted directly with genes in brown modules (shown in orange text boxes) are presented within each enriched KEGG pathway (framed in orthogonal polygons). The numbers in orthogonal polygons represent the KEGG ID, 00010: glycolysis/gluconeogenesis; 00250: alanine, aspartate, and glutamate metabolism; 00260: glycine, serine, and threonine metabolism; 00270: cysteine and methionine metabolism; 00290: valine, leucine, and isoleucine biosynthesis; 04152: AMPK signaling pathway; 04964: proximal tubule bicarbonate reclamation
Hub Genes Selections
In network biology, a hub gene is a good representative of a module. All the top 2 % hub genes of each salinity stress-related module were searched against the genome of C. gigas. Their correlation were visualized in Fig. 2, and the related information of hub genes were listed in Table S3. For instance, brown module, as the only module significantly correlated with salinity, has huntingtin-interacting protein 1 (HIP1), involved in cytoskeleton, Protein iolS, Betaine aldehyde dehydrogenase (BADH); 2-aminoethanethiol dioxygenase (ADO) and eosinophil peroxidase (EPX), involved in oxidation-reduction process; and myosin-Vb, organic cation transporter protein (OCT), and metal transporter CNNM2 involved in transport and so on.
Discussion
In this study, we constructed a gene co-expression network of C. gigas in response to different salinity, and identify salinity stress-related modules by transcriptome data of gills using WGCNA analysis. Importantly, we used the data from the same article to avoid batch effects when constructing the networks from expression data from multiple profiling techniques. Although differential expression analysis had been used to study the oyster transcriptome of euryhaline adaptation, gene co-expression network analysis focuses on the strong correlation patterns between the genes (Miller et al. 2008).
WGCNA considers not only the correlation between two genes but also the connectivity of their shared neighbors across the whole network. Hierarchical average linkage clustering based on topological overlap matrix (TOM) was used to genes with highly similar co-expression patterns into modules. Based on the cluster result, salt 5 was identified as an outlier. In the previous analysis, salt 5 was considered to be special with previous principal component analysis (PCA) of transcriptomes (Zhang et al. 2012a). This results indicated that 5 ‰ may exceed the range of oyster salinity tolerance. For increasing the confidence of results, data of the other six groups were used to perform the gene co-expression network analysis. As a result, we identified 22 highly correlated modules, and only five modules were significantly correlated with salinity based on the results of gene significance and module membership.
The most interested module is the brown module, which is the only one module positively correlated to salinity and almost all FAAs contents. It consisted of 1718 genes, of which there are 165 DEGs. As the previous studies reported, osmolytes play a primary role in the osmotic activities of the osmoconformer, such as the large amount of nitrogenous solutes, amino acids, and inorganic ions (Berger and Kharazova 1997; Pierce 1982; Yancey et al. 1982). Genes in these enriched GO terms might be correlated with the metabolism and biosynthesis of the osmolytes. Based on the results of KEGG pathways enrichment, “selenocompound metabolism,” “biosynthesis of amino acids,” and “glycine, serine, and threonine metabolism” were enriched, which verified amino acids playing important roles in osmoregulation in C. gigas again. In parallel, the hypothetical pathways network showed that signals transmitted down by AMPK signaling pathways and triggered the production of a series of FAAs. The other enriched pathway “methane metabolism” belongs to “energy metabolism,” which took part in oxidation to CO2 for energy source for energy supply. Functional annotations and enrichments found that “cellular metabolic process,” “nucleoside monophosphate biosynthetic process,” and “single-organism biosynthetic process” were the top three enriched GO terms. As listed in Table S1, “nitrogen compound metabolic process,” “cellular amino acid metabolic process,” “RNA metabolic process,” “oxidation-reduction process,” “protein dephosphorylation,” and others were involved in GO-enrichment analysis. This proved that it is a complex cellular process in response to osmotic stress. FAAs and amine were the important participants in osmotic equilibrium. Other biological process might help the biosynthesis, metabolic and transport of FAAs and amine, and signal transduction.
