
Abstract
Parkinson’s disease (PD) is a chronic and progressive neurodegenerative disorder that affects 1% of the population worldwide. Etiology of PD is likely to be multi-factorial such as protein misfolding, mitochondrial dysfunction, oxidative stress, and neuroinflammation that contributes to the pathology of Parkinson’s disease (PD), numerous studies have shown that mitochondrial dysfunction may play a key role in the dopaminergic neuronal loss. In multiple ways, the two most important are the activation of neuroinflammation and mitochondrial dysfunction, while mitochondrial dysfunction could cause neuroinflammation and vice versa. Thus, the mitochondrial proteins are the highly promising target for the development of PD. However, the limited amount of dopaminergic neurons prevented the detailed investigation of Parkinson’s disease with regard to mitochondrial dysfunction. Both genetic and environmental factors are also associated with mitochondrial dysfunction and PD pathogenesis. The induction of PD by neurotoxins that inhibit mitochondrial complex I provide direct evidence linking mitochondrial dysfunction to PD. A decrease of mitochondrial complex I activity is observed in PD brain and in neurotoxin- or genetic factor-induced in vitro and in vivo models. Moreover, PINK1, Parkin, DJ-1 and LRRK2 mitochondrial PD gene products have important roles in mitophagy, a cellular process that clear damaged mitochondria. This review paper would discuss the evidence for the mitochondrial dysfunction and neuroinflammation in PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is categorized by the attenuation of dopaminergic neurons in brain particularly substantia nigra region and the existence of Lewy bodies (LBs) is the common symptoms of PD [24]. Lewy body is produced in the presynaptic region by the α-syn expression and gets aggregated. Factors such as oxidative stress, inflammation, aging, genetic mutations, and environmental toxins may also cause PD. Off all factors, inflammation plays a major part in the introduction of PD [30]. Neuroinflammation is caused by infectious agents or neurotoxins with proinflammatory action properties that lead to the pathogenesis of PD. Previous reports have shown that dopamine-induced oxidative stress contributed to the inflammatory response in PD patients [6]. Production of ROS enhances the chronic inflammatory response by altering different biomolecules leading to the destruction of neurons [3]. Infectious substances or neurotoxins that alter glial cells include astrocytes and microglia which liberate neurotoxic factors like phagocyte oxidase (PHOX)-induced H2O2 and cytokines (TNFα, IL-1β, etc.) [90].
Cytokines activate receptor-mediated pro-apoptotic pathways in dopaminergic neurons and up-regulate inducible nitric oxide synthase (iNOS) and cycloxygenase 2 (COX2) by stimulating microglia. Thus the increase in generation of ROS and NO leads to DNA damage, lipid peroxidation, and protein disruption [19]. In knockout mice studies, auto-recurrent factors such as PARKIN, PINK1, and DJ-1 suggested that these factors could be involved in negative regulation of neurosystem. Although mitochondrial dysfunction is well known in the dopaminergic neurons of idiopathic and familial PD, the mechanisms involved are unclear. Mitochondrial dysfunction is primarily due to the formation of reactive oxygen species (ROS), ATP depletion, cytochrome c-release, a reduction in mitochondrial complex I enzyme activity, and caspase 3 activation [46]. The disorder of the systemic mitochondrial complex I has been associated for a long time in the diagnosis of idiopathic PD that produces an improved level of OS (Oxidative Stress) in nigral neurons. Reduced mitochondrial activity enhances OS and ROS, accelerating the degeneration of neurons to worsen the integrity of neurons. The OS or ROS causes cellular damage and also implements signaling pathways which result in cell death [62].
Pathways and mechanisms for neuroinflammation
Cytokines
Surface markers present in both microglia and peripheral macrophages are similar which are difficult to differentiate cell types in postmortem PD brain tissue. Lipopolysaccharide (LPS) stimulated peripheral macrophages from PD patients to produce fewer TNF-α, IL-1 a/b, IFN-α, and IL-6 than healthy regulation and extent associated with disability, indicating the decreased cytokine which can develop in tandem with a disease. In different levels of multiple cytokines like TNF-α, IL-1b, IL-3, and IL-6, in postmortem striatum, SN and cerebral spinal fluid (CSF) in PD patients [21] are high, and increased levels of TNF-α receptor R1 (TNF-R1, p55), bcl-2, soluble Fas (sFas), caspase-1, and caspase-3 [55] indicate the presence of a proinflammatory/apoptotic microenvironment in PD patients. However, other regulatory cytokines, including IL-4, transforming growth factor (TGF)-α, TGF-β1, and TGF-β2 [10], also got elevated that shows its ability to control the environment during inflammation. In addition, hippocampal tissues possessed the increased level of IL-2 from PD patients compared to controls indicating that IL-2 receptors (IL-22R) on cells contained in the hippocampus is also up-regulated in PD patients [39]. Although probably expressed by both neuronal and glial cells, the location of IL-2 and IL-2R primarily for frontal cortex, septum, striatum, hippocampal formation, hypothalamus, locus coeruleus, cerebellum and pituitary, and corpus callosum fibers suggested possible regulatory interactions between peripheral tissue and CNS [57]. Most often, IL-2 acts in an auto- and the paracrine manner in the brain as in the peripheral immune system, but that reveals the characteristics of a neuroendocrine modulator under various physiological conditions. For example, IL-2 regulates neuronal and glial growth and differentiation during development and also has its effects in the modulation of sleep/excitement, memory, and cognition, dementia, and neuropsychology [9] (Fig. 1).

