
Abstract
Direct combustion of sulfur-enriched liquid fuel oil causes sulfur oxide emission, which is one of the main contributors to air pollution. Biodesulfurization is a promising and eco-friendly method to desulfurize a wide range of thiophenic compounds present in fuel oil. Previously, numerous bacterial strains from genera such as Rhodococcus, Corynebacterium, Gordonia, Nocardia, Mycobacterium, Mycolicibacterium, Paenibacillus, Shewanella, Sphingomonas, Halothiobacillus, and Bacillus have been reported to be capable of desulfurizing model thiophenic compounds or fossil fuels. In the present study, we report a new desulfurizing bacterium, Tsukamurella sp. 3OW, capable of desulfurization of dibenzothiophene through the carbon–sulfur bond cleavage 4S pathway. The bacterium showed a high affinity for the hydrocarbon phase and broad substrate specificity towards various thiophenic compounds. The overall genome-related index analysis revealed that the bacterium is closely related to Tsukamurella paurometabola species. The genomic pool of strain 3OW contains 57 genes related to sulfur metabolism, including the key dszABC genes responsible for dibenzothiophene desulfurization. The DBT-adapted cells of the strain 3OW displayed significant resilience and viability in elevated concentrations of crude oil. The bacterium showed a 19 and 37% reduction in the total sulfur present in crude and diesel oil, respectively. Furthermore, FTIR analysis indicates that the oil's overall chemistry remained unaltered following biodesulfurization. This study implies that Tsukamurella paurometabola species, previously undocumented in the context of biodesulfurization, has good potential for application in the biodesulfurization of petroleum oils.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The three primary fossil fuels are coal, petroleum oil, and natural gas. After food, fossil fuels constitute mankind’s most essential source of energy. Among these, the most prevalent fossil fuel is oil, which accounts for around 40% of the total worldwide use, followed by coal (24%) and natural gas (22%) (Gupta et al. 2005). The existence of sulfur compounds is one of the leading causes of pollution generated by the combustion of petroleum oils. The sulfur content of crude oil ranges from 0.03 to 7.89% depending on its source (Kilbane II and Le Borgne 2004). Aliphatic and aromatic thiols and disulfides (their oxidation products), thioethers, heterocyclic compounds, i.e., thiophenes, benzothiophene, dibenzothiophene, and their substituted derivatives, and various other complex molecules are reported to be found in crude oil fractions. Amongst organic sulfur content, the thiophenic form is the most abundant, and alkyl-substituted DBTs and BTs are found in the highest proportion of organosulfur compounds (Kilbane II and Le Borgne 2004; Monticello and Finnerty 1985; Carvajal et al. 2017; Saleh 2020). The conventional hydrodesulfurization (HDS) process is inefficient in removing sulfur from some highly HDS-resistant thiophenic compounds like benzothiophene, dibenzothiophene, and their alkyl-substituted derivatives. Therefore, there is a dire need to explore more powerful strategies with the potential to combat the destructive effects of sulfur compound consumption and their by-products.
A potential complementary alternative to HDS is biodesulfurization (BDS) which offers a high degree of selectivity while operating at mild temperature and pressure conditions. In comparison to HDS, an industrial-scale BDS is considered to have lower operating costs as it can reduce energy requirements. Furthermore, CO2 emissions are estimated to decrease by up to 70–80% (Debabov 2010), while requiring 50% less capital costs to set up the process (Bachmann et al. 2014). In BDS, specific microorganisms having efficient thiophenic sulfur-extracting capabilities are utilized. These bacteria harness a carbon–sulfur (C–S) bond cleaving metabolic pathway called the “4S” pathway (known as 4S because of the four sulfur-containing intermediates, i.e., sulfoxide–sulphone–sulfinate–sulphite) which is controlled by three plasmid-born genes dszABC (encoding for monooxygenase and desulfinase) and one chromosomal gene dszD (encoding for oxidoreductase) (Piddington et al. 1995; Aggarwal et al. 2013). The biodesulfurization trait has been found in various microorganisms of which Rhodococcus erythropolis IGTS8 (ATCC 53968) is the first microorganism known to possess BDS activity and is thoroughly studied (Gray et al. 1996). Other bacterial strains showing BDS activity belong to Corynebacterium, Gordonia, Nocardia, Mycobacterium, Paenibacillus, Shewanella, Sphingomonas, Halothiobacillus, and Bacillus genera (Parveen et al. 2020). The sulfur-removal capacity of the published bacterial species against thiophenic compounds varies; most of the bacterial species can desulfurize either DBTs or BTs, while only a few species are capable of desulfurizing both BTs and DBTs (Aggarwal et al. 2013; Tanaka et al. 2002; Alves et al. 2005; Kilbane II 2006; Wang et al. 2013; Akhtar et al. 2016; Akram et al. 2021).
Biodesulfurization technology has not yet been able to meet the standards for its industrial-scale applications and the rate of desulfurization is one of the major hurdles in this respect. This fact urges for the acquisition of an efficient biocatalyst. Isolation of new active microbial strains following sulfur-specific 4S pathway having highly hydrophobic cell surfaces and wide substrate specificity could be helpful to speed up the desulfurization process (Chen et al. 2021). Similarly, the bacteria that can tolerate and survive in the two-phase oil–water reaction system are also potential candidates for the development of an efficient desulfurization process (Mohamed et al. 2015). In the present study, a new C–S bond cleaving bacterium Tsukamurella sp. (coded as 3OW) was first characterized, and the pathway of sulfur removal from DBT was validated using genomic, chromatographic, and mass spectrometric techniques. The bacterium hydrophobicity and substrate specificity towards various thiophenic sulfur-containing compounds were evaluated. Moreover, crude oil was used to evaluate the hydrocarbon tolerance potential of the bacterium. Finally, the bacterium was exposed to desulfurize actual forms of petroleum oil, laying the groundwork for its potential utilization in future biodesulfurization processes.
Materials and methods
Microorganisms and culture media used
The dibenzothiophene desulfurizing bacterial isolate Tsukamurella sp. (designated as 3OW) isolated from an oil-contaminated soil sample was used in the current study. The bacterial isolation and culturing were carried out in a mineral glucose (MG) medium as described earlier (Akram et al. 2021). The dibenzothiophene (DBT) was used as an exclusive source of thiophenic sulfur for bacterial growth.