Hub genes were considered a good representative of a module in network biology. HIP1 contains clathrin-binding domains and actin-binding TALIN homology domains, and its domains can assist clathrin-mediated endocytosis (Ross 2012). Comparative genomic analyses indicated that HIP1 was similar with a protein involved in organization of cortical actin cytoskeleton in yeast (Hackam et al. 2000). However, the function of HIP1 has been studied for Huntington’s disease; it was not known completely, especially in invertebrates. HIP1, as the hub gene with max connectivity, might be involved in amino acids transport and assembly of cytoskeleton. TRIM2 was also found to be under positive selection in adaptation to hypo-osmotic environment in a previous study (Zhao et al. 2014). Myosin-Vb is annotated as transmembrane transport in mammals. Apparently, myosin-Vb should participate in the transport of osmolytes to ensure osmotic equilibrium. As is reported, mammalian TRIM2 and TRIM3 are more similar to each other, and both predominantly expressed in brain compartments and interact with myosin-Vb (El-Husseini and Vincent 1999; Meroni 2012). In this study, TRIM2 has been found to co-express with myosin-Vb which might be implicated in osmolytes transport and regulation in C. gigas. ADO is the key enzyme in the pathway, taurine and hypotaurine metabolism, and was important in the conversion of cysteamine to hypotaurine (Coloso et al. 2006; Goto et al. 2001). This suggested that ADO might participate in FAAs biosynthesis for osmoregulation in C. gigas.
Among the top 2 % hub genes in brown module, there are five DEGs, and of which three DEGs are annotated. EPX differentially expressed in both salt 10 and salt 15 significantly and was annotated as “oxidation-reduction process.” Besides EPX, there were several genes among the top 2 % hub genes of brown module participating in oxidation reduction. Changes in temperature, oxygen levels, and salinity can cause the stress in natural and artificial conditions via induction of disbalance between reactive oxygen species (ROS) production and elimination (Lushchak 2011). The downregulation of EPX in hypo-osmotic conditions may represent a protective strategy to reduce cellular stress and decrease the ROS. Similar changes in oxidation reduction associated genes have been observed in crabs and Japanese eels exposed to osmotic stress (Paital and Chainy 2010; Tse et al. 2013). Compared with the results of differentially expressed genes (Meng et al. 2013), ROS signal transduction pathways were identified as one of salt stress signal transduction pathway.
Pink module was negatively correlated with taurine significantly. Taurine is well known that during adaptation to environmental salinity, levels of taurine in invertebrate and amphibian tissues will change to maintain osmotic equilibrium (Thurston et al. 1980). In the Pacific oyster, taurine, as the primary osmolyte, accounts for approximately 80 % of the total amino-acid content (Hosoi et al. 2003). Considered the enriched pathways, “ECM-receptor interaction” and “cytokine-cytokine receptor interaction” belonged to “signaling molecules and interaction” module and “proteasome” participated in signal transduction pathways and stress signaling and so on. It implicated that pink module might be involved in signal transduction. “Cysteine and methionine metabolism” was enriched in both pink and brown modules. In bivalves, taurine metabolism occurs mainly via synthesis from cysteine and a high-affinity transport system. “Cysteine and methionine metabolism” manipulated the content of the sulfur-containing amino acid to play important roles in taurine metabolism. Only two hub genes in pink module had been annotated: cytochrome P450 2G1 (CYP2G1) and innexin unc-9 (UNC-9). Cytochromes P450 family are membrane proteins and require a source of electrons from an electron transfer chain to function (Nelson and Nebert 2011). CYP2G1 was found olfactory specific in the rabbit and have distinct substrate specificities against endogenous compounds or nasal toxicants (Maïbèche-Coisne et al. 2002; Maïbèche-Coisne et al. 2005). This gene may take part in the sense of osmotic pressure in C. gigas, which were also identified as salt stress effectors under low osmotic stress (Meng et al. 2013). While UNC-9 regulated gap junctions to connect and communicate between two cells. In mammals’ central nervous system, taurine was a neurotransmitter (Wu and Prentice 2010). Taken together, taurine may be both an important osmolyte and a signal factor or key molecule regulating signal transduction.
