
Abstract
The native Asian oyster, Crassostrea ariakensis is one of the most common and important Crassostrea species that occur naturally along the coast of East Asia. Molecular species diagnosis is a prerequisite for population genetic analysis of wild oyster populations because oyster species cannot be discriminated reliably using external morphological characters alone due to character ambiguity. To date there have been few phylogeographic studies of natural edible oyster populations in East Asia, in particular this is true of the common species in Korea C. ariakensis. We therefore assessed the levels and patterns of molecular genetic variation in East Asian wild populations of C. ariakensis from Korea, Japan, and China using DNA sequence analysis of five concatenated mtDNA regions namely; 16S rRNA, cytochrome oxidase I, cytochrome oxidase II, cytochrome oxidase III, and cytochrome b. Two divergent C. ariakensis clades were identified between southern China and remaining sites from the northern region. In addition, hierarchical AMOVA and pairwise Φ ST analyses showed that genetic diversity was discontinuous among wild populations of C. ariakensis in East Asia. Biogeographical and historical sea level changes are discussed as potential factors that may have influenced the genetic heterogeneity of wild C. ariakensis stocks across this region.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The native Asian oyster, Crassostrea ariakensis, is a large flat oyster that occurs naturally in intertidal areas and muddy creeks of warm water estuaries (An et al. 2006; Yoon et al. 2008), and is one of the most common and important Crassostrea species in Asia. It occurs naturally along the coast of eastern Asia (Wang et al. 2004) with the species’ ecological traits having been described in a number of previous studies (Zhou and Allen 2003). C. ariakensis, known locally as “Jinjiang” or “near-river” oyster (China), “Suminoe” or “Ariake” oyster (Japan) and “Kang-gul” oyster (Korea), has recently been the subject of intense scientific interest, with the focus in particular on its ecology, taxonomy and population genetics (Reece et al. 2008; Wang et al. 2008a; Xiao et al. 2010; Zhang et al. 2005). Organised field surveys of non-indigenous oyster species, primarily focusing on C. ariakensis, have also been conducted by the Virginia Institute of Marine Science (VIMS) USA since the late 1990s in an attempt to identify alternative culture species for the Eastern oyster, C. virginica (Zhou and Allen 2003). This is to address a continued decline of C. virginica numbers observed over the past decade in the USA (Wilberg et al. 2011). Previous studies of the taxonomy and geographical distribution of wild stocks of C. ariakensis have focused mainly on Japanese and Chinese populations owing to a lack of information and systematic data from other regional neighbours (Cordes et al. 2008; Zhang et al. 2005). Although several authors have described C. ariakensis’s distribution in Asia including in Korea (Cahn 1950; Coan et al. 2000), and have addressed C. ariakensis identity (Jung et al. 2006; Wang and Guo 2008a, b), few studies have been directed at assessments of population genetic structure of wild populations and hatchery stocks of C. ariakensis, and in particular, Korean stocks are poorly known. While Xiao et al. (2010) reported on the population genetic structure of C. ariakensis in Asia that included some Korean samples, the study was based on nuclear DNA (microsatellite) genetic markers, which can be inefficient for identifying deeper historical evolutionary relationships.
Due to the significant ecological and economic importance of C. ariakensis, attention has been directed to date at documenting the levels and patterns of genetic variation in wild C. ariakensis populations in East Asia and investigating historical and contemporary processes that have influenced population structure. Field observations suggest that C. ariakensis exists as three natural large and discrete populations in Korea (Yoon et al. 2008). Natural beds of C. ariakensis in Korea are also considered to have experienced substantial recent declines, attributed to over-harvesting and increased seawater flow into estuaries (Yoon et al. 2008). Existing studies that have applied molecular markers for species identification that include mitochondrial 16S rRNA (Kwun et al. 2013; Lee et al. 2000) and CO I (Wang et al. 2004), and nuclear 28S rRNA (Wang et al. 2008a), internal transcribed spacer regions (Kim et al. 2009), single nucleotide polymorphism (SNP) (Jung et al. 2006), microsatellites (Xiao et al. 2010), restriction fragment length polymorphism (RFLP) (Cordes et al. 2008) and random amplification of polymorphic DNA (Kim et al. 2009). Results suggest that a sharp genetic differentiation exists among C. ariakensis samples in East Asian wild populations, identified as northern (N-type) and southern (S-type) genetic groups (Xiao et al. 2010). To our knowledge the Korean C. ariakensis taxa have not been assessed using multiple mtDNA markers in any of the previous studies (i.e. limited only to 16S rRNA and CO I) that have documented the distribution of the two C. ariakensis clades in this region.