Composition and procedure to prepare MG medium
Commercially available high-quality chemicals (manufactured by Bio Basic Inc., TCI, Merck, Riedel-de Haen, MP Biomedicals, Acros Organics, Sigma Aldrich, etc.) were used to prepare the MG medium. The MG medium is composed of mineral salt solution, glucose, metal solution, and vitamin mixture, supplemented with dibenzothiophene to a final concentration of 0.2 mM. The mineral salt solution contained 2.0 g/L potassium phosphate (monobasic) KH2PO4, 4.0 g/L potassium phosphate (dibasic) K2HPO4, 1.0 g/L ammonium chloride (NH4Cl), 0.2 g/L magnesium chloride (MgCl2), 5.0 g/L glucose, 10.0 mL/L metal solution, and 1.0 mL/L vitamin solution (Akhtar et al. 2009). The pH of the mineral salt solution was adjusted with NaOH or HCl solution. The mineral salt and glucose solutions were sterilized by autoclaving at standard conditions of temperature and pressure (121 °C and 15 psi) for 20 min. The metal and vitamin solutions were filter sterilized by using a vacuum filtration assembly (LF30, Rocker Scientific, Taiwan) equipped with a 0.2-μm nylon membrane filter. After filter sterilization, the solutions were kept at 4 °C in pre-autoclaved reagent bottles of suitable size. In a laminar airflow hood, mineral salt solution and glucose solution were mixed under aseptic conditions and then vitamin and metal solutions were pipetted to the mixture. The medium was transferred to pre-autoclaved 250-mL Erlenmeyer flasks for further experiments. A suitable volume of the already prepared stock solution of DBT (50 mM in ethanol) was poured aseptically as the sole thiophenic sulfur source to revive the culture. To prepare a solid MG medium, 0.75% of Gellan gum was added to it.
Analytical procedures for the determination of the DBT desulfurization metabolites
Metabolites of the DBT desulfurization were explored by HPLC (Dionax 3000 Ultimate, Thermofisher, Germany) and LC/MS (Linear Ion Trap Mass Spectrometer; Thermo Scientific LTQ XL, USA) analyses. For the extraction of DBT metabolites, 50 mL of the growing culture was taken out, and the pH was adjusted to 2.0 by the addition of 6 N HCl. The acidified culture broth was centrifuged, and the cell-free supernatant was transferred to a separating funnel. The extraction was carried out with half of the volume of ethyl acetate, and the extract was passed through a 0.2-μm pore-size PTFE filter to remove the unwanted materials. In HPLC, the elution was performed with acetonitrile/water (60:40 v/v) as mobile phase at the flow rate of 0.6 mL/min, and the metabolites’ peaks were detected at 245 nm. For LC/MS analysis, the filtered extract was first concentrated/dried by a rotary evaporator. The sample was prepared using HPLC-grade methanol and then analyzed in electrospray ionization-mass spectrometry (ESI-MS) with a spray voltage of 3.7 KV in negative and positive modes. The sheath and auxiliary gas flow rates were adjusted at 20 and 5 arbitrary units, respectively. The capillary temperature was 280 °C. The data was collected in a mass scan range of 50–500 m/z.
Evaluation of bacterium substrate specificity and hydrophobicity
The substrate specificity of the new strain was evaluated by growing the cells in a 0.2 mM concentration of different alkylated and non-alkylated forms of Benzothiophene (BT) and Dibenzothiophene (DBT). The 50 mM stock solutions of the thiophenic compounds were prepared in ethanol, and an appropriate volume of the stock solution was added to the media to get a final concentration of 0.2 mM. The pH of the growing cultures was measured using a multiparameter analyzer (InoLab IDS Multi 9430, WTW, Germany). The growth was measured using a double-beam spectrophotometer (Camspec MC50, UK) by taking the OD of the culture broth at 660 nm. The Gibbs assay was used for the estimation of the 2-HBP/2-HBP equivalent (eq) in the culture broth (Akhtar et al. 2009). The glucose estimation was done by Dinitrosalicyclic acid (DNS) colorimetric method as follows: The growing culture was centrifuged at 10,000 rpm for 10 min to remove the cells. Subsequently, 3 mL of 1% DNS reagent was added to 3 mL of the cell-free supernatant in a lightly capped glass test tube. The contents were heated at approximately 90 °C in a water bath for 5–15 min until a red-brown color was developed. Following this, 1 mL of a 40% potassium sodium tartrate solution was added to stabilize the color. The resulting mixture was then cooled to room temperature, and the absorbance was recorded using a spectrophotometer (Camspec MC50, UK) at 575 nm. The concentration of glucose was determined by comparing the OD values with a standard glucose curve.
The bacterial hydrophobicity was tested by the microbial adhesion to hydrocarbons (MATH) test. The strain was cultured in MG medium until the mid-logarithmic phase (48-h growth), centrifuged, and washed (twice) with modified potassium phosphate buffer containing (per liter): 17 g K2HPO4, 7.26 g KH2PO4, 1.8 g urea, and 0.2 g MgSO4·7H2O. The washed cells were resuspended in the buffer to an optical density value of 1.0–1.2. Aliquots (2 mL) of this cell suspension were transferred to a set of test tubes, to which were added increasing volumes (range 0–0.25 mL) of hexadecane. The test tubes were shaken for 2–3 min and allowed to stand for 2 h to facilitate phase separation. The OD660 of the aqueous suspension was measured. The cell-free buffer served as the blank.
Genome sequencing
The extraction of the genomic DNA and whole genome sequencing (WGS) of the bacterial strain was carried out through the commercial services provided by Macrogen Inc. Korea. The library and the sequencing platform details are as follows:
-
Library: Illumina TruSeq Nano DNA Library (350 bp insert)
-
Platform: HiSeq; 150 bp paired-end reads
Genome assembly
The SPAdes assembler (version 3.13.0) was used to assemble the raw reads by setting the following assembly parameters: k automatic selection based on the read length, repeat resolution enabled, mismatch corrector skipped, mismatch careful mode, and coverage cutoff turned OFF.