Schematic representation of neuroinflammation in PD
Free radicals and reactive oxygen species
Inflammatory responses are induced by reactive microglia, macrophages, and proinflammatory T cells that provide a primary source of free radicals with the ability to modify proteins, lipids, and nucleic acids. It increases a state of antioxidant stress that increases the production of high intracranial organisms, which in turn reduces the free radical capture that leads to greater changes to survival and reductions in damaged macromolecules [81]. The most reactive nature and short half-lives of reactive species, combined with the limiting nature of neuroinflammatory foci for clinical sampling, avoid direct measurement in the pathogenic processes of these reactive species. However, changes in proteins, lipids, and nucleic acids offer substitute biomarkers, which is used to measure the extent of oxidative stress [2, 57]. Postmortem analyses of patients with PD have indicated the presence of these biomarkers for oxidative stress. Protein modifications are also a biomarker which is exhibited in the brains of PD patients. Contrasted with minds from control benefactors, elevated dimensions of nitrated proteins was found in cerebrum and CSF of PD patients [56]. Most eminent are adjustments of proteins that include Lewy bodies (LB), the neuronal considerations that are viewed as the signs of PD and comprises fundamentally of α-synuclein, ubiquitin, and lipids. Additionally, S-nitrosylated types of Parkin, an E3 ubiquitin ligase engaged with protein ubiquitination, has been separated from the transient cortex from PD patients, however not from minds of HD or AD patients [1].
In vitro and in vivo, S-nitrosylation of Parkin actuates an underlying increment in ligase action prompting autoubiquitination of Parkin, inevitable hindrance of ubiquitin ligase movement, and diminished activity in the E3 ligase–ubiquitin–proteasome degradative pathway [87]. Carbonyl adjustments, which are intelligent of protein oxidation, are expanded by more prominent than twofold in the SN contrasted with the basal ganglia and prefrontal cortex of ordinary subjects [83]. Increments in protein carbonyls have been found in SN, basal ganglia, globus pallidus, substantia innominata, cerebellum, and frontal post, yet not in patients with accidental LB ailment (ILBD), a putatively presymptomatic PD issue. The inclusion of the last two mind locales are surprisingly dependent on the limited neuropathology of PD, yet may mirror a result of L-DOPA treatment or an increasingly worldwide outcome of the incendiary spread of oxidative worry in PD. Other proof for oxidative harm to proteins in PD is the expanded articulation of neural heme oxygenase-1 [71] and expanded immunostaining of glycosylated proteins by nigral neurons [7]. Free radicals and nucleic corrosive adjustments change of nucleic acids by free radicals and responsive species can actuate chromosomal deviations with increase effectiveness [20], proposing that chromosomal harm showed in neurons of PD patients may be identified with a strangely increased oxidative pressure.
Among the most encouraging biomarkers of oxidative harm to nucleic acids is nucleoside 8-hydroxyguanosine (8-OHG) for RNA or 8-hydroxy-20-deoxyguanosine (8-OHdG) for DNA. 8-OHG is an oxidized base delivered by free extreme assault on DNA by C-8 hydroxylation of guanine and is a standout among the most regular nucleic corrosive adjustments seen under states of oxidative pressure [95]. The immunohistochemical portrayal of these adjustments demonstrates that the most abnormal amounts of 8-OHG changes are present in neurons of the SN and to a lesser degree in neurons of the core raphe dorsalis and oculomotor core, and at times in glial cells [95]. 8-OHG nucleic corrosive changes are once in a while recognized in the atomic region and for the most part limited to the cytoplasm, and (2) immunoreactivity is fundamentally reduced by RNase or DNase and removed with the two proteins, [95] propose that objectives of oxidative assault incorporate both cytoplasmic RNA and mitochondrial DNA. Specific notable are the discoveries that convergences of 8-OHG in CSF of PD patients are higher than in age-coordinated controls,nonetheless, serum groupings of 8-OHG show deep factor [43, 95].