While highly polymorphic markers such as microsatellite and SNPs have been widely applied in oysters (Hubert and Hedgecock 2004; Varney et al. 2009) and have been used for population genetic studies in the past, these approaches suffer from the fact that they tend to be both time consuming and relatively expensive (Fleury et al. 2009; Quilang et al. 2007). Furthermore, it has been demonstrated that, high levels of non-amplification and PCR-null alleles are often problems in oysters (Xiao et al. 2010; Yu and Guo 2005). There is also a lack of expressed sequence tag (EST) data for this species (Kim et al. 2011). mtDNA has widely been used to assess population delineation because of the molecule’s simple non-recombinant structure, maternal mode of inheritance, and relatively rapid evolutionary rate (Obata et al. 2008), as well as the capacity to compare data with the ready availability of seven complete Crassostrea mitochondrial genomes (Wu et al. 2010). Knowledge of the ecology and biology of C. ariakensis in Asia, has been restricted largely due to confusion with nomenclature and difficulties with accurate identification of the species in the field (Reece et al. 2008; Xiao et al. 2010). The main aim of the current study therefore was to assess levels and patterns of molecular genetic variation in East Asian C. ariakensis populations using DNA sequence analysis of five mtDNA gene regions. Different mtDNA markers with slow to moderate rates of evolution can be used for species discrimination even at fine geographical scales of population genetic differentiation (Reeb and Avise 1990). Here we used concatenated data from five mtDNA markers (i.e. 16S rRNA, CO I, CO II, CO III, and Cyt b) that resulted in a 3,020 bp fragment used to discriminate the species status of samples collected and to describe natural population structure across the study area. Identification of genetic heterogeneity among C. ariakensis populations across this region and documentation of their relative levels of genetic diversity within and among wild populations will provide important biological information that can assist development of appropriate management strategies for the regional oyster mariculture industry where C. ariakensis is farmed or has been under consideration as a new culture species.
Materials and methods
Sampling area
A total of 89, C. ariakensis individuals, were collected from six locations as follows; Chinese samples from Fujian (FJ, n = 10) and Shandong (SD, n = 10) were provided by Prof. Ximing Guo at Rutgers University, Japanese samples from the Itoki River (IR, n = 8) were provided by Prof. Kimberly Reece at Virginia Institute of Marine Science, and Korean samples from the Seomjin River (SR, n = 20), Kawha Sacheon River (KSR, n = 21) and Kangwha-Do (KD, n = 20) were collected by the authors in 2003 (Fig. 1; Table 1). All tissue samples were stored in 95 % ethanol prior to DNA extraction.
DNA extraction and amplification
Total genomic DNA was extracted using a DNeasy tissue kit (Qiagen, Germantown, MD, Germany). Each partial fragment of mitochondrial DNA fragment was amplified using specific primer sets (Table 2). Initial primer sets for CO II, CO III and cytochrome b were designed based on the complete mitochondrial genome sequences of C. gigas (Ren et al. 2010) and C. virginica (Milbury and Gaffney 2005). Since none of the Chinese samples amplified successfully with these primers, new primers were designed based on the complete mitochondrial genome sequence of C. ariakensis (Wu et al. 2010). 30 μl PCR reaction volumes were employed containing 20 mM Tris–HCl (pH 8.3), 20 mM MgCl2, 100 mM KCl, 10 pmol of each primer, 200 µM of dNTPs, 0.5 units Ex-taq DNA polymerase (Takara, Otsu, Japan), and 50 ng of genomic DNA. PCR was performed in a PTC-220 thermal cycler (MJ Research, Waltham, MA, USA) programmed for 3 min at 95 °C, and 35 cycles of; 30 s at 94 °C, 30 s at 48–50 °C, and 30 s at 72 °C, with a final extension period of 10 min at 72 °C. Electrophoresis was visualized after staining with ethidium bromide to confirm clean amplification of each PCR product. PCR products were purified using Qiagen PCR purification kits. Purified products were used as templates directly in cycle sequencing reactions with dye-labeled terminators (Applied Biosystems, Foster City, CA, USA). Cycle sequencing reactions were performed in a PTC-100 thermocycler (MJ Research) according to the manufacturer’s protocol. Nucleotide sequences were obtained from both strands using an ABI 3130 Genetic Analyzer (Applied Biosystems).
Sequence data and alignment
Eighty-nine C. ariakensis individuals were sequenced for each mtDNA gene region (16S, CO I, CO II, CO III, and Cytb) using an ABI3130 Genetic Analyzer (Applied Biosystems). Sequence alignment was performed with Lasergene V. 10.1 (DNASTAR, Madison, WI). For clarity and convenience with analysis, first all sequences of each gene region were edited to identical lengths after alignment and then all sequences were concatenated for further analyses. All sequences were deposited in GenBank and the most common haplotype/sequence (accession number FJ743504 for 16S rRNA, FJ743527 for CO I, FJ743561 for CO II, FJ743582 for CO III, and FJ743572 for Cyt b) identified was designated as the reference sequence (not each sequence fragment but all five concatenated sequence fragments from the same individuals) for further genetic analyses.