Genome annotation and overall genome-related index (OGRI) analysis
The Classic RAST version 2.0 of rapid annotation using subsystem technology (Aziz et al. 2008) server with default settings was used for sequence and function-based genome annotation studies. Moreover, the bacterial genomic annotation was also carried out by the NCBI PGPA (Prokaryotic Genome Annotation) pipeline. A combination of 16S rRNA gene homology and overall genome-related index, which include the ANI and dDDH estimation, were used in a systematic way for genome-based taxonomic evaluation of the bacteria. The closely related strains to the isolate were selected by submitting the 16S rRNA gene sequence of the isolate 3OW to the EzBioCloud database, which contains the bacterial and archaeal taxonomic hierarchy, represented by high-quality genome and 16S rRNA gene sequences (https://www.ezbiocloud.net/identify) (Yoon et al. 2017). The genome sequences of bacteria, whose 16S rRNA gene sequences showed ≥ 97.8% similarity to the 16S rRNA gene sequence of the isolate 3OW, were downloaded from the EzBioCloud database. The Genome-to-Genome Distance Calculator (GGDC) (Meier-Kolthoff and Göker 2019) version 2.1 was used to calculate the genomic digital DNA–DNA hybridization (dDDH) values by submitting the genome sequences to DSMZ (http://ggdc.dsmz.de). The Kostas web server (http://enve-omics.ce.gatech.edu/ani/) was used to measure the Average Nucleotide Identity (ANI) values between any two genomes.
Data availability
The Whole Genome Shotgun (WGS) project of the bacterial isolate 3OW was deposited to the GenBank/ENA/DDBJ under the accession JAGXOE000000000. The raw genome sequence data was also deposited in the NCBI SRA database, and the accession number PRJNA726364 was obtained.
Biodesulfurization of petroleum oils
The crude and diesel oil samples for biodesulfurization studies were acquired from Attock Refinery Limited, Rawalpindi, Pakistan.
Preparation of DBT-adapted cells
The inoculum of dibenzothiophene (DBT) adapted cells of 3OW was prepared in Erlenmeyer flasks of 250 mL capacity having 100 mL MG medium, supplemented with 0.2 mM of DBT as the sole source of thiophenic sulfur. The growth was obtained by incubating the flasks at 30 °C and 180 rpm. After 4–5 days of growth, the cells were centrifuged at 5000 rpm for 10 min and transferred to fresh MG media without DBT. The second time growth procedure in the MG medium without DBT was adopted to make the DBT-adapted cells completely free from 2-HBP. Again, after 4–5 days of growth, the supernatant was tested for the presence of 2-HBP by Gibbs test. The Gibbs test negative cells were centrifuged at 5000 rpm for 10 min and transferred to a fresh 30 mL MG medium without DBT. This final cell suspension was used as inoculum in all the desulfurization experimental procedures.
Tolerance and viability of bacterium in crude oil
To check the tolerance/viability of the bacterial isolate in crude oil, the colony-forming unit (CFU) method and Gibbs test were used. The bacterial viability studies were carried out at three pulp densities of crude oil (10, 30, and 50% v/v). For this purpose, an appropriate volume of MG medium was taken in conical flasks of 250 mL capacity, and crude oil was added in each flask to a final pulp density of 10, 30, and 50% v/v with a glass pipette. The control flasks were also run in parallel with the experimental flasks having DBT but no crude oil. Each flask was inoculated with 1.0 mL of DBT-adapted cell cultures. The experimental flasks were put in a gyratory shaker with an agitation speed of 180 rpm and 30 °C temperature. Samples were taken after each 48 h to check the bacterial viability in crude oil by estimating the increase/decrease in growth by counting the CFUs. The experiments were carried out twice in duplicates.
Biodesulfurization of crude and diesel oils
The crude and diesel oil biodesulfurization activity of the isolate 3OW was tested in 1.0 L capacity conical flasks using the DBT-adapted cells as inoculum. The experiment was run with a working volume of 500 mL containing 10% v/v oil and 10% of inoculum (1 × 108 cells/mL). Control was also run in parallel to the experimental flasks containing only the oil in the MG medium. The experimental flasks were put at 30 °C in a gyratory shaker with an agitation speed of 180 rpm for 30 days. The oil was separated from the culture medium by centrifugation at 5000 rpm for 10 min. The total sulfur contents of the biotreated and untreated oil samples were estimated on an EDX-RF sulfur analyzer. Moreover, the FTIR analysis of the biotreated and untreated oil samples was carried out to understand the functional group changes that happened before and after the biodesulfurization. Spectra were recorded by using the transmittance mode between 600 and 4000 cm−1.
Data analysis
Data were collated and presented in Excel. The results presented in the graphs are mean values derived from triplicate experiments. The percent standard error is represented as error bars.
Results and discussion
Biodesulfurization is a method that employs bacteria capable of metabolizing organo-sulfur residues bound to petroleum hydrocarbon chains via a sulfur-specific metabolic pathway (Chen et al. 2021; Le Borgne and Quintero 2003). Throughout the formation and maturation of crude oil, microorganisms have been living in close proximity to it, and they have adapted metabolic pathways for extracting sulfur from such harsh conditions for their survival (Mohamed et al. 2015; Agarwal and Sharma 2010). Biodesulfurization of liquid fuels is a potential and viable alternative to the standard hydrodesulfurization processes. The new dibenzothiophene (DBT) desulfurizing bacterial isolate 3OW, belonging to Tsukamurella species on the basis of 16S rRNA gene analysis was used in this study. It formed off-white rough-shaped colonies on the MG media plate (Fig. 1A) and showed rod-shaped morphology under the phase-contrast microscope. The bacterium was able to use DBT as the sole source of sulfur. The culture broth of the isolate 3OW gave a permanent blue color with Gibbs reagent (Fig. 1B), showing that DBT was solely used as a sulfur source and presumably converted to 2-HBP via the C–S bond cleavage 4S pathway as reported previously (Mohamed et al. 2015).

The growth of pure bacterial isolate 3OW on MG media plate (A). The blue color of the complex formed between 2-HBP produced in the culture broth of the isolate 3OW and Gibbs reagent (B)
Figure 2 shows the time-course profile of growth, glucose consumption, 2-HBP production, and changes in pH during biodesulfurization of DBT. The cells of the isolate 3OW started to desulfurize at 24 h and produced a maximum of 0.18 mM of 2-HBP after 144 h of incubation. A full consumption of glucose was observed within 120 h of growth, and the highest growth OD660 of 9.5 was achieved after 72 h. A decrease in pH (from 7.0 to 6.2) was observed during the biodesulfurization process (Fig. 2). The decrease in pH may be due to the production of bacterial metabolic byproducts like organic acids, owing to the breakdown of glucose (Wei et al. 2021).