Lipid peroxidation
Lipid peroxidation often occurs in response to oxidative stress, and a large variety of aldehydes are formed when lipid hydroperoxides break down into biological systems. 4-Hydroxy-2-nominal (HNE) is a responsive α, β-unsaturated aldehyde that is significant amid the oxidation of film lipid polyunsaturated fats and structures stable adducts with nucleophilic bunches on proteins [26]. HNE change of film proteins shapes stable adducts that can be utilized as biomarkers of cell harm because of oxidative pressure. Immunochemical recoloring on enduring dopaminergic nigral neurons in the midbrains of PD patients demonstrate the nearness of HNE-changed proteins on 58% of the neurons contrasted with 9% of those in control subjects. The powerless or non-coloring on oculomotor neurons was observed in a similar midbrain areas of PD patients and the nearness of HNE altered proteins in LB from PD patients [14]. HNE species are regularly more steady than oxygen species, they can without much of a stretch spread from the site of creation to impact alterations at a far off site [96]. HNE adjustments of DNA, RNA, and proteins have different unfavorable organic impacts, for example, obstruction with enzymatic responses and enlistment of warmth stun proteins, and are viewed as to a great extent in charge of the cytotoxic impacts under states of oxidative pressure [78, 92]. The cytotoxic impacts of HNE changes might be established to some extent because of the restraint of edifices I and II of the mitochondrial respiratory chain; enlistment of caspase-8, caspase-9, and caspase-3; cleavage of poly (ADP–ribose) polymerase (PARP) with consequent DNA fracture; inhibition of NFκB-intervened signalling pathways [51], and reduction of glutathione levels [25, 65]. Fixations that initiate no intense change in cell in vitro at first reason is an abatement in the proteasomal reactant action to the degree that it incites collection of ubiquitinated and nitrated proteins, decreases in glutathione levels and mitochondrial action, and expanded dimensions of oxidative stress to DNA, RNA, proteins, and lipids [37]. Another receptive aldehyde species created from the peroxidation of lipids is malondialdehyde (MDA), which is shaped from the breakdown of endoperoxides amid the last phases of the oxidation of polyunsaturated fats; especially powerless are those containing at least three or more bonds. MDA can exist as free aldehydes or respond with essential amine gatherings of macromolecules to shape adducts with cell structures [2, 31]. Proof of expanded dimensions of MDA-adjusted proteins are found in the SN and CSF of PD patients, but not in controls which is a characteristic feature of expanded lipid peroxidation and supportive presence of perpetual reactions in the patients [68]. F2-isoprostane (F2-IsoP) and isofuran (IsoF) are different results of lipid peroxidation, and both are entrenched as explicit biomarkers of in vivo oxidative pressure [66]. This particular increment in IsoF species in PD patients demonstrates that the microenvironmental oxygen strain is ordinarily more noteworthy in PD than different clusters and proposes a one of a kind method of oxidant damage in PD, which might be characteristic of an expanded intracellular oxygen pressure coming about because of mitochondrial brokenness or a more prominent force of incendiary reaction in PD. This information positively show that oxidative worry in the SNpc area in PD, however, yet to be determined is whether innate immune cell activation of microglia and/or astrocytes during the progression of PD shifts the homeostatic balance towards increased protection from or exacerbation of ROS damage and whether this dynamically changes with disease progression.
Mitochondrial dysfunction
During mitochondrial respiration, the electrons of nicotinamide adenine dinucleotide (NADH) are transferred to complex I (NADH: ubiquinone oxidoreductase) and form a final product, H2O, via the activities of complexes III and IV. During the course of electron transfer, recovery of energy is done through ATP formation which in turn enhances the production of ROS and RNS which triggers an apoptotic cascade. Previous studies shown that oxidative damage in the catalytic subunits of complex I in the frontal cortex region of PD patients, which is correlated with complex assembly and dysfunction of the complex [42]. Due to the influence of high proton motive force, the electron supplied from complex II to ubiquinone is reversibly transferred to complex I and reduces NAD+ to NADH, resulting in an alkaline pH or high membrane, which influences the production of ROS. ROS promote single electron transfer across complexes and form a superoxide anion leading to overproduction of free radicals [85]. Increased ROS production in the mitochondria or decreased mitochondrial defense suppression results in DNA, protein, and mitochondrial lipid damage. This damage compromises the respiratory chain, thereby establishing a new link between oxidative stress and bioenergetic failure [46, 91] (Fig. 2).

Schematic representation of mitochondrial dysfunction in PD
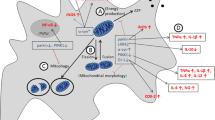
Mitochondrial familial in Parkinson’s disease
Mitochondria are very dynamic organelles which perform a profuse function. Apart from the versatile role of mitochondria, it possesses various cellular processes like regulation of calcium homeostasis, cell death pathway, and stress response [84]. Thus, deterioration of mitochondrial function results in cellular damage leading to neurodegenerative disorder. Many researchers evidenced that mitochondrial dysfunction plays an important role in the pathogenesis of Parkinson’s disease and this might be due to the inhibition of complex I of the ETC which can induce Parkinsonism. PD-associated genes such as α-synuclein and LRRK2 are responsible for autosomal dominant forms, and Parkin, PINK1, and DJ-1 mediate autosomal recessive PD [67].
Autosomal dominant PD
α-Synuclein
α-Synuclein, a 140 amino acid polypeptide encoded by SNCA, is the main component of Lewy bodies. α-Syn present in the mitochondrial membrane had shown its effect on mitochondrial structure and functions [59]. A mutation in α-Syn with PD-related mutations such as A53T, A30P, E46K, H50Q, and G51D leads to mitochondrial fragmentation, proteasomal and lysosomal protein degradation, ER stress, and Golgi fragmentation. Recent studies showed that biogenesis of mitochondria was influenced through the regulation of PGC1α [75, 89]. Cytosolic acidification is helpful to enhance the binding of α-Syn to mitochondria [67]. The mitochondrial alteration was found in the mouse model by overexpressing wild-type or mutant α-synuclein [88]. This is due to increased α-synuclein aggregation that leads to decreased COX, complex IV activity, and mitochondrial membrane potential. α-Synuclein knockout mice were reported for abnormal mitochondrial lipid with decrease in cardiolipin content which was related with a decrease in complex I/III activity [38]. Increased α-synuclein expression and deficiency lead to mitochondrial abnormalities, thus from the above evidence that α-syn has effects on mitochondria indirectly influencing its mitochondrial function.