Phylogenetic analyses
Only concatenated sequences were employed for phylogenetic, evolutionary network, and population structure analyses to infer the species’ phylogenetic status, evolutionary relationships among individuals, and genetic population structure, respectively, for Asian oyster samples across this region. To reconstruct phylogenetic trees, only gene fragments from complete mitochondrial genome sequences of Crassostrea species available from GenBank (Table 1) were applied as references. The Eastern oyster, C. virginica, was used as the outgroup in all phylogenies. Three different inference methods were employed to construct phylogenies for each marker region, namely neighbour-joining (NJ), maximum likelihood (ML) and Bayesian analyses (BA). NJ analyses were undertaken with MEGA 5 (Tamura et al. 2011) applying the Poisson method, uniform rates and pairwise deletion. Optimum DNA substitution model was selected as the Tamura-Nei plus Gamma model in Modeltest (Posada and Crandall 1998) but if necessary the next-most-complex model was applied in the ML and BA analyses. In addition, since the five molecular markers selected from different mitochondrial locations have heterogeneous rates of nucleotide substitution, a general likelihood-based mixture model of gene sequence evolution based on General Time Reversible model (GTR) (Pageland Meade 2004) was applied. ML tree building was performed with RAxML 7.0.3 (Stamatakis 2006). Non-parametric bootstrapping with 1,000 pseudoreplicates was used to estimate confidence intervals for tree topologies. BA analyses were performed using the Mr. Bayes 3.1.2 package (Ronquist and Huelsenbeck 2003).
Haplotype network
A median-joining network was generated for all unique mitochondrial haplotypes, identified using the program Network v. 4.6.1.1 (http://www.fluxus-engineering.com). Network files were generated from aligned DNA sequence data using the program DNA alignment (http://www.fluxus-engineering.com). Median-joining networks are commonly used to depict relationships among closely related mitochondrial or nuclear haplotypes for which traditional phylogenetic approaches yield multiple plausible trees (Bandelt et al. 1999). Haplotype networks reconstruct the genealogical history of haplotypic variation and illustrate the evolutionary relationship among unique haplotypes. MtDNA median joining network can provide information on the demographic and geographical history of a population including inference about past population expansions, and bottlenecks. Frequency and site information therefore were incorporated into the Asian oyster network here to illustrate the distribution of haplotypes among sampled locations.
Population genetic analyses
Genetic variation estimates were calculated using several standard diversity indices including the number of haplotypes (H), the number of polymorphic sites (S), halpotype (gene) diversity (Hd), and molecular diversity indices including nucleotide diversity (π) (Nei 1987), the average number of pairwise nucleotide differences (k) (Tajima 1983), and expected heterozygosity based on number of segregating sites (θS) (Watterson 1975) implemented in Arlequin v.3.11 (Excoffier et al. 2005) for the C. ariakensis sample collections. Population genetic analyses were conducted in Arlequin v.3.11 and DnaSP v.5.1 (Rozas et al. 2003). Differentiation among sites was estimated using the fixation index (Φ ST) (Excoffier et al. 1992) that includes information on mtDNA haplotype frequency (Weir and Cockerham 1984) and genetic distances among unique haplotypes applying the 10,000 iterations of a nonparametric bootstrap permutation procedure to test for significant differentiation among samples. For all Φ ST analyses, the Tamura and Nei (1993) distance method was used as it accounts for mutational rate heterogeneity among nucleotides within the gene fragment. In order to provide more detailed information about hierarchical population structure, first, an analysis of molecular variance (AMOVA) was performed for concatenated mtDNA data of the entire sample collection, with the allowed level of missing data set at 5 %. AMOVA, as implemented in Arlequin v.3.11, evaluates the pattern of molecular variation among groups within a species, taking into account sequence divergence of molecular haplotypes as well as their relative frequencies (Excoffier et al. 2005). Secondly, genetic variation between the clade containing northern population and the clade containing southern population was examined. Then the genetic variation between each pair of collections was examined via pair wise Φ ST analysis. As there was a discontinuous genetic variation even within the clade containing northern population, an AMOVA and a pairwise Φ ST analysis were carried out for the clade containing northern population only. The significance of variance components for each hierarchical comparison was tested using 1,000 permutations. Significance of pairwise site comparisons was tested using a permutation process, as above. In all instances with multiple tests, P values were adjusted using Bonferroni correction (Rice 1989). The pattern of genetic differentiation among sites was tested using a Mantel test (Mantel 1967; Smouse et al. 1986) employed in Arlequin v.3.11 to determine if there was a significant ‘isolation by distance’ (IBD) pattern.
Demographic analyses
Since neutrality tests can provide clues regarding population demographic events, sequence data sets were tested for deviation from neutral expectations using both Tajima’s D (Tajima 1989), and Fu’s FS (Fu 1997) tests implemented in Arlequin v.3.11 (Excoffier et al. 2005).
Results
Molecular diversity and neutral evolution
Sequence data (3,020 bp) from five mtDNA gene fragments (16S, CO I, CO II, CO III, and Cyt b) were obtained for 89 individuals represented in six sampled populations. Sampling sites, sample sizes, and GenBank accession numbers of haplotypes for each gene fragment are presented in Table 1. Molecular diversity and descriptive statistics for concatenated data from each sampling site and for each clade are detailed in Table 3.
Phylogenetic and evolutionary relationships
All 89 individuals screened in the study were subjected to phylogenetic analysis based on the concatenated mtDNA gene region to confirm that they were C. ariakensis with high bootstrap and posterior probability values (>99) separating them from other Crassostrea species. According to the ML, BA and NJ analyses, phylogenetic analysis of the concatenated mtDNA region compared with other Crassostrea species sequences available on GenBank confirmed that all individuals screened here were C. ariakensis, and that they constituted a monophyletic group (Fig. 2). In addition, the phylogenetic tree clearly separated populations geographically into the clade containing southern population (Fujian) and the clade containing northern population (remaining sites). The level of divergence between the two clades was significant and supported by high bootstrap and posterior probability values (>88) in all ML, BA and NJ analyses. Haplotype networks for concatenated mtDNA data also showed two genetic clades consisting of the Fujian samples as one clade (southern), and remaining sample sites forming a second clade (northern) with producing a star shaped haplotype network. Figure 3 presents the haplotype network for concatenated mtDNA data.