Time-course profile for bacterial growth, glucose consumption, 2-HBP production, and pH of Tsukamurella sp. 3OW
Analysis of the DBT metabolites
The aerobic microorganisms metabolize the DBT in three different ways (Gupta et al. 2005): in type I metabolism, the microorganisms partially oxidize the carbon skeleton of DBT while the C–S bond remains intact; in type II metabolism, the DBT serves as a source of carbon, sulfur, and energy, i.e., complete mineralization of DBT takes place; and in type III metabolism, the DBT serves as a sole source of sulfur. Moreover, in type III metabolism, the DBT is desulfurized by selective cleavage of the C–S bond (sulfur-specific 4S pathway). In the 4S pathway, consecutive oxidation of DBT sulfur results in the formation of sulfoxide (DBTO), sulfone (DBTO2), hydroxyphenyl benzene sulfinate (HPBS), and 2-hydroxybiphenyl (2-HBP) (Gray et al. 1996; Parveen et al. 2020; Akhtar et al. 2009).
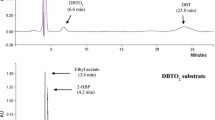
To confirm the sulfur-specific nature of the isolate, the ethyl acetate extract of the 72 h old culture broth was analyzed by HPLC and LC/MS for the presence of key DBT metabolites. Clear peaks of 2-HBP (retention time 10.1 min) and DBT (retention time 6.5 min) were observed in HPLC analysis (Fig. S1) when compared with chromatograph obtained from standards. Moreover, the LC/MS analysis (ESI in the negative ion mode) confirmed the presence of the deprotonated molecular ion [M-H]¯ peaks of 2-HBP (m/z = 169.08) and DBTO (199.08) in the culture extract of 3OW (Fig. 3). In addition, a very clear peak for hydroxyphenyl benzene sulfinate HPBS (m/z = 233) was also detected in the extract. The observation of 2-HBP, DBTO, and HPBS in the culture broth of the isolate 3OW confirmed that DBT desulfurization by this bacterium belongs to type III metabolism, carrying out transformation through an oxidative, C–S bond cleaving 4S pathway. This pathway keeps the carbon skeleton intact (Aggarwal et al. 2013), thus, preserving the fuel’s calorific value. These results show a well-characterized 4S pathway at the biochemical (DBT conversion to 2-HBP detected via Gibbs assay) and molecular levels (metabolites analysis) in the isolate 3OW.

The MS spectra (ESI in the negative ion mode) of the dibenzothiophene (DBT) desulfurization metabolites. The x-axis represents the increasing m/z ratio. The y-axis represents the relative abundance of each ion, which is related to the number of times an ion of that m/z ratio strikes the detector
Substrate specificity and hydrophobicity of the isolate 3OW
Understanding the substrate specificity of desulfurizing bacteria is important for optimizing biodesulfurization processes, as it allows for the selection of suitable strains and the design of targeted approaches for the removal of sulfur from petroleum oils. Crude oil or its derivatives are rich in different organic sulfur-containing compounds, and a major portion of sulfur heteroatom is bound within an aromatic structure which is singly or doubly hindered (Khan and Al-Sayed 2007). The process of hydrodesulfurization has very little reactivity towards the aromatically hindered sulfur compounds such as benzothiophene (BT), dibenzothiophene (DBT), or their alkylated forms. The key to reducing the sulfur contents of petroleum oils is the removal of sulfur from these compounds (Song 2003; Mohebali and Ball 2016).
The isolate 3OW was able to use DBT (the most copious form of organic sulfur present in fuel oil) as the sole source of sulfur. Besides, the bacterium was able to use various other types of more recalcitrant organosulfur compounds present in petroleum oils as a sulfur source (Fig. 4A and B). Very less number of bacteria are capable of removing sulfur simultaneously from alkyl BTs and alkyl DBTs. Moreover, the DBTs with alkyl substitutions at the 4- and/or 6-positions are difficult to desulfurize by reported bacterial strains (Chen et al. 2021). The isolate 3OW was not only able to desulfurize the alkyl BTs and alkyl DBTs but the DBT at the 4,6-position was also desulfurized by the bacterium (Fig. 4B). This depicted that the biodesulfurization substrate specificity of the bacterium was comprehensively enough to act on the complex organosulfur compounds found in petroleum oils. The substrate specificity of desulfurizing bacteria is often linked to the specific desulfurization enzymes they produce (Parveen et al. 2020; Silva et al. 2020). These enzymes have evolved to recognize and act upon specific sulfur-containing structures. The isolate 3OW possesses different sets of enzymes which might be contributing towards its potential to biodesulfurize a range of sulfur-containing compounds.

Time-course profile for bacterial growth (A) and desulfurization curve (2-HBP or 2-HBP equivalent (eq) production) (B) of isolate 3OW in the presence of 5 g/L glucose but, different sulfur sources (0.2 mM)
Petroleum oil is mainly a mixture of hydrocarbons. The hydrocarbons found in crude oils have properties similar to organic solvents which are highly toxic to bacteria (Parveen et al. 2020; Chen et al. 2021). It is reported that hydrophobic desulfurization bacteria could be helpful to speed up the desulfurization process by improving solvent adaptability as well as by overcoming the mass-transfer limitations (Monticello 2000). The MATH assay showed that the isolate 3OW has a high affinity toward the hydrocarbon phase as the cells were adhered to the hexadecane (oil)–water interface and formed an upper emulsion layer, which might be caused by the hydrophobic surface of the bacteria. The more hydrophobic the bacterial cells, the greater their affinity for the hydrocarbon that can impact the desulfurization rate. The adhesion of the 3OW cells to the hexadecane was evident even at the lowest 50 μl concentration of the hydrocarbon, resulting in a reduction of more than 80% in the turbidity of the culture after 2 h (Fig. 5A and B). High cell-surface hydrophobicity enables microorganisms to attach to hydrocarbon droplets on the surface of cells and to move from water to the organic/hydrocarbon phase, where biosurfactants and enzymes attack the thiophenic compounds (Kaczorek et al. 2008). This fact contributes to their being solvent-tolerant (De Carvalho et al. 2004) which facilitates the biological desulfurization reaction of alkyl DBTs/BTs by the transport of these compounds from the oil phase to the cell membrane. The highly hydrophobic nature of the isolate 3OW may have also contributed towards the broader substrate specificity due to the excellent uptake ability of sulfur compounds.