LRRK2
LRRK2 (leucine-rich repeat kinase 2) is a multirole protein kinase that exerts its pathogenic role by enhancing the kinase activity [67]. PD-associated mutations in LRRK2 include p.G2019S, p.R1441C/G/H, p.Y1699C, p.I2020T, and p.N1437H [60]. By overexpressing WT or mutant LRRK2 showed increased vulnerability to mitochondrial toxins that cause defects in mitochondrial dynamics and increased ROS production. Several proteins (mitochondrial fission protein, dynamin-related protein 1, mitofusin (MFN) 1/2, and optic atrophy 1) interact with LRRK2 which are responsible for pathological effects on mitochondria. UCP2 and UCP4 up-regulation lead to increased proton leak and loss of mitochondrial membrane potential [70]. Overexpression of LRRK2 leads to loss of DA neurons in Drosophila and C. elegans. PD can contribute to the immune system by directly disabling LRRK2 immune system cells. The strong stimulation of the LRRK2 protein is found in microglia cells of the mouse [54]. However, in LRRK2 p.R1441G knock-in mice, LPS-activated microglia cells showed the highest exposure of pro-inflammatory cytokines and decreased expression of anti-inflammatory cytokines compared with wild-type control microglia cells [48, 64].
Parkin
Parkin, a gene that encodes a cytosolic 465 amino acid protein with the ubiquitin-like domain at N-terminal and RBR domain of C-terminus, has two RING finger motifs which flanks a cysteine rich between RING finger domain [45]. Mutation in Parkin leads to autosomal recessive PD. Mitochondrial morphology and their functions are greatly affected by Parkin deficiency. Major Parkin targets located in mitochondria were found by ubiquitous examination. Parkin has a different function in managing healthy mitochondria by organizing their biogenesis and degeneration through mitophagy [77]. In the early phase of mitochondrial deterioration, Parkin leads to damage of mitochondria, and PTEN is stimulated [8]. Mitophagy removes the process to mitochondria from the healthy mitochondrial pool and enhances their degradation through the autophagy-lysosomal pathway. In the recent past, the in vitro models revealed the pathophysiological significance of Parkin-mediated mitophagy in PD [27]. Under stable conditions, Parkin mediates the destruction of Parkin interacting molecule, a repression of the PGC1α activity, leads to nuclear transfer of PGC1α and transcriptional implementation of mitochondrial related genes. Loss of Parkin function permits PARIS to accumulate and suppress mitochondrial biogenesis and results in decreased mitochondrial mass and functional defects [15]. These findings focus on the key role of Parkin which plays in restructuring the balance of mitochondrial production and destruction.
Autosomal recessive PD
PINK1
Mutation in PINK1 is the main cause of autosomal recessive PD [29]. PINK1 is a mitochondrial serine/threonine kinase which plays an important role in managing mitochondrial homeostasis. Defects in PINK1 may cause deformities in mitochondria like a change in morphology, trafficking etc. [40]. PINK1 activate the Parkin by direct phosphorylation of Parkin at S65 and transactivation by phosphorylation of ubiquitin at S65 thus in turn helpful in clearance of damaged mitochondria [58]. Nuclear Dot protein and optineurin help PINK1 to mediate mitophagy. Similarly, LRRK2 and PINK1 promote mitophagy by aborting the mitochondrial trafficking through phosphorylation [4]. Various researchers have proved that loss of PINK1 leads to mitochondrial dysfunction in vitro and in vivo models (Drosophila and mice) [61]. PINK1 regulates mitochondrial homeostasis, for example, PINK1 deficiency is found to be effective in mitochondrial Ca2+ overload and a specific depletion in mitochondrial complexes I and III [32]. Increased mitochondrial fission has increased protein kinase A (PKA)-mediated DRP1 activation and modified mitochondrial biogenesis through Parkin-mediated destruction of PARIS [72].
DJ-1
DJ-1 protein is largely present in the cytosol and a little part in the nucleus and mitochondrial matrix and intermembrane space and decreases degradation [34]. Upon OS, DJ-1 transfer easily to mitochondria and nucleus and plays a vital role and act as a neuroprotector [79]. A decrease in mt DNA, ATP level, and respiratory control ratio was observed in DJ-1 knockout in fly and mice [76]. Further, DJ-1 and its mutant are highly related to Hsp 70 in mitochondria. During OS, wild-type DJ-1 with mitochondrial Hsp 70 was increased, and translocation happens with the help of mitochondrial chaperones [97]. DJ-1 interacts with Parkin and PINK1 under OS conditions and activates Mn-SOD gene responsible for the mitochondrial antioxidant enzyme. Furthermore, decreased DJ-1 leads to reduced MMP, increased mitochondrial fragmentation, autophagy, OS, and mitochondrial fusion [86].
Mitophagy in PD
Autophagy is a highly conserved cellular degradative pathway that is essential for survival, differentiation, development, and homeostasis [94]. The specific autophagic elimination of mitochondria is defined as mitophagy [94]. Previous studies have reported that autophagosomes accumulate due to aggregation of misfolded proteins in the brains of patients leading to diverse neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease [73]. Defects in mitophagy have been shown to summarize a number of reported PD features, namely impaired motor coordination, tremor, and the accumulation of protein aggregates/inclusion bodies in residual neurons. In general, the autophagy process involved in the suppression of many neurodegenerative processes by degrading unfolded proteins and also inhibits a few types of PD by degrading damaged mitochondria. Parkin (PARK2) and Pink1 (PARK6) are the genes involved in eliminating damaged mitochondria by autophagy in familial PD [62]. During the process of mitophagy, first Pink1 accumulates on the outer mitochondrial membrane from the cytosol succeeding mitochondrial depolarization. Later, Parkin, a cytosolic E3-like ligase, binds with Pink1 on the outer mitochondrial membrane, which then leads to ubiquitination of depolarized mitochondria by the ubiquitin ligase activity of the Parkin followed by employing numerous autophagy components such as p62 [41]. Autophagic molecules recognize ubiquitinated mitochondria and are digested by autophagy. Thus, genetic mutations in these genes Parkin and Pink1in familial and sporadic PD becomes inefficient in the elimination of damaged mitochondria, which results in the degeneration of dopaminergic neurons.