Population structure
Hierarchical AMOVA analysis was carried out using Tamura and Nei’s (1993) genetic distance, (i) for the entire collection, (ii) for the clade containing northern population and the clade containing southern population, and (iii) for the clade only containing northern population. AMOVA analysis for the entire collection shows a significant genetic structure over the entire collection area (Table 4a; F ST = 0.642, P < 0.001). Table 4b shows that there was significant genetic variation for hierarchical grouping of the ‘clade group containing northern population’ and ‘clade group containing southern population’ (F CT = 0.839, P < 0.001), and also among populations within groups (F SC = 0.869, P < 0.001). In addition, significant genetic differentiation was observed for sample populations within the clade containing northern population (Table 4c; F CT = 0.199, P < 0.001). Pairwise Φ ST analyses were conducted applying Tamura and Nei’s (1993) genetic distance (i) for the entire collection (Table 5a), and (ii) for the clade containing northern population only, to examine genetic variation within the clade containing northern population (Table 5b). While the Fujian site was highly differentiated from all other sites, IR, SR and KSR sites were differentiated from most “clade containing northern population” sites (Table 5a). The SR site was the most differentiated site among northern sampled populations (Table 5b) which was also observed in the median-joining haplotype network (Fig. 3). While a Mantel test was carried out to test whether the genetic variation showed a pattern of isolation by distance (IBD), no significant IBD pattern was observed (R2 = 0.425, P = 0.139).
Population history and demographic patterns
Fu’s Fs and Tajima’s D, neutrality tests results for each clade and sample collections are presented in Table 6. Neutrality tests showed high negative Fu’s Fs and Tajima’s D values with significant P values for the clade containing northern population, while the clade containing southern population showed only low and non-significant Fu’s Fs and Tajima’s D values (Table 6). This suggests that the clade containing northern population has undergone a sudden population expansion in recent evolutionary time. In a similar way, some northern sample populations, including KD, KSR, and SR appear to have experienced recent population expansions. Mismatch distribution analyses provide support for these population expansions. The mismatch distribution including both clades was bimodal (Fig. 4a).The mismatch distribution for the clade containing northern population was examined to test whether there had been a recent population expansion within this clade. A single smooth peak of pairwise differences suggests that there had been a recent population expansion within the clade containing northern population (Fig. 4b). Furthermore, the haplotype network also showed a star-like pattern around the central common haplotype in the clade containing northern population, adding further support for suggestions for a recent population expansion (Fig. 3).
Discussion
Phylogenetic and evolutionary relationships
Diagnosis of species using molecular markers is often a prerequisite for population genetic analysis of wild oyster populations because many oyster species cannot be easily discriminated reliably using external morphological characters alone due to a lack of key discriminating characters (Reece et al. 2008). Here we confirmed the species status of all of our sampled individuals via phylogenetic analysis applying concatenated mtDNA data from five different mtDNA gene regions. Phylogenetic analyses confirmed consistently that all samples examined here were C. ariakensis and this finding was consistent with previous molecular taxonomic studies that have reported clear relationships among Asian Crassostrea oysters (Reece et al. 2008; Wang et al. 2008a; Zhou and Allen 2003). Concatenated mtDNA data from multiple mtDNA gene regions produced a robust data set for intraspecific population structure assessment.
Phylogenetic analyses clearly show that C. ariakensis in the study region consisted of two divergent clades (Fujian site-clade containing southern population, and remaining sites-clade containing northern population) with high bootstrap values. The clade containing northern population and the clade containing southern population have been detected in previous studies of the same species based on 16S and CO I gene sequences (Wang et al. 2004), RFLP profiles of mtDNA and nuclear DNA (Zhang et al. 2005) microsatellites (Xiao et al. 2010). Previous studies have suggested that long term isolation and subsequent population subdivision have produced structure between the two geographical groups, and further, have suggested that a major biogeographical barrier may exist around the Yangzi River estuary (Wang et al. 2004). Large fresh water plumes from the Yangzi River in the summer potentially may create a barrier to dispersal north and south of this area for local bivalve species (Xu 1997). The northern Yellow Sea is characterized by cold, low salinity water compared with warmer and more saline water to the south in the East China Sea (Xiao et al. 2010). An alternative explanation for the apparent sharp genetic boundary between the clade containing northern population and the clade containing southern population involves sea level changes in the paleo-East China and Yellow Seas, which is described in more detail below.