Hydrophobicity of bacterial isolate 3OW determined by the microbial adhesion to hydrocarbon test. Cells were provided with increasing concentrations of hexadecane. Tubes (A); data (B)
Genomic features of bacterium
The genomic features of Tsukamurella sp. 3OW are listed in Table 1. The removal of short contigs (< 200 bp) and sequences with possible contamination resulted in 906 (N50 of 20,544 bp) contigs in the isolate 3OW. The size of the assembled draft genome sequence was 5.2 Mb. The RAST server quality report showed that Tsukamurella sp. 3OW contains 4939 features (PEGs + RNAs) in its genome sequence. The PEG refers to protein-encoding genes and is equivalent to CDS.
The RAST genomic analysis showed that a total of 404 subsystems were present in the genome sequence of 3OW. These subsystems were linked to different categories like metabolism of amino acids and derivatives, carbohydrates metabolism, RNA/DNA metabolism, aromatic compounds metabolism, phosphorous metabolism, fatty acids/lipids/isoprenoids metabolism, stress response, membrane transport, iron acquisition and metabolism, nitrogen metabolism, cofactors/vitamins/prosthetic groups/pigments, protein metabolism, and miscellaneous functions (Fig. S2). The subsystem feature count analysis showed that a total of 57 genes in 3OW were associated with sulfur metabolism (Fig. S2). The comparison of the genomic features of the isolate 3OW with other Tsukamurella species used in this study is listed in Table 2. The genome size of Tsukamurella species was in the range of 4 to 5.3 Mb.
Phylogenomic and overall genome-related index (OGRI) analysis
Bacterial populations are routinely characterized based on microscopic examination, colony morphology, and biochemical tests. However, in the recent past, bacterial identification, classification, and nomenclature have been influenced by the genome sequence information (Meier-Kolthoff and Göker 2019). Genome-based phylogenetic analysis was carried out by using a type strain genome server (TYGS) (https://tygs.dsmz.de/) that determines the closely related type strain genomes. Figure 6 shows the phylogenetic tree of the isolate 3OW based on its whole genome sequence. The phylogenomic inference yielded 8 species clusters, and the provided query strain was assigned to species clusters highlighted in gray (Fig. 6). Delta statistics of the phylogenomic tree range from 0.122 to 0.244. The lower delta value indicated the that phylogenomic tree has higher accuracy. As evident from Fig. 6, the isolate 3OW forms a separate clad with Tsukamurella paurometabola NCTC 10741 at 100 bootstrap value which suggests that the isolate has a maximum genomic identity to this type strain. In the current study, the overall genome-related index (OGRI), which includes the ANI and dDDH estimation, was also used in a systematic way for genome-based taxonomic evaluation of the bacteria. The proposed and generally accepted species boundary for ANI and dDDH values are 95–96% and 70% respectively. In the EzBioCloud database, on the basis of 16S rRNA gene analysis, the isolate 3OW showed > 97% identity to different types of strains of the genus Tsukamurella. The top hit was Tsukamurella paurometabola NCTC 10741(T) (16S identity 99.86%). The dDDH and ANI values were 92.20% and 99.30%, respectively, between 3OW and Tsukamurella paurometabola NCTC 10741(T), showing them as similar species (Table 3). The phylogenomic analysis, dDDH, and ANI values suggested that Tsukamurella sp. 3OW is closely related to Tsukamurella paurometabola NCTC 10741.

Whole genome-based phylogenetic tree of Tsukamurella sp. 3OW generated by the Type Strain Genome Server (TYGS). Tree inferred with FastME 2.1.6.1 from genome blast distance phylogeny (GBDP), distances calculated from genome sequences. The branch length is scaled in terms of the GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values > 60% from 100 replications, with an average branch support of 70%. The tree was rooted at the midpoint. The different attributes of the genome sequences are as follows: Percent G + C (68.4–71.11), delta statistics (0.122–0.244), genome size (4,109,250–5,281,118 bp), and number of proteins (3827–5809)
Genomic analysis for sulfur metabolism
Microorganisms use sulfur for their growth and to carry out many important biological activities. Mostly, it is used for the synthesis of essential biomaterials like sulfur-containing amino acids (cysteine and methionine), prosthetic groups, and vitamins (biotin, thiamin) (Parveen et al. 2020). The genome annotation using ClassicRAST showed that 57 genes are involved in the sulfur metabolism of 3OW. The sulfur metabolism in the genome of 3OW was categorized into three sub-categories (Table S1). The first category was inorganic sulfur assimilation, which included different genes with the assigned role/function of adenylyl sulfate kinase, sulfate- and thiosulfate-binding enzyme, adenylyl sulfate reductase beta-subunit, sulfate adenylyltransferase subunit 2, sulfate transport system permease protein, sulfate adenylyltransferase subunit 1, phosphoadenylyl-sulfate reductase [thioredoxin], ferredoxin-sulfite reductase actinobacterial type, adenylyl-sulfate reductase [thioredoxin], sulfate transporter CysZ-type, sulfate and thiosulfate import ATP-binding protein, and ferredoxin and ferredoxin-NADP(+) reductase actinobacterial FPR (eukaryote-like) type proteins (Table S1). The second subcategory of sulfur metabolism was organic sulfur assimilation, which was divided into two subsystems, i.e., alkanesulfonate assimilation and alkanesulfonate utilization. The roles of the genes in subsystems of alkanesulfonate assimilation and alkanesulfonate utilization were almost similar. The third subcategory of sulfur metabolism was named “no subcategory” and was divided into two subsystems, i.e., thioredoxin-disulfide reductase and galactosylceramide and sulfatide metabolism (Table S1).