Lightyellow module was positively correlated with glycine, alanine, and proline significantly. Based on the result of GO and KEGG enrichment, lightyellow module was involved in “ATP metabolic process,” “actin filament organization,” and “protein export.” The hub genes in lightyellow module were integral component of membrane. Neuronal acetylcholine receptor subunit beta-3 (CHRNB3) binds acetylcholine and then leads to opening of an ion-conducting channel across the plasma membrane (Kormelink and Luyten 1997). Sodium-dependent phosphate transporter 1 (SLC17A1) is known to co-transport sodium and phosphate, with a capacity to also transport organic anions (Sreedharan et al. 2010). The results showed that genes in this module increased energy metabolism and altered membrane fluidity, which initiated transport of organic and inorganic osmolytes.
Cyan and royalblue modules were correlated with glutamate. In cyan module, histone-lysine N-methyltransferase SETD8-A (SETD8-A) is involved in methyltransferase activity. SETD8 was found to be conserved in zebrafish and mammals and take part in epidermal differentiation to help epithelium survive in a hypotonic external environment (Thompson 2011). SETD8-A was probably conserved between oyster and zebrafish to ensure the balance between internal environments and external environments. Deoxyribonucleoside 5′-monophosphate N-glycosidase (DNMP) was another target gene of c-Myc, as the same as SETD8, and involved in epidermal differentiation (Driskell et al. 2012; Ghiorghi et al. 2007). According to the GO and KEGG enrichment analysis, royalblue module was enriched in carbohydrate metabolism. The function of glutamate was similar with alanine to form ammonia to regulate the osmotic pressure. While glutamate also participated in carbohydrate metabolism (Brosnan 2000). We speculated that during the salinity stress, cells might increase energy demand to compensate for osmoregulation.
Based on the significant correlation between certain modules and certain FAAs, the over-representation analysis of DEGs at different osmotic stress was conducted in four salinity-related modules (pink, lightyellow, cyan, and royalblue). Salt 10 and salt 15 were enriched with DEGs in pink module, indicating that pink module responded specifically at extreme hypo-osmotic conditions, where taurine might be the primary osmolyte to maintain osmotic equilibrium. Owing to the negative correlation between pink module and taurine content, the genes in pink module may be related to inhibition of taurine biosynthesis. Lightyellow module responded specifically at salt 40. Alanine was involved predominantly in reductive amination reactions to transiently produce the ammonia for osmotic equilibrium with glutamate. It was reported that ammonia changed little with an abrupt decrease in salinity but increased with an abrupt increase in salinity (Livingstone et al. 1979). Alanine, glycine, and proline were found to be accumulated, and the activity of the cilia may be relatively insensitive to changes in cytoplasmic osmolality during hyperosmotic volume regulation in mussels (Deaton 2001). It indicated that amino acids export and ammonia production were the primary reaction in response to hyperosmotic stress. Cyan module responded specifically at salinity 20. Ribosome was enriched in this module. One characteristic shared by the DEGs in this module is that all genes were upregulated at salinity 20. It was found that repressed protein synthesis may be a self-protection mechanism against heat stress that is commonly existed in various animals (Fu et al. 2014). It indicated that salinity 20 may not be an acute stress to C. gigas. DEGs were enriched in salt 25 and salt 40 in royalblue module, which suggested that energy metabolism was the highlight process during moderate salinity stress.
Meng et al. (2013) confirmed the important roles of FAAs for oyster low salinity adaptation using physiological experiment and differential gene expression analyses, and described the salt stress signal transduction network in oyster. Compared with these results, organic substances such as FAAs and quaternary amines were verified to play dominant roles in oysters in our analyses. Organic osmolytes are able to provide osmotic bulk under high osmotic stress without the direct physiological trade-off that inorganic ions would have (Yancey 2005). Therefore, organic osmolytes have an advantage over inorganic ions in the study. In differential genes expression analyses, ROS signal transduction pathways were identified to be induced by osmotic stress, which was confirmed by gene co-expression network analysis. Meanwhile, the hypothetical pathways network indicated that salt stress induced cytosolic Ca2+ oscillations and excited AMPK signal pathway. Furthermore, amine was also found to be an important osmolyte in response to high salinity.