Population genetic structure
Levels and patterns of genetic differentiation were assessed among wild samples of C. ariakensis collected from six sites in China, Korea, and Japan. Significant genetic differentiation was evident between the clade containing northern population and the clade containing southern population, and also among sample populations within the clade containing northern population, confirming genetic heterogeneity of C. ariakensis across this region. It is widely accepted that the Tsuchima current (TC) is a major driver producing discrete population demographic effects on aquatic species in East Asia and has two sources (Cho et al. 2013); one is a branch of the Kurioshio Current (KC) that flows on shore across the shelf break in the East China Sea (Lie et al. 1998) and the other is a continuation of the Taiwan Warm Current (TWC) that originates in the Taiwan Strait and enters the Korea Strait (KS) as the TC (Zhu et al. 2004). Hierarchical AMOVA and pairwise Φ ST analyses further showed that the Fujian population was significantly genetically differentiated from all other C. ariakensis sampled populations. Potentially, the Yangzi River estuary oscillation may influence the phylogenetic and evolutionary relationships and produce a sharp genetic boundary among C. ariaknesis populations examined here identified as the clade containing northern population vs the clade containing southern population. In addition, the IR site in Japan, SR and KSR sites at the southern tip of Korea were significantly genetically differentiated from most other sites within the clade containing northern population as shown by pairwise Φ ST analyses. While the overall genetic pattern of C. ariakensis across the three regions (China, Korea and Japan), may involve the KC and TC currents having a major influence, the local KS current may also play a role as has been suggested for a number of aquatic organisms studied across these regions (Kang et al. 2012; Kwan et al. 2012; Yang et al. 2009). The KD and SD sites are situated within a closed bay area (Fig. 1), and were not significantly genetically differentiated. The close genetic relationship between the two sites can potentially be explained by a hydrographic influence from the current in the Yellow Sea (Yang et al. 2009). On the western coast of the Korean Peninsula, the Yellow Sea Warm Current, a branch of the Tsushima Current, flows counter clockwise to the western coast of the Korean Peninsula and towards the entrance to the Gulf of Bohai (Pang et al. 1992). Water current patterns within the bay essentially create a closed eddy, and C. ariakensis in this area may be locally adapted to a cold, low saline water mass within the bay that is separated from the relatively warmer, high saline water mass outside of the bay area (An et al. 2013). Thus, C. ariakensis at the KD and SD sites may be essentially homogenized with larvae largely retained within the bay due to the closed circulation pattern. This hypothesis is also consistent with previous findings for pelagic larvae of other aquatic organisms (i.e. fish and octopus) in this geographical region that are often influenced by ocean currents (Kang et al. 2012; Wang et al. 2008b).

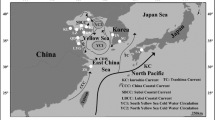
Sampling locations for C. ariakensis in Korea: (1) Seomjin River [SR], (2) KawhaSacheon River [KSR], (3) Kangwha-Do [KD], (4) Itoki River [IR], (5) Shandong [SD] and (6) Fujian [FJ]. Generated from Google Earth Map

Maximum likelihood phylogenetic tree of five concatenated mtDNA fragments. The numbers above the internal branches represent percent bootstrap support values (below 70 marked as either not presented or dash) based on 1,000 pseudoreplicates by the maximum likelihood method, the posterior probability with Bayesian method, and neighbour-joining distance

Median-joining network of C. ariakensis haplotypes for five concatenated mtDNA gene regions showing the evolutionary relationships among haplotypes. Each circle represents a unique haplotype in the sample, and the size of each circle represents the relative frequency of each haplotype. Colours and their percentage in each circle represent the presence of each haplotype at different sites and their relative abundance at each site. Red small circles indicate a missing haplotype. Lines connecting haplotypes represent single mutations unless otherwise indicated in underline parentheses (Blue-KSR, Black-KD, Yellow-SD, Green-IR, Gray-SR, and Pink-FJ)

a Observed and expected population model mismatch distribution for all pairwise combinations of C. ariakensis for the entire northern and southern concatenated mtDNA data set. b Observed and expected population model mismatch distribution for all pairwise combinations of C. ariakensis northern type concatenated mtDNA data set
While the SR and KSR sites in Korea are in close proximity compared with geographical distances between other sites, they were significantly genetically differentiated from each other, and with other sites as well, as indicated in pairwise Φ ST analysis. Genetic differentiation evident between the geographically close SR and KSR sites can potentially be explained by geographical characteristics and associated ecological attributes of the two sites and/or paleo sea level changes across this region. The SR site is directly open to the bay area and the sea while the KSR site is isolated from the open ocean (Yoon et al. 2008). Thus, the geographical arrangement of these two sites may restrict larval dispersal between the two sites. A previous genetic study of C. ariakensis samples that included samples from the same KSR and SR sites, and assessed using microsatellite markers had reported genetic differentiation between these two sites, and also with other sites (Xiao et al. 2010). C. ariakensis populations in South-East Asia therefore show a pattern of patchy genetic population structure that is not associated with isolation by distance effect. Similar discontinuous patchy distributions of genetic differentiation have been reported in the same species in China (Guo et al. 2006), and also for other estuarine bivalve species across this region. For example, the Kumamoto oyster (C. sikamea) sampled across the same geographical region showed discontinuous, high genetic differentiation among sites (Sekino et al. 2012). The adult phase of oysters is sedentary, with their pelagic larvae requiring 2–3 weeks duration before they must settle (Thorson 1950). This is considered to be one of the major factors that can influence patterns of dispersal and produce genetic population structure (DeWoody and Avise 2000). In particular, contemporary events involving Yangzi River estuary freshwater plumes in summer during the spawning season of C. ariakensis from June until September across East Asia (Bushek et al. 2006; Yoo et al. 2004) could influence movement patterns of pelagic larvae of this species that may use the Yangzi River event along with connected currents in this region. Given however, results here and those for other estuarine bivalves with similar life history traits (Sekino et al. 2012), it is likely that local larval retention (Heads 2005) may influence the observed discontinuous, patchy population structure distributions with elevated F ST values. Thus, genetic isolation among populations, which allows enhanced genetic drift of mtDNA as proposed in teleost fishes (Liu et al., 2006) may also impact distributions of genetic diversity in C. ariakensis across the sampled regions. The pattern of genetic differentiation in C. ariakensis (and other estuarine bivalve species in this region) may also potentially be associated with geographical history of the ocean(s). The majority of sea floors in the paleo-Eastern China Sea and Yellow Sea (<200 m in depth) were either dry land or very shallow sea areas during the last glacial maxima (Lambeck et al. 2002) because of lowered sea levels estimated at 15 kyr BP (Xiao et al. 2004). Isolated river mouths and estuaries during lower sea levels may have contributed to genetic differentiation among sites.