Genome-based identification of dszABC genes
The DBT is known to be used as a sulfur source by a variety of bacterial species through the 4S pathway. This is simply a metabolic pathway that converts thiophenic compounds into bioavailable sulfur for microbial development. The DBT desulfurization activity of Rhodococcus erythropolis IGTS8 and several other reported bacterial strains has been accredited to the existence of the dsz operon consisting of three desulfurization genes called dszA, B, and C (Yu et al. 2006; Li et al. 1996). To identify the dszABC genes involved in DBT metabolism, the genome of 3OW was annotated by ClassicRAST. Moreover, the DszABC enzymes, which are involved in DBT desulfurization, and associated proteins were also annotated using the SEED database and NCBI blastn tool. The dsz genes were connected to subsystem alkanesulfonate assimilation (Table S1). The analysis indicated that the dszABC operon and the associated genes are present on contig 41 in the genome of Tsukamurella paurometabola 3OW (Fig. 7). The dsz genes retrieved from the genome of the isolate 3OW showed 85–95% (for dszA), 84–95% (for dszB), and 85–97% (for dszC) similarity to the dszABC genes of different DBT desulfurizing bacteria in blastn analysis (Table 4). The dsz operon-associated proteins, in the genome of the isolate 3OW were identified as mobile element proteins/IS256 family transposase, hypothetical protein, transposase B from transposon Tn554, and site-specific recombinase XerD/integrase (Fig. 7). The identification of transposase like proteins in the up/downstream of the dsz operon indicates that these genes can be transferred through horizontal gene transfer among different bacterial genera (Parveen et al. 2020), making them adaptable to stressful environments like oil-contaminated environments enrich with thiophenic compounds. Furthermore, the presence of alkanesulfonates transport system permease and ABC transporter ATP-binding proteins required for the uptake of thiophenic compounds were predicted in the genome of 3OW (Table S1). This arsenal of genes coding for DBT desulfurization enzymes and associated proteins in the genome of 3OW represent a key indicator of this bacterium’s adaptation to the oil-contaminated niches; as these genes are involved in the metabolism and transport of thiophenic sulfur-containing compounds.

Schematics of the operon dszABC and the associated genes detected (Contig 41) in the genome of Tsukamurella paurometabola 3OW. Proteins DszABC which are involved in DBT desulfurization were annotated using the SEED database and NCBI blastn tool
Biodesulfurization of petroleum oils
The direct burning of high-sulfur fossil fuels like petroleum oils releases a vast amount of sulfur dioxide (SO2) into the atmosphere, which results in the formation of sulfates. This sulfate is one of the main components of atmospheric fine particulate matter, having a diameter of less than 2.5 microns (PM 2.5). The PM 2.5 particulate matter may penetrate the lungs and serve as a vector for transferring toxic metals and other air pollutants into the body (ATSDR 1998; Ostro et al. 2011). The severe health impacts of PM 2.5 inhalation may range from minor irritation to chronic disease or even death (Ostro et al. 2011). Due to a direct correlation between the sulfur level of fuel oil and SO2 release, the removal of sulfur from fuel oil can have considerable impacts on SO2 emissions and thus PM 2.5 formation.
Crude oil contains various toxic and inhibitory compounds, such as aromatic hydrocarbons, olefins (Wu et al. 2020), and heavy metals, which can be detrimental to bacterial viability. To evaluate the potential of the isolate 3OW to desulfurize the petroleum oils, first, the viability of the bacterium was tested in a crude oil environment. The crude oil tolerance studies showed that the number of CFUs of the isolate 3OW was increased with the elapse of time at all concentrations of the crude oil. The count of the colony-forming units after 10 and 20 days of incubation was much higher than the corresponding numbers of CFUs obtained after 2 days of incubation. The 3OW cells showed better viability initially at 10%. However, with the elapse of time, the viability at 30 and 50% concentrations of crude oil also increased as compared to the control (Fig. 8). Moreover, it was observed that bacterial cells were forming a tight oil/media emulsion with the increase of time and were more conspicuous in the experimental flasks containing more crude oil.

Viability of 3OW cells in crude oil compared to control cultures having DBT. Viability is determined as a percent of cells recovered (CFU after 48 h on DBT-agar plates) from assays containing different crude oil concentrations
The crude oil–tolerant bacteria typically possess specific transporters that play a crucial role in their ability to survive and function in crude oil environments (Hamamura et al. 2006). The PGAP genomic analysis of the isolate 3OW showed that the transport system of the bacterium included, 354 proteins (Table S2). Among them, 50 were ATP-binding Cassette (ABC) related proteins with the capacity to transport a wide array of substances, such as inorganic ions, metal ions, lipids, and hydrocarbons. Efflux transporters are membrane proteins that actively pump out toxic compounds from the bacterial cell, reducing their intracellular concentration and protecting the bacteria from harm (Pal et al. 2017). The transporter system of the isolate 3OW contains 12 different types of efflux transporters (Table S2) including ACR3 (Arsenic Resistance) family arsenite efflux transporter, multidrug efflux MFS (Major Facilitator Superfamily) transporter, MATE (Multidrug and Toxic Compound Extrusion) family efflux transporter, DHA2 (drug/H+ antiporter) family efflux MFS transporter permease subunit, QacE (Quaternary Ammonium Compound Efflux) family SMR (Small Multidrug Resistance) transporter, and Cmx/CmrA family chloramphenicol efflux MFS transporter. The abundance of varied nature of transporters in the isolate 3OW reflected its capacity to persist in the crude oil environment.
The total sulfur content of the crude oil was estimated on the EDX-RF sulfur analyzer which showed that the sulfur contents of the oil were decreased by 19% (initial total sulfur content of 3270 ppm) (Fig. 9A). The decrease in sulfur contents suggests that the sulfur needed for growth might be delivered from desulfurization of thiophenic compounds, which constitute the majority of sulfur-containing compounds present in oil. Fourier-transform infrared (FTIR) spectroscopy was used as an analytical tool to obtain detailed information on the hydrocarbon chemistry and organic sulfur-related functional groups of crude oil samples. The different organic sulfur-containing compounds show different stretching frequencies in FT-IR analysis. The C–S linkage stretching vibrations occur in the range of 700–600 cm−1. The FT-IR spectra of biotreated crude oil samples showed an increase in the transmittance value in the region of 700–600 cm−1 as compared to the control. This increase in transmittance value suggests a decrease in organic sulfur-containing molecules having C-S linkages in their structure. Similarly, the transmittance values of the sulfones at 1350 cm−1, sulfoxide at 1070 cm−1, and S–H stretching at 2600 cm−1 were also increased in the biotreated crude oil samples indicating a decrease in the concentration of molecules having such stretching (Fig. 10). The FT-IR spectrum of biotreated and untreated crude oil samples showed no change in the stretching vibrations assigned to the C–H linkages occurring in the region of 2925 and 2856 cm−1 which suggests that the overall chemistry of the crude oil was not affected after biodesulfurization. The C–C bending vibrations occur at very low frequencies (below 500 cm−1), and therefore, they did not appear in the FT-IR spectra.

