
Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by motor disturbances, appearance of Lewy bodies and dopaminergic neuronal death. The etiology of PD is unknown, although aging and neurotoxins are established risk factors. The activation of glial cells in the brain is the first defense mechanism against pathological events in neurodegenerative diseases, and neuroinflammation is suggested to play an important role in PD disease progression leading to dopaminergic neuronal degeneration. Gene mutations in several PD-related genes may affect up to 15 % of the PD cases. These gene mutations can cause either loss or gain of function in their respective proteins leading to autosomal recessive and autosomal dominant PD, respectively. Most of the identified genes play a role in mitochondrial activity and integrity, and this was demonstrated mostly in neuronal cells. In this review, we aim to describe the link between PD-related genes, which are involved in mitochondrial function, and deleterious neuroinflammation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by resting tremor, muscular rigidity and gait disturbances (Fahn et al. 1998; Mayeux 2003), affecting more than 1 % individuals over 55 years old and more than 3 % of those over the age of 75 years (Mayeux 2003). PD pathology is characterized by the progressive loss of dopaminergic neurons in the substantia nigra (SN) pars compacta and their termini in the dorsal striatum (Thomas 2009). The pathological hallmark of PD is the presence of intracellular inclusions of aggregated α-synuclein known as Lewy bodies (Croisier et al. 2005; Spillantini et al. 1998). The etiology of PD is unknown, although neurotoxins and older age are known risk factors. In recent years, several genes and susceptibility factors have been identified, implicating abnormal handling of misfolded proteins by the autophagy–lysosomal and ubiquitin–proteasome systems, mitochondrial and lysosomal dysfunctions, increased oxidative stress and other pathogenic dysfunctions, as contributing factors for PD (Lesage and Brice 2009). Neuroinflammatory mechanisms might contribute to the cascade of events leading to neuronal degeneration (Hirsch and Hunot 2009), and several reports suggest an important role of neuroinflammation in triggering dopaminergic neuronal death in PD (Hirsch 2000; Jenner 2003; Koutsilieri et al. 2002).
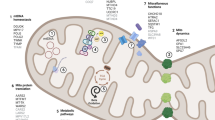
Interestingly, while the cause of sporadic PD is still unclear, the percent of gene mutation in PD can reach to approximately 15 % with genes still being identified (Wood-Kaczmar et al. 2006; Xiromerisiou et al. 2010). To date, 17 genes have been identified as related to PD (Singleton et al. 2013) including α-synuclein and several mitochondrial genes (see Fig. 1), and 10–25 % of patients with early onset (before age 50) PD carry mutations in these genes. These gene mutations can cause either loss or gain of function in their respective proteins, and can result in autosomal recessive and autosomal dominant PD (Wood-Kaczmar et al. 2006; Xiromerisiou et al. 2010).

PD gene mutations impair mitochondrial activity and might increase pro-inflammatory response. A Mitochondrial energy production is impaired by αSyn over-expression, by down-regulation or loss of function of parkin, PINK1 or DJ-1, and by expression of mutant LRRK2. B Mitochondrial morphology, controlled by fission and fusion, is disrupted by αSyn over-expression; by down-regulation or loss of function of parkin or PINK1; and by expression of mutant LRRK2 or over-expression of wild-type LRRK2. C Turnover of mitochondria through mitophagy is affected by down-regulation or loss of function of parkin and PINK1. D Expression of pro-inflammatory cytokines, such as TNFα, IL-1β and IL-6 as well as oxidative stress markers, such as iNOS, NO and NADPH oxidase, due to differential expression of PD-related genes, is marked with arrows to the respective genes