Collectively, we postulate that C. gigas might have triggered several cellular processes to acclimate to osmotic stress. Amino acids and ammonia are the primary regulated factor to maintain osmotic equilibrium accompanying ion transport. Genes participating in oxidation reduction were possibly to decrease production of ROS that can potentially cause damage to all cellular components. ROS signal transduction pathways and AMPK signal pathway were identified to participate in the oyster’s responses. We speculate that different osmolytes regulated osmotic pressure in different salinity. At low salinity (salt 10, salt 15), genes related to regulation of transcription were enriched that might indicate that DNA transcription was active in response to hypo-osmotic stress, and signal transduction and cellular communication were also active. Meanwhile, sulfur amino acid metabolic process was enriched, thereby assisting in regulation of taurine contents for isotonicity. At moderate low salinity (salt 20, salt 25), cellular macromolecule biosynthesis and carbohydrate metabolism were enriched. Energy supply and cell differentiation were triggered that indicated salt 20 and salt 25 were the relatively suitable salinity. At moderate high salinity (salt 40), ammonia was the primary osmolyte for osmotic equilibrium assisting with glycine and proline. Overall, our study provides the first systematic insights into molecular mechanisms underlying the physiological changes of C. gigas in response to salinity stress.
References
Anders S (2010) Analysing RNA-seq data with the DESeq package. Mol Biol 43:1–17
Barnes DK (1999) High diversity of tropical intertidal zone sponges in temperature, salinity and current extremes. Afr J Ecol 37:424–434
Berger VJ, Kharazova A (1997) Mechanisms of salinity adaptations in marine molluscs. Hydrobiologia 355:115–126
Brosnan JT (2000) Glutamate, at the interface between amino acid and carbohydrate metabolism. J Nutr 130:988S–990S
Coloso RM, Hirschberger LL, Dominy JE, Lee JI, Stipanuk MH (2006) Cysteamine dioxygenase: evidence for the physiological conversion of cysteamine to hypotaurine in rat and mouse tissues. Adv Exp Med Biol 583:25–36
Deaton LE (2001) Hyperosmotic volume regulation in the gills of the ribbed mussel, Geukensia demissa: rapid accumulation of betaine and alanine. J Exp Mar Biol Ecol 260:185–197
Driskell I, Oda H, Blanco S, Nascimento E, Humphreys P, Frye M (2012) The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J 31:616–629
Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. PNAS 95:14863–14868
El-Husseini AE-d, Vincent SR (1999) Cloning and characterization of a novel RING finger protein that interacts with class V myosins. J Biol Chem 274:19771–19777
Fu X et al. (2014) Sequencing-based gene network analysis provides a core set of gene resource for understanding thermal adaptation in Zhikong scallop Chlamys farreri. Mol Ecol Resour 14:184–198
Fuller T, Langfelder P, Presson A, Horvath S (2011) Review of weighted gene coexpression network analysis. In: Lu HH-S, Schölkopf B, Zhao H (ed) Handbook of Statistical Bioinformatics. Springer-Verlag, Berlin, Heidelberg, pp 369–388
Ghiorghi YK, Zeller KI, Dang CV, Kaminski PA (2007) The c-Myc target gene Rcl (C6orf108) encodes a novel enzyme, deoxynucleoside 5′-monophosphate N-glycosidase. J Biol Chem 282:8150–8156
Goto T, Matsumoto T, Takagi S (2001) Distribution of the hepatic cysteamine dioxygenase activities in fish. Fish Sci 67:1187–1189
Guo X (2009) Use and exchange of genetic resources in molluscan aquaculture. Rev Aquac 1:251–259
Hackam AS, Yassa AS, Singaraja R, Metzler M, Gutekunst C-A, Gan L, Warby S, Wellington CL, Vaillancourt J, Chen N (2000) Huntingtin interacting protein 1 induces apoptosis via a novel caspase-dependent death effector domain. J Biol Chem 275:41299–41308
Hosoi M, Kubota S, Toyohara M, Toyohara H, Hayashi I (2003) Effect of salinity change on free amino acid content in Pacific oyster. Fish Sci 69:395–400