Mitochondrial fission and fusion in Parkinson’s disease
Parkinson’s disease (PD) is a neurodegenerative disorder with characteristic symptoms. The most compelling research platform in PD or any other neurodegenerative disorders is the underlying pathology as they are potential therapeutic targets and beacon for the better understanding of the disease. In the case of PD, dysfunctional mitochondrial dynamics and the resultant incapacitation form the basic driving force for the cause and progress of disease as indicated by scientific studies [5, 12, 18]. The most notable and compelling evidence on the role of mitochondrial dysfunction in PD is the exposure of the chemical 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and its metabolite MPP+ on the complex I activity of mitochondria. MPTP mice model is one of the most widely used mice models for PD research and scientific study for therapeutic interventions [63].
Within the nerve cell, the mitochondria (Mt) generate energy for the active functioning of the cell. These mitochondria often undergo specific dynamic changes with respect to the number, size, and shape by the process of mitochondrial fission or fusion. Fission of Mt results in multiple smaller organelles and fusion results in on larger Mt. These processes are supported by the fission/fusion proteins and by the cells demand. Dysfunctions in mitochondria will disrupt the integrity of the nerve cell network and metabolic efficiency. Genetic and biochemical studies on the model organisms such as Drosophila melanogaster and Saccharomyces cerevisiae along with human cells have vividly accounted for the regulation of Mt fission and fusion mechanics by conserved proteins and dynamin-GTPases [16, 52]. Also, the Mt biogenesis is not de novo which means that Mt cannot be made; hence, the fission and fusion process are quintessential for the biogenesis process.
The implications of Mt fission and fusion in PD pathogenesis are described as follows. Pink1 is a serine/threonine kinase that is present in the mitochondria and cytosol, and Parkin is a cytosolic E3 ubiquitin ligase. These two proteins are mostly associated with PD. Scientific research on these proteins have given solid evidence that loss of Pink1 in the neuronal cell cultures resulted in abnormal Mt morphology [28]. Further, the knockdown of Pink1 in SH-SY5Y cells can lead to the fragmentation of Mt and is reversible by the overexpression of Parkin [22]. This evidence suggest that a strong interaction exists between these two protein and act downstream of each other [44, 93]. A genetic study on the Pink1/Parkin mutant Drosophila flies has proven that overexpression of Marf/Mfn2 (a fly homolog of mammalian Mfns) or Opa1 can ameliorate the phenotypes such as the flight muscle degeneration, defects in flying, or abnormal wing position [23]. Taken together, the mitochondrial fission and fusion have quite a significant effect over the functionality and quality of the nerve cell integrity, subsequently contributing to the pathogenesis of PD. Currently, the genetic testing kits for the proteins, SNCA, Parkin, PINK1, and LRRK2 are available commercially [33] along with animal models. Given the heterogeneity and complexity of the disease, more suitable underlying principles and their respective biomarkers have to be investigated deeply to win over PD.
The experimental model of neuroinflammation and mitochondrial dysfunction
There is widespread evidence that PD is caused by neurotoxins particularly 6-hydroxydopamine, rotenone, paraquat, diquat, and MPTP. All these chemicals act through various mechanism (Fig. 3).

Schematic representation of various toxin-induced animal model of PD
6-Hydroxydopamine
6-Hydroxydopamine serves as a well-formed animal model for PD to induce neurodegeneration and stimulation. 6-OHDA is the neurotoxic effects of entering the cytosol via the complex electron transporter chain, the complex I-preventing DA transporter, which enhances the ROS productivity, where it can automatically carry oxidation. Despite the 6-OHDA, it does not penetrate into the blood–brain barrier, which requires its local injection in SNpc or the striatum. The local injection of 6-OHDA in SNpc was first reported by Ungerstedt [80]. The researchers used a bone sample to stimulate the neurons-containing disorder in the rodent brain, in which the SNpc or brain was used to decompose the dopamine levels in the tyrosine after using the stereotaxic injection of this compound, which is the TH positive terminals of the striatum [11]. This functional procedure, like human, provides a gradual improvement of the neurological process. Therefore, 6-OHDA stimulates the most repetitive and reliable brain SNpc injury, which is shown to be very helpful in finding novel treatment techniques for neurological effects.