Biodesulfurization of crude (A) and diesel (B) oils using Tsukamurella paurometabola 3OW

FT-IR spectra of Tsukamurella sp. 3OW bio-treated and untreated crude oil samples
In the case of diesel oil, the DBT-adapted cells of the isolate 3OW showed a considerable decrease in the total sulfur content of the hydrodesulfurized diesel oil (initial sulfur 245 ppm). After treatment with bacterial cells, the total sulfur contents of the diesel oil were 154 ppm, corresponding to a reduction of 37.1% (Fig. 9B). A high desulfurization of hydrodesulfurized diesel oil suggests that bio-available organic/thiophenic sulfur needed for the growth of the bacterial cells was readily available in hydrodesulfurized diesel oil than the crude oil. In the FT-IR analysis of diesel oil, the spectral region of interest, showed many broad vibration bands which were overlapping; therefore, true interpretation of the biotreated and untreated FTIR spectra at these points was difficult.
Biodesulfurization of diesel and crude oils has been reported in various bacterial species/strains (Kilbane II and Le Borgne 2004; Carvajal et al. 2017; Saleh 2020; Mohamed et al. 2015; Agarwal and Sharma 2010; Yu et al. 2006; Sadare et al. 2017). Although the results obtained in the current study are comparable to the already reported studies, however, the exact assessment is not possible due to the variation in the experimental setup used by various researchers. These variations involve the nature and concentration of the crude oil feedstock, the types of sulfur compounds present in the oil, the nature/type of the microorganism used, the scheme/design of the desulfurization process (growing cell/resting cell conditions), and the method of the recovery of the biodesulfurized oil and its analysis. The sulfur-specific 4S pathway, highly hydrophobic cell surfaces, wide substrate specificity, and viability in the crude oil environment of the isolate 3OW are the crucial traits that make it a potential candidate for an efficient desulfurization process.
Conclusions
To the best of our knowledge, this is the first report of the identification of Tsukamurella paurometabola species as a thiophenic sulfur removing and petroleum oil desulfurizing bacterium. The HPLC and LC/MS analysis of the culture extract showed that DBT was solely used as a sulfur source via the 4S pathway (C–S bond cleavage). The utilization of different types of organosulfur compounds, potentially present in petroleum oils, imparts to the isolate 3OW a broad spectrum of biodesulfurization advantages. Identification of key DBT desulfurization and transporter proteins proved that strain 3OW has the capacity for sulfur removal and mechanisms to survive in a petroleum oil environment. It is concluded that the Tsukamurella paurometabola sp. 3OW has the necessary desulfurization characteristics such as utilization of the sulfur specific 4S pathway, hydrophobic cell surfaces, broad substrate specificity, robust viability within the crude oil environment, and good recycling performance, so, the implementation of the 3OW-based microbial desulfurization process along with HDS process can be beneficial in removing HDS-refractory sulfur moieties from the petroleum oils.
Data availability
The data presented in the study are deposited in the NCBI SRA repository (BioProject number PRJNA726364) and GenBank/ENA/DDBJ genetic sequence database (WGS accession JAGXOE000000000).
References
Agarwal P, Sharma DK (2010) Comparative studies on the bio-desulfurization of crude oil with other desulfurization techniques and deep desulfurization through integrated processes. Energy fuels 24:518–524
Agency for Toxic Substances and Disease Registry (ATSDR), Toxicological profile for sulfur dioxide. (1998) ttps://wwwn.cdc.gov/TSP/ToxProfiles/ToxProfiles.aspx?id=253&tid=46
Aggarwal S, Karimi IA, Ivan GR (2013) In silico modeling and evaluation of Gordonia alkanivorans for biodesulfurization. Mol Biosyst 9:2530–2540
Akhtar N, Ghauri MA, Akhtar K (2016) Dibenzothiophene desulfurization capability and evolutionary divergence of newly isolated bacteria. Arch Microbiol 198:509–519
Akhtar N, Ghauri MA, Anwar MA, Akhtar K (2009) Analysis of the dibenzothiophene metabolic pathway in a newly isolated Rhodococcus spp. FEMS Microbiol Lett 301:95–102
Akram J, Parveen S, Akhtar N (2021) Isolation and characterization of thiophenic sulfur metabolizing bacteria from oil contaminated soil samples. In: 2021 International Bhurban conference on applied sciences and technologies (IBCAST). IEEE, pp 458–463
Alves L, Salgueiro R, Rodrigues C, Mesquita E, Matos J, Gírio FM (2005) Desulfurization of dibenzothiophene, benzothiophene, and other thiophene analogs by a newly isolated bacterium, Gordonia alkanivorans strain 1B. Appl Biochem Biotechnol 120:199–208
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Zagnitko O (2008) The RAST server: rapid annotations using subsystems technology. BMC genomics 9:1–15
Bachmann RT, Johnson AC, Edyvean RG (2014) Biotechnology in the petroleum industry: an overview. Int Biodeterior Biodegradation 86:225–237
Carvajal P, Dinamarca MA, Baeza P, Camú E, Ojeda J (2017) Removal of sulfur-containing organic molecules adsorbed on inorganic supports by Rhodococcus rhodochrous spp. Biotechnol Lett 39:241–245
Chen S, Zang M, Li L, Chen J, Liu Q, Feng X, Zhao C (2021) Efficient biodesulfurization of diesel oil by Gordonia sp. SC-10 with highly hydrophobic cell surfaces. Biochem Eng J 174:108094
De Carvalho CC, Da Cruz AA, Pons MN, Pinheiro HM, Cabral JM, Da Fonseca MMR, Fernandes P (2004) Mycobacterium sp., Rhodococcus erythropolis, and Pseudomonas putida behavior in the presence of organic solvents. Microsc Res Tech 64:215–222
Debabov VG (2010) Microbial desulfurization of motor fuel. Appl Biochem Microbiol 46:733–738
Gray KA, Pogrebinsky OS, Mrachko GT, Xi L, Monticello DJ, Squires CH (1996) Molecular mechanisms of biocatalytic desulfurization of fossil fuels. Nat Biotechnol 14:1705–1709