Previous studies analyzed only a single mtDNA fragment (either 16S or CO I) or only microsatellite data (Xiao et al. 2010). While the sample size here could limit the current study’s ability to draw a definitive conclusion, the current mtDNA phylogeographic survey was the first conducted using multiple mtDNA fragments and, thus, should improve our understanding of the processes underlying the spatial and temporal dimensions of genetic variation in C. ariakensis, and also verified previous results (presence of two distinct clades) obtained from a microsatellite phylogeographic study (Xiao et al. 2010). Since the genetic and phylogeographic relationships observed for mitochondrial and nuclear gene fragments often differ, comparing the current mtDNA data with available results from microsatellite data (Xiao et al. 2010) should improve our understanding of intraspecific structure, demographic history and gene flow patterns of C. ariakensis belonging to different geographical and biogeographical regions. Therefore, the consensus between the current study and the microsatellite inference of Xiao et al. (2010), together with our implied agreement with regard to potential explanations of the observed pattern (i.e., fresh water plumes from the Yangzi River and sea level changes in the paleo-East China and Yellow Seas), provides the most comprehensive phylogeographic investigation of C. ariakensis, to date. A lack of comprehensive information about C. ariakensis identification, abundance, distribution and genetic resources could potentially hinder successful conservation and development of appropriate management practices for Asian oysters in the future. However, a broader scale analysis of the extent of genetic differentiation in C. ariakensis undertaken across the whole natural geographical distribution, including distant areas in China and Japan would assist long term conservation of this important local aquatic resource. In addition, development of additional molecular markers that possess different properties and sensitivity (mitochondrial and nuclear DNA) will be required to fully understand the overall pattern of genetic structure in the C. ariakensis across this region.
Limitations and contributions of the current study
While a few earlier studies had indicated presented two distinct clades (the clade containing northern population and the clade containing southern population) of C. ariakensis using mitochondrial and nuclear markers, the current investigation is the first population genetic study applying multiple mtDNA fragments and a concatenated approach to confirm the genetic discontinuity identified earlier from microsatellite markers. In combination mtDNA and nuclear datasets, provide basic biological information for developing effective management strategies for oyster mariculture industry across this region of northern Asia.
References
An YK, Yoon HS, Choi SD (2006) Effects of temperature, salinity on the growth of Crassostrea ariakensis in Seomjin River. Korean J Environ Biol 24:60–66
An HS, Lee JW, Park JY, Jung HT (2013) Genetic structure of the Korean black scraper Thamnaconus modestus inferred from microsatellite marker analysis. Mar Biol Rep 40:3445–3456
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Bushek D, Kornbluh A, Debrosse G, Wang H, Guo X (2006) Oyster sex wars: evidence for a gamete sink if Crassostrea virginica and Crassostrea ariakensis spawn synchronously. J Shellfish Res 25:716
Cahn AR (1950) Oyster culture in Japan (Fishery leaflet). Fish and Wildlife Service, US
Cho YK, Seo GH, Kim CS, Choi BK, Shaha DC (2013) Role of wind stress in causing maximum transport through the Korea Strait in autumn. J Mar Syst 115–116:33–39
Coan EV, Scott PV, Bernard FR (2000) Bivalve seashells of western North America: marine bivalve mollusks from Arctic Alaska to Baja California. Santa Barbara Museum of Natural History, Santa Barbara
Cordes JF, Xiao J, Reece KS (2008) Discrimination of nine Crassostrea oyster species based upon restriction fragment-length polymorphism analysis of nuclear and mitochondrial DNA markers. J Shellfish Res 27:1155–1161
DeWoody JA, Avise JC (2000) Microsatellite variation in marine, freshwater and an adromous fishes compared with other animals. J Fish Biol 56:461–473
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Larval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Fleury E, Huvet A, Lelong C, de Lorgeri J, Boulo V, Gueguen Y et al (2009) Generation and analysis of a 29,745 unique expressed sequence tags from the Pacific oyster (Crassostrea gigas) assembled into a publicly accessible database: the Gigas Database. BMC Genom 10:341