Rotenone
It is a natural compound present in various plant species such as Derris, Lonchocarpus, Tephrosia, and Mundulea. It is broadly used as an insecticide and pesticides which possess a neurotoxic effect. Rotenone is lipophilic and has the tendency to cross the blood–brain barrier, neuronal cells, and organelles such as mitochondria without the help of transporters. It blocks the mitochondrial ETC via inhibition of complex I that lead to free radical generation in the mitochondrial matrix and ROS formation. The mechanism of action of rotenone is unclear, but evidence from in vitro and in vivo PD models suggest that this may cause delayed decrease in glutathione leading to oxidative damage to protein and DNA. Administration of rotenone modify the DJ-1 and leads to α-syn aggregation causes PD. Rotenone exposure to rats leads to cytoplasmic inclusions in the brain; thus it has linked with mitochondrial dysfunction and PD. This may also cause damage to neurons in the striatum.
Paraquat and diquat
After the identification of MPTP, MPP+ as a functioning metabolite of MPTP model and analysts have discovered a structure like MPP+ in forming synthetic substances. Subsequently, herbicide paraquat was recognized as a specialist that used to think about PD in mice. Paraquat was infused into mice to initiate engine shortfalls and loss of nigral dopaminergic neurons in a portion subordinate way. Uversky [82] revealed that paraquat administrated fundamentally, have been appeared to repeat highlights of PD in rodents. Somayajulu-Niţu et al. [74] also detailed that paraquat-led neurodegeneration speaks to a beneficial rodent model of PD that is reasonable for thought-out and neuroprotective studies to recognize new medication that focuses on the treatment of PD.
MPTP
MPTP is a primary toxin that connected with mitochondrial complex I inhibition and PD. MPTP is produced as a byproduct of the synthesis of meperidine simple with heroin-like properties [36]. It has additionally been appeared to intently repeat the DA degeneration and manifestations of PD in different models and has been the most generally utilized toxin-induced models of PD [47]. MPTP promptly crosses the blood–brain barrier and is changed over to the lethal 1-methyl-4-phenyl-2,3-dihydropyridium particle (MPP +) (Fig. 3) by monoamine oxidase B in astrocytes toxin [17]. Furthermore, it is then taken up into DA neurons by DAT decrease in MPTP danger in DAT lacking mice [53]. MPP+ is taken up into mitochondria by means of uninvolved transport because of the expanded mitochondrial transmembrane angle, where MPP + restrains mitochondrial complex I [13, 29]. This restraint of complex I prompts cell passing by means of vitality deficiencies, free radical, and ROS age [49], and perhaps excitotoxicity [35]. In an MPTP mouse model of PD, α-synuclein is nitrated [50], giving another connection between MPTP and PD. In any case, regardless of the majority of the proof of connections among MPTP and PD, there are contrasts between MPTP models of PD and idiopathic PD with varieties in sickness movement, an intense beginning, and the absence of normal LB development [69].
Conclusion
PD is a complex neurodegenerative disorder with various etiological factors involved in the pathogenesis. There are many pathways that play a vital role in modulating pathogenic events that lead to the death of the dopaminergic neurons in PD. Curiously, these pathways affect the neuronal dopaminergic cell function and survival. The role of neuroinflammation and mitochondrial dysfunctions is the overall inability of available animal models to predict accurately the outcomes of trials that test neuroprotection in human beings.
References
Abrams AJ, Farooq A, Wang G (2011) S-nitrosylation of ApoE in Alzheimer’s disease. Biochemistry 50:3405–3407
Al-Kuraishy HM, Al-Gareeb AI (2020) Citicoline improves human vigilance and visual working memory: the role of neuronal activation and oxidative stress. Basic Clin Neurosci 11:423
Al-Kuraishy HM, Al-Gareeb AI, Naji MT, Al-Mamorry F (2020) Role of vinpocetine in ischemic stroke and poststroke outcomes: a critical review. Brain Circ 6:1
Angeles DC et al (2011) Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum Mutat 32:1390–1397
Archer SL (2013) Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med 369:2236–2251
Barnum CJ, Tansey MG (2010) Modeling neuroinflammatory pathogenesis of Parkinson’s disease. In: Progress in brain research, vol 184. Elsevier, pp 113–132
Beal MF (2002) Oxidatively modified proteins in aging and disease. Free Radical Biol Med 32:797–803
Beal MF (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58:495–505
Bilbo SD, Schwarz JM (2012) The immune system and developmental programming of brain and behavior. Front Neuroendocrinol 33:267–286
Bjarnadóttir K, Benkhoucha M, Merkler D, Weber MS, Payne NL, Bernard CC, Molnarfi N, Lalive PH (2016) B cell-derived transforming growth factor-β1 expression limits the induction phase of autoimmune neuroinflammation. Scientific reports 6(1):1–14
Blandini F, Armentero M-T, Martignoni E (2008) The 6-hydroxydopamine model: news from the past. Parkinsonism Relat Disord 14:S124–S129
Bose A, Beal MF (2016) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139:216–231
Burté F, De Girolamo LA, Hargreaves AJ, Billett EE (2011) Alterations in the mitochondrial proteome of neuroblastoma cells in response to complex 1 inhibition. J Proteome Res 10:1974–1986
Castellani RJ et al (2002) Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett 319:25–28
Castillo-Quan JI (2011) Parkin’control: regulation of PGC-1α through PARIS in Parkinson’s disease. Dis Mod Mechan 4:427–429
Cerveny KL, Tamura Y, Zhang Z, Jensen RE, Sesaki H (2007) Regulation of mitochondrial fusion and division. Trends Cell Biol 17:563–569