Hosoi M, Shinzato C, Takagi M, Hosoi-Tanabe S, Sawada H, Terasawa E, Toyohara H (2007) Taurine transporter from the giant Pacific oyster Crassostrea gigas: function and expression in response to hyper- and hypo-osmotic stress. Fish Sci 73:385–394
Iancu OD, Colville A, Oberbeck D, Darakjian P, McWeeney SK, Hitzemann R (2015) Cosplicing network analysis of mammalian brain RNA-seq data utilizing WGCNA and mantel correlations. Front Genet 6:174
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36
Kormelink PJG, Luyten WH (1997) Cloning and sequence of full-length cDNAs encoding the human neuronal nicotinic acetylcholine receptor (nAChR) subunits β3 and β4 and expression of seven nAChR subunits in the human neuroblastoma cell line SH-SY5Y and/or IMR-32. FEBS Lett 400:309–314
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559
Lee I, Date SV, Adai AT, Marcotte EM (2004) A probabilistic functional network of yeast genes. Science 306:1555–1558
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12:323
Liu W, Ye H (2014) Co-expression network analysis identifies transcriptional modules in the mouse liver. Mol Gen Genomics 289:847–853
Livingstone D, Widdows J, Fieth P (1979) Aspects of nitrogen metabolism of the common mussel Mytilus edulis: adaptation to abrupt and fluctuating changes in salinity. Mar Biol 53:41–55
Lushchak VI (2011) Environmentally induced oxidative stress in aquatic animals. Aquat Toxicol 101:13–30
Maïbèche-Coisne M, Jacquin-Joly E, Francois M, Meillour NL (2002) cDNA cloning of biotransformation enzymes belonging to the cytochrome P450 family in the antennae of the noctuid moth Mamestra brassicae. Insect Mol Biol 11:273–281
Maïbèche-Coisne M, Merlin C, François M-C, Porcheron P, Jacquin-Joly E (2005) P450 and P450 reductase cDNAs from the moth Mamestra brassicae: cloning and expression patterns in male antennae. Gene 346:195–203
Malki K et al. (2013) Integrative mouse and human mRNA studies using WGCNA nominates novel candidate genes involved in the pathogenesis of major depressive disorder. Pharmacogenomics 14:1979–1990
Meng J et al. (2013) Genome and transcriptome analyses provide insight into the euryhaline adaptation mechanism of Crassostrea gigas. PLoS One 8:e58563
Meroni G (2012) TRIM/RBCC Proteins. Springer, Trieste
Miller JA, Oldham MC, Geschwind DH (2008) A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J Neurosci 28:1410–1420
Morgan M, Falcon S, Gentleman R (2008) GSEABase: gene set enrichment data structures and methods. R package version 1. Availabe at http://www.bioconductor.org. Accessed 17 Oct 2015
Nelson DR, Nebert DW (2011) Cytochrome P450 (CYP) gene superfamily. ELS. doi:10.1038/npg.els.0005667
Paital B, Chainy G (2010) Antioxidant defenses and oxidative stress parameters in tissues of mud crab (Scylla serrata) with reference to changing salinity. Comp Biochem Physiol C 151:142–151
Pauley GB, Van Der Raay B, Troutt D (1988) Species profiles: life histories and environmental requirements of coastal fishes and invertebrates (Pacific Northwest), Pacific Oyster. In: Fish and Wildlife Service Biological Report. San Francisco: US Fish and Wildlige Service
Pierce SK (1982) Invertebrate cell volume control mechanisms: a coordinated use of intracellular amino acids and inorganic ions as osmotic solute. Biol Bull 163:405–419
Ross TS (2012) Huntingtin Interacting Protein 1. In: Schwab M (ed) Encyclopedia of Cancer. Springer Berlin Heidelberg, Berlin, Heidelberg, pp 1761–1764
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Sreedharan S, Shaik JH, Olszewski PK, Levine AS, Schiöth HB, Fredriksson R (2010) Glutamate, aspartate and nucleotide transporters in the SLC17 family form four main phylogenetic clusters: evolution and tissue expression. BMC Genomics 11:17
Thompson SR (2011) Cloning and expression of the zebrafish gene, setd8, a histone methyl-tranferase required for cell proliferation. Kalamazoo College
Thurston JH, Hauhart RE, Dirgo JA (1980) Taurine: a role in osmotic regulation of mammalian brain and possible clinical significance. Life Sci 26:1561–1568