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Fu YX (1997) Statistical tests for neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Guo X, Zhang G, Qian L, Wang H, Liu X, Wang A (2006) Oysters and oyster farming in China: a review. J Shellfish Res 25:734
Heads M (2005) Towards a panbiogeography of the seas. Biol J Linn Socy 84:675–723
Hubert S, Hedgecock D (2004) Linkage maps of microsatellite DNA markers for the Pacific oyster Crassostrea gigas. Genetics 168:351–362
Jung H, Kim WJ, Gaffney PM (2006) Development of single nucleotide polymorphisms (SNPs) in Crassostrea ariakensis and related Crassostrea species. J Shellfish Res 25:742
Kang JH, Park JY, Choi TJ (2012) Genetic differentiation of octopuses from different habitats near the Korean Peninsula and eastern China based on analysis of the mDNA cytochrome C oxidase 1 gene. Genet Mol Res 11:3988–3997
Kim WJ, Lee JH, Kim KK, Kim YO, Nam BH, Kong HJ, Jung HT (2009) Genetic relationships of four Korean oysters based on RAPD and nuclear rDNA ITS sequence analyses. Korean J Malacol 25:41–49
Kim WJ, Jung H, Gaffney PM (2011) Development of type I genetic markers from expressed sequence tags in highly polymorphic species. Mar Biotechnol 13:127–132
Kwan YS, Song HK, Lee HJ, Lee WO, Won YJ (2012) Population genetic structure and evidence of demographic expansion of the Ayu (Plecoglossus altivelis) in East Asia. Anim Syst Evol Div 28:279–290
Kwun HJ, Song YS, Myoung SH, Kim JK (2013) Two new records of juvenile Oedalechilus labiosus and Ellochelon vaigiensis (Mugiliformes: Mugilidae) from Jeju island, Korea, as revealed by molecular analysis. Fish Aquat Sci 16:109–116
Lambeck K, Esat TM, Potter EK (2002) Links between climate and sea levels for the past three million years. Nature 419:199–206
Lee SY, Park DW, An HS, Kim SH (2000) Phylogenetic relationship among four species of Korean oysters based on mitochondrial 16S rDNA and COI gene. Korean J Syst Zool 162:203–211
Lie HJ, Cho CH, Lee JH, Niller P, Hu JH (1998) Separation of the Kuroshio water and its penetration onto the continental shelf west of Kyushu. J Geophys Res 103:2963–2976
Liu JX, Gao TX, Yokogawa K, Zhang YP (2006) Differential population structuring and demographic history of two closely related fish species, Japanese Sea bass (Lateolabrax japonicus) and spotted sea bass (Lateolabrax maculatus) in Northwestern Pacific. Mol Phylogenet Evo 39:799–811
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Can Res 27:209–220
Milbury CA, Gaffney PM (2005) Complete mitochondrial DNA sequence of the eastern oyster Crassostrea virginica. Mar Biotech 7:697–712
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Obata M, Shimizu M, Sano N, Komaru A (2008) Maternal inheritance of mitochondrial DNA (mtDNA) in the Pacific oyster (Crassostrea gigas): a preliminary study using mtDNA sequence analysis with evidence of random distribution of MitoTracker-stained sperm mitochondira in fertilized eggs. Zool Sci 25:248–254
Pagel M, Meade A (2004) A phylogenetic mixture model for detecting pattern heterogenetity in gene sequence or character-state data. Syst Biol 53:571–581
Palumbi SR (1994) Genetic divergence, reproductive isolation, and marine speciation. Annu Rev Ecol Syst 25:547–572
Pang IC, Rho HK, Kim TH (1992) Seasonal variation of water mass distributions and their causes in the Yellow Sea, the East China Sea and the adjacent seas of Cheju Island. Bull Korean Fish Soc 25:151–163
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Quilang J, Wang S, Li P, Abernathy J, Peatman E, Wang Y, Wang L, Shi Y, Wallace R, Guo X, Liu Z (2007) Generation and analysis of ESTs from the eastern oyster, Crassostrea virginica Gmelin and identification of microsatellite and SNP markers. BMC Genom 8:157
Reeb CA, Avise JC (1990) A genetic discontinuity in a continuously distributed species: mitochondrial DNA in the American oyster, Crassostrea virginica. Genetics 124:397–406
Reece KS, Cordes JF, Stubbs JB, Hudson KL, Francis EA (2008) Molecular phylogenies help resolve taxonomic confusion with Asian Crassostrea oyster species. Mar Biol 153:709–721
Ren J, Liu X, Jiang F, Guo X, Liu B (2010) Unusual conservation of mitochondrial gene order in Crassostrea oysters: evidence for recent speciation in Asia. BMC Evol Biol 10:394
Rice WJ (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rozas J, Sanchez-Delbarro JC, Messegue X, Rozas R (2003) DnaSp, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Sekino M, Sato S, Hong JS, Li Q (2012) Contrasting pattern of mitochondrial population diversity between an estuarine bivalve, the Kumamoto oyster Crassostrea sikamea, and the closely related Pacific oyster C. gigas. Mar Biol 159:2757–2776