Chao YX, He BP, Tay SSW (2009) Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson’s disease. J Neuroimmunol 216:39–50
Chen H, Chan DC (2009) Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet 18:R169–R176
Costa G (2014) Vulnerability to cognitive, neurotoxic and neuroinflammatory effects of toxins that induce Parkinson’s disease after administration of amphetamine-related drugs in mice. Universita'degli Studi di Cagliari, Cagliari
Culbertson CT, Mickleburgh TG, Stewart-James SA, Sellens KA, Pressnall M (2013) Micro total analysis systems: fundamental advances and biological applications. Anal Chem 86:95–118
Cunningham C (2013) Microglia and neurodegeneration: the role of systemic inflammation. Glia 61:71–90
Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, Chu CT (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284:13843–13855
Deng H, Dodson MW, Huang H, Guo M (2008) The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci 105:14503–14508
Dexter DT, Jenner P (2013) Parkinson disease: from pathology to molecular disease mechanisms. Free Radical Biol Med 62:132–144
Dickinson DA, Forman HJ (2002) Cellular glutathione and thiols metabolism. Biochem Pharmacol 64:1019–1026
Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biol Med 11:81–128
Exner N, Lutz AK, Haass C, Winklhofer KF (2012) Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J 31:3038–3062
Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci 105:11364–11369
Geisler S et al (2010) The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6:871–878
Gu X-L, Long C-X, Sun L, Xie C, Lin X, Cai H (2010) Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol Brain 3:12
Hartley DP, Kroll DJ, Petersen DR (1997) Prooxidant-initiated lipid peroxidation in isolated rat hepatocytes: detection of 4-hydroxynonenal-and malondialdehyde-protein adducts. Chem Res Toxicol 10:895–905
Heeman B et al (2011) Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci 124:1115–1125
Henchcliffe C, Beal MF (2008) Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis Nature Reviews. Neurology 4:600
Hong Z et al (2010) DJ-1 and α-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133:713–726
Hsieh M-H et al (2012) Effects of MK-801 on recognition and neurodegeneration in an MPTP-induced Parkinson’s rat model. Behav Brain Res 229:41–47
Hu X et al (2008) Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J Immunol 181:7194–7204
Hyun DH, Lee MH, Halliwell B, Jenner P (2002) Proteasomal dysfunction induced by 4-hydroxy-2, 3-trans-nonenal, an end-product of lipid peroxidation: a mechanism contributing to neurodegeneration? J Neurochem 83:360–370
Jomova K, Vondrakova D, Lawson M, Valko M (2010) Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 345:91–104
Kang SS, McGavern DB (2009) Inflammation on the mind: visualizing immunity in the central nervous system. In: Visualizing Immunity. Springer, pp 227–263
Kawajiri S, Saiki S, Sato S, Hattori N (2011) Genetic mutations and functions of PINK1. Trends Pharmacol Sci 32:573–580
Kazlauskaite A, Muqit MM (2015) PINK1 and Parkin–mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J 282:215–223
Keeney PM, Xie J, Capaldi RA, Bennett JP (2006) Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 26:5256–5264
Kikuchi A et al (2002) Systemic increase of oxidative nucleic acid damage in Parkinson’s disease and multiple system atrophy. Neurobiol Dis 9:244–248
Kim Y et al (2008) PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun 377:975–980
Kitada T et al (1998) Mutations in the Parkin gene cause autosomal recessive juvenile Parkinsonism. Nature 392:605
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787
Lin X et al (2012) Conditional expression of Parkinson’s disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J Neurosci 32:9248–9264
Liu D, Wang Z, Liu S, Wang F, Zhao S, Hao A (2011) Anti-inflammatory effects of fluoxetine in lipopolysaccharide (LPS)-stimulated microglial cells. Neuropharmacology 61:592–599
Lu M, Su C, Qiao C, Bian Y, Ding J, Hu G (2016) Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol 19:pyw047
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y (2012) Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953
Ma H, Li D, Rengarajan T, Manokaran K (2018) Effect of simvastatin on neuroinflammation in microglial cells via mitogen-activated protein kinase and nuclear factor κB pathways. Pharmacogn Mag 14:237
Martens S, McMahon HT (2008) Mechanisms of membrane fusion: disparate players and common principles. Nat Rev Mol Cell Biol 9:543
Masoud S et al (2015) Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and l-DOPA reversible motor deficits. Neurobiol Dis 74:66–75
Moehle MS et al (2012) LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 32:1602–1611
Mogi M et al (2000) Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J Neural Transm 107:335–341
Morris G, Berk M (2015) The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med 13:68
Mosley RL et al (2006) Neuroinflammation, oxidative stress, and the pathogenesis of Parkinson’s disease. Clin Neurosci Res 6:261–281
Nguyen TN, Padman BS, Lazarou M (2016) Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol 26:733–744
Norris EH, Uryu K, Leight S, Giasson BI, Trojanowski JQ, Lee VM-Y (2007) Pesticide exposure exacerbates α-synucleinopathy in an A53T transgenic mouse model. Am J Pathol 170:658–666
Paisán-Ruiz C, Lewis PA, Singleton AB (2013) LRRK2: cause, risk, and mechanism. J Parkinsons Dis 3:85–103
Patel AS et al (2015) Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PloS one 10:e0121246
Pickrell AM, Youle RJ (2015) The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273