Gupta N, Roychoudhury PK, Deb JK (2005) Biotechnology of desulfurization of diesel: prospects and challenges. Appl Microbiol Biotechnol 66:356–366
Hamamura N, Olson SH, Ward DM, Inskeep WP (2006) Microbial population dynamics associated with crude-oil biodegradation in diverse soils. Appl Environ Microbio. 72:6316–6324
Kaczorek E, Pijanowska A, Olszanowski A (2008) Yeast and bacteria cell hydrophobicity and hydrocarbon biodegradation in the presence of natural surfactants: rhamnolipides and saponins. Bioresour Technol 99:4285–4291
Khan MR, Al-Sayed E (2007) Hydrocarbon desulfurization to clean fuels by selective oxidation versus conventional hydrotreating. Energy sources, part A: recovery, Util Environ Eff 30:200–217
Kilbane JJ II (2006) Microbial biocatalyst developments to upgrade fossil fuels. Curr Opin Biotechnol 17:305–314
Kilbane JJ II, Le Borgne S (2004) Petroleum biorefining: the selective removal of sulfur, nitrogen, and metals. Stud Surf Sci Catal 151:29–65
Le Borgne S, Quintero R (2003) Biotechnological processes for the refining of petroleum. Fuel Process Technol 81:155–169
Li MZ, Squires CH, Monticello DJ, Childs JD (1996) Genetic analysis of the dsz promoter and associated regulatory regions of Rhodococcus erythropolis IGTS8. J Bacteriol 178:6409–6418
Meier-Kolthoff JP, Göker M (2019) TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun 10:2182
Mohamed MES, Al-Yacoub ZH, Vedakumar JV (2015) Biocatalytic desulfurization of thiophenic compounds and crude oil by newly isolated bacteria. Front Microbiol 6:112
Mohebali G, Ball AS (2016) Biodesulfurization of diesel fuels–past, present and future perspectives. Int Biodeterior Biodegradation 110:163–180
Monticello DJ (2000) Biodesulfurization and the upgrading of petroleum distillates. Curr Opin Biotechnol 11:540–546
Monticello DJ, Finnerty WR (1985) Microbial desulfurization of fossil fuels. Annu Rev Microbiol 39:371–389
Ostro B, Tobias A, Querol X, Alastue A, Amato F, Pey J, Perez N, Sunyer J (2011) The effects of particulate matter sources on daily mortality: a case-crossover study of Barcelona, Spain. Environ Health Perspect 119:1781–1787
Pal S, Kundu A, Banerjee TD, Mohapatra B, Roy A, Manna R et al (2017) Genome analysis of crude oil degrading Franconibacter pulveris strain DJ34 revealed its genetic basis for hydrocarbon degradation and survival in oil contaminated environment. Genomics 109:374–382
Parveen S, Akhtar N, Ghauri MA, Akhtar K (2020) Conventional genetic manipulation of desulfurizing bacteria and prospects of using CRISPR-Cas systems for enhanced desulfurization activity. Crit Rev Microbiol 46:300–320
Piddington CS, Kovacevich BR, Rambosek J (1995) Sequence and molecular characterization of a DNA region encoding the dibenzothiophene desulfurization operon of Rhodococcus sp. strain IGTS8. Appl Environ Microbiol 61:468–475
Sadare OO, Obazu F, Daramola MO (2017) Biodesulfurization of petroleum distillates—current status, opportunities and future challenges. Environ 4:85
Saleh TA (2020) Environmental concerns and the importance of desulfurization. In: Nanocomposites for the desulfurization of fuels. IGI Global, pp 284–294
Silva TP, Alves L, Paixao SM (2020) Effect of dibenzothiophene and its alkylated derivatives on coupled desulfurization and carotenoid production by Gordonia alkanivorans strain 1B. J Environ Manage 270:110825
Song C (2003) An overview of new approaches to deep desulfurization for ultra-clean gasoline, diesel fuel and jet fuel. Cat Today 86:211–263
Tanaka Y, Matsui T, Konishi J, Maruhashi K, Kurane R (2002) Biodesulfurization of benzothiophene and dibenzothiophene by a newly isolated Rhodococcus strain. Appl Microbiol Biotechnol 59:325–328
Wang W, Ma T, Lian K, Zhang Y, Tian H, Ji K, Li G (2013) Genetic analysis of benzothiophene biodesulfurization pathway of Gordonia terrae strain C-6. PLoS One 8:e84386
Wei SN, Li YF, Jeong EC, Kim HJ, Kim JG (2021) Effects of formic acid and lactic acid bacteria inoculant on main summer crop silages in Korea. J Anim Sci Technol 63:91
Wu P, Lu L, He J, Chen L, Chao Y, He M, Zhu F, Chu X, Li H, Zhu W (2020) Hexagonal boron nitride: a metal-free catalyst for deep oxidative desulfurization of fuel oils. Green Energy Environ 5:166–172
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613
Yu B, Xu P, Shi Q, Ma C (2006) Deep desulfurization of diesel oil and crude oils by a newly isolated Rhodococcus erythropolis strain. Appl Environ Microbiol 72:54–55
Acknowledgments
The authors are grateful to Dr. Muhammad Farooq (Principal Scientist, NIBGE) for his assistance in assembling the bacterial genome sequence.
Funding
This research work was supported by the International Foundation for Science (IFS), Stockholm, Sweden (agreement no. F-5379-2) and the Higher Education Commission (HEC) of Pakistan (research project no. NRPU-11570).
Author information
Authors and Affiliations
Contributions
Javeria Akram: Experimentation, Formal analysis, Visualization, Writing. Muhammad Umar Hussain: Experimentation, Visualization, Metabolite extraction, HPLC/MS analysis, Writing. Asma Aslam: Experimentation, Formal analysis, Data interpretation, Writing-basic draft. Kalsoom Akhtar: Visualization, Review &; Editing. Munir Ahmad Anwar: Proof reading- Review &; Editing. Mazhar Iqbal: LC/MS analysis &; data interpretation. Muhammad Tahir Hussain: Sampling, Investigation, Crude and diesel oil analysis, Nasrin Akhtar: Conceptualization, Supervision, Project administration, Funding acquisition, Resources, Writing-original draft, Writing-review &; editing.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 404 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Akram, J., Hussain, M.U., Aslam, A. et al. Genomic analysis and biodesulfurization potential of a new carbon–sulfur bond cleaving Tsukamurella sp. 3OW. Int Microbiol (2024). https://doi.org/10.1007/s10123-024-00484-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10123-024-00484-z