Smouse PE, Long JC, Sokal RR (1986) Multiple regression and correlation extensions of the Mantel test of matrix correspondence. System Zoo 35:627–632
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Tamura K, Peterson D, Peterson N, Nei G, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Thorson G (1950) Reproductive and larval ecology of marine bottom invertebrates. Biol Rev 25:1–45
Varney RL, Galindo-Sánchez CE, Cruz P, Gaffney PM (2009) Population genetics of the eastern oyster Crassostrea virginica (Gmelin, 1791) in the Gulf of Mexico. J Shellfish Res 28:855–864
Wang H, Guo X (2008a) Identification of Crassostrea ariakensis and related oysters by multiplex species-specific PCR. J Shellfish Res 27:481–488
Wang H, Guo X (2008b) ITS length polymorphism in oyster and its use in species identification. J Shellfish Res 27:489–494
Wang H, Guo X, Zhan G, Zhang F (2004) Classification of jinjiang oysters Crassostrea rivularis (Gould, 1861) from China, based on morphology and phylogenetic analysis. Aquaculture 242:137–155
Wang H, Zhang G, Liu X, Guo X (2008a) Classification of common oysters from North China. J Shellfish Res 27:495–504
Wang M, Zhang X, Yang T, Han Z, Yanagimoto T, Gao T (2008b) Genetic diversity in the mtDNA control region and population structure in the Sardinella zunasi Bleeker. African J Biotechnol 7:4384–4392
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Pop Biol 7:256–276
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wilberg MJ, Livings ME, Barkman JS, Morris BT, Robinson JM (2011) Overfishing, disease, habitat loss, and potential extirpation of oysters in upper Chesapeake Bay. Mar Ecol Prog Ser 436:131–144
Wu XY, Xu XD, Yu ZN, Wei ZP, Xia JJ (2010) Comparison of seven Crassostrea mitogenomes and phylogenetic analyses. Mol Phylogenet Evol 57:448–454
Xiao S, Li A, Jiang F, Li T, Wan S, Huang P (2004) The history of the Yangtze River entering sea since the Last Glacial Maxima: a review and look forward. J Coast Res 20:599–604
Xiao J, Cordes JF, Wang H, Guo X, Reece KS (2010) Population genetics of Crassostrea ariakensis in Asia inferred from microsatellite markers. Mar Biol 157:1767–1781
Xu F (1997) Bivalves from China Seas. Science Press, Beijing
Yang EC, Lee SY, Lee WJ, Boo SM (2009) Molecular evidence for recolonization of Ceramium japonicum (Ceramiaceae, Rhodophyta) on the west coast of Korea after the last glacial maximum. Bot Mar 52:307–315
Yoo SK, Lim HK, Jang YJ (2004) Growth and spawning of Crassostrea rivularis from the southern sea of Korea. Korean J Malacol 20:131–134
Yoon HS, Jung H, Choi SD (2008) Suminoe oyster (Crassostrea ariakensis) culture in Korea. J Shellfish Res 27:505–509
Yu Z, Guo X (2005) Genetic analysis of selected strains of eastern oyster (Crassostrea virginica Gmelin) using AFLP and microsatellite markers. Mar Biotechnol 6:575–586
Yu Z, Wei Z, Kong X, Shi W (2008) Complete mitochondrial DNA sequence of oyster Crassostrea hongkongensis: a case of “Tandem duplication random loss” for genome rearrangement in Crassostea? BMC Genom 9:477
Zhang Q, Allen SK, Reece KS (2005) Genetic variation in wild and hatchery stocks of Suminoe oyster (Crassostrea ariakensis) assessed by PCR-RFLP and microsatellite markers. Mar Biotech 7:1–13
Zhou M, Allen SK (2003) A review of published work on Crassostrea ariakensis. J Shellfish Res 22:1–20
Zhu J, Chen C, Ding P, Li C, Lin H (2004) Does the Taiwan warm current exist in winter? Geophys Res Lett 31:L12302
Acknowledgments
The authors would like to acknowledge the help provided by Dr. Peter Mather, Dr. David Hurwood and Dr. Patrick Gaffney in regard to constructive comments and valuable suggestions on the manuscript. The authors also thank Dr. Ximing Guo and Dr. Kimberly Reece for providing priceless samples from China and Japan, and Dr. Patrick Gaffney for sequencing of a few Chinese and Japanese samples. This study was supported by Korea Science and Engineering Foundation (Grant No. 2005-215-F00009), by a partial grant from the National Fisheries Research and Development Institute (RP-2014-BT-013), and by a partial grant from the Ministry of Maritime Affairs and Fisheries (Technical Development for Higher Productivity of Kang-gul), Korea.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
W.-J. Kim, S. T. Dammannagoda and H. Jung contributed equally to this work.
Rights and permissions
About this article

Cite this article
Kim, WJ., Dammannagoda, S.T., Jung, H. et al. Mitochondrial DNA sequence analysis from multiple gene fragments reveals genetic heterogeneity of Crassostrea ariakensis in East Asia. Genes Genom 36, 611–624 (2014). https://doi.org/10.1007/s13258-014-0198-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-014-0198-5