Prorok T, Jana M, Patel D, Pahan K (2019) Cinnamic acid protects the nigrostriatum in a mouse model of Parkinson’s disease via peroxisome proliferator-activated receptorα. Neurochemical Research:1–12
Raivich G, Jones L, Werner A, Blüthmann H, Doetschmann T, Kreutzberg G (1999) Molecular signals for glial activation: pro-and anti-inflammatory cytokines in the injured brain. In: Current Progress in the Understanding of Secondary Brain Damage from Trauma and Ischemia. Springer, pp 21–30
Raza H, John A, Brown EM, Benedict S, Kambal A (2008) Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol Appl Pharmacol 226:161–168
Roberts LJ, Fessel JP, Davies SS (2005) The biochemistry of the isoprostane, neuroprostane, and isofuran pathways of lipid peroxidation. Brain Pathol 15:143–148
Ryan BJ, Hoek S, Fon EA, Wade-Martins R (2015) Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem Sci 40:200–210
Sanders LH, Greenamyre JT (2013) Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radical Biol Med 62:111–120
Schulz-Schaeffer WJ (2010) The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol 120:131–143
Serviddio G et al (2008) Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia–reperfusion injury. Gut 57:957–965
Shadrina M, Slominsky P, Limborska S (2010) Molecular mechanisms of pathogenesis of Parkinson’s disease. In: International review of cell and molecular biology, vol 281. Elsevier, pp 229–266
Sharp WW et al (2014) Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J 28:316–326
Shimizu S (2018) Association between autophagy and neurodegenerative diseases. Front Neurosci 12:255
Somayajulu-Niţu M, Sandhu JK, Cohen J, Sikorska M, Sridhar TS, Matei A, Borowy-Borowski H, Pandey S (2009) Paraquat induces oxidative stress, neuronal loss in substantia nigra region and Parkinsonism in adult rats: neuroprotection and amelioration of symptoms by water-soluble formulation of coenzyme Q 10. BMC Neurosci 10(1):1–2
Song X, Rahimnejad S, Zhou W, Cai L, Lu K (2019) Molecular characterization of peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC1α) and its role in mitochondrial biogenesis in blunt snout bream (Megalobrama amblycephala). Front Physiol 9:1957
Su B, Wang X, Zheng L, Perry G, Smith MA, Zhu X (2010) Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochimica et Biophysica Acta (BBA)-Mol Basis Dis 1802:135–142
Sun N, Youle RJ, Finkel T (2016) The mitochondrial basis of aging. Mol Cell 61:654–666
Toyokuni S, Uchida K, Okamoto K, Hattori-Nakakuki Y, Hiai H, Stadtman ER (1994) Formation of 4-hydroxy-2-nonenal-modified proteins in the renal proximal tubules of rats treated with a renal carcinogen, ferric nitrilotriacetate. Proc Natl Acad Sci 91:2616–2620
Trempe J-F, Fon EA (2013) Structure and function of Parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front Neurol 4:38
Ungerstedt U (1968) 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol 5(1):107–110
Uttara B, Singh AV, Zamboni P, Mahajan R (2009) Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 7:65–74
Uversky VN (2004) Neurotoxicant-induced animal models of Parkinson’s disease: understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissue Res 318(1):225–241
Uversky VN (2008) α-synuclein misfolding and neurodegenerative diseases. Curr Protein Pept Sci 9:507–540
Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E (2017) The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun 482:426–431
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39:44–84
van der Merwe C, Jalali Sefid Dashti Z, Christoffels A, Loos B, Bardien S (2015) Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur J Neurosci 41:1113–1125
Vandiver MS et al (2013) Sulfhydration mediates neuroprotective actions of parkin. Nat Commu 4:1626
Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L (2011) Pathological roles of α-synuclein in neurological disorders. Lancet Neurol 10:1015–1025
Wareski P, Vaarmann A, Choubey V, Safiulina D, Liiv J, Kuum M, Kaasik A (2009) PGC-1α and PGC-1β regulate mitochondrial density in neurons. J Biol Chem 284:21379–21385
Whitton P (2007) Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br J Pharmacol 150:963–976
Winklhofer KF, Haass C (2010) Mitochondrial dysfunction in Parkinson’s disease. Biochimica et Biophysica Acta (BBA)-Mol Basis Dis 1802:29–44
Yamagami K, Yamamoto Y, Kume M, Ishikawa Y, Yamaoka Y, Hiai H, Toyokuni S (2000) Formation of 8-hydroxy-2′-deoxyguanosine and 4-hydroxy-2-nonenal-modified proteins in rat liver after ischemia-reperfusion: distinct localization of the two oxidatively modified products. Antioxid Redox Signal 2:127–136
Yang Y et al (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci 103:10793–10798
Yin Z, Pascual C, Klionsky DJ (2016) Autophagy: machinery and regulation. Microbial cell 3:588
Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ (1999) Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154:1423–1429
Zhang S, Eitan E, Wu T-Y, Mattson MP (2018) Intercellular transfer of pathogenic α-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol Aging 61:52–65
Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, Freed CR (2011) Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J Biol Chem 286:14941–14951
Acknowledgements
Authors sincerely acknowledge Malarvizhi R, Meenakshi B, and Vidhushini Sekar for their help in literature collection
Author information
Authors and Affiliations
Contributions
SM, AK, and SS drafted the manuscript MS and SM reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mani, S., Sevanan, M., Krishnamoorthy, A. et al. A systematic review of molecular approaches that link mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurol Sci 42, 4459–4469 (2021). https://doi.org/10.1007/s10072-021-05551-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-021-05551-1