Tse WKF et al. (2013) Transcriptomic and iTRAQ proteomic approaches reveal novel short-term hyperosmotic stress responsive proteins in the gill of the Japanese eel (Anguilla japonica. J Proteome 89:81–94
Wu J-Y, Prentice H (2010) Role of taurine in the central nervous system. J Biomed Sci 17:S1
Yancey PH (2005) Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J Exp Biol 208:2819–2830
Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN (1982) Living with water stress: evolution of osmolyte systems. Science 217:1214–1222
Yang Y, Han L, Yuan Y, Li J, Hei N, Liang H (2014) Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat Commun 5:3231
Zhang B, Horvath S (2005) A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4:Article 17
Zhang G, Fang X, Guo X, Li L, Luo R, Xu F, Yang P, Zhang L, Wang X, Qi H, Xiong Z, Que H, Xie Y, Holland PW, Paps J, Zhu Y, Wu F, Chen Y, Wang J, Peng C, Meng J, Yang L, Liu J, Wen B, Zhang N, Huang Z, Zhu Q, Feng Y, Mount A, Hedgecock D, Xu Z, Liu Y, Domazet-Loso T, Du Y, Sun X, Zhang S, Liu B, Cheng P, Jiang X, Li J, Fan D, Wang W, Fu W, Wang T, Wang B, Zhang J, Peng Z, Li Y, Li N, Chen M, He Y, Tan F, Song X, Zheng Q, Huang R, Yang H, Du X, Chen L, Yang M, Gaffney PM, Wang S, Luo L, She Z, Ming Y, Huang W, Huang B, Zhang Y, Qu T, Ni P, Miao G, Wang Q, Steinberg CE, Wang H, Qian L, Liu X, Yin Y (2012a) The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490:49–54
Zhang L, Hou R, Su H, Hu X, Wang S, Bao Z (2012b) Network analysis of oyster transcriptome revealed a cascade of cellular responses during recovery after heat shock. PLoS One 7:e35484
Zhang Y et al. (2014) Proteomic basis of stress responses in the gills of the Pacific oyster Crassostrea gigas. J Proteome Res 14:304–317
Zhao X, Yu H, Kong L, Li Q (2012) Transcriptomic responses to salinity stress in the Pacific oyster Crassostrea gigas. PLoS One 7:e46244
Zhao X, Yu H, Kong L, Liu S, Li Q (2014) Comparative transcriptome analysis of two oysters, Crassostrea gigas and Crassostrea hongkongensis provides insights into adaptation to hypo-osmotic conditions. PLoS One 9:e111915
Zhou Y, Xu J, Liu Y, Li J, Chang C, Xu C (2014) Rat hepatocytes weighted gene co-expression network analysis identifies specific modules and hub genes related to liver regeneration after partial hepatectomy. PLoS One 9:e94868
Acknowledgments
This study was supported by the grants from the National Natural Science Foundation of China (31372524), Shandong Seed Project, and Shandong Province (2014GHY115002).
Author Contributions
XZ carried out the molecular genetic studies, participated in the data analysis, and drafted the manuscript. HY participated in the data analysis. LK participated in the design of the study and performed the statistical analysis. QL conceived of the study, participated in experimental design and coordination, and contributed to t the manuscript preparation. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Electronic supplementary material
Online Resource 1
Fig. S1 Clustering dendrogram of samples based on their gene expressions (TIFF 1.39 mb)
Online Resource 2
Fig. S2 Analysis of network topology for various soft-thresholding powers. The left panel shows the scale-free fit index (y-axis) as a function of the soft-thresholding power (x-axis). The right panel displays the mean connectivity (degree, y-axis) as a function of the soft-thresholding power (x-axis) (TIFF 1.08 mb)
Online Resource 3
Table S1 GO enrichment results of 5 stress-related modules (P-value > 0.05) (XLSX 43.8 kb)
Online Resource 4
Table S2 KEGG pathways enrichment results of 5 stress-related modules (P-value > 0.05) (XLSX 13.7 kb)
Online Resource 5
Table S3 The connectivity and annotation of top 2 % hub genes in 5 stress-related modules (XLSX 12.6 kb)
Rights and permissions
About this article

Cite this article
Zhao, X., Yu, H., Kong, L. et al. Gene Co-Expression Network Analysis Reveals the Correlation Patterns Among Genes in Euryhaline Adaptation of Crassostrea gigas . Mar Biotechnol 18, 535–544 (2016). https://doi.org/10.1007/s10126-016-9715-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10126-016-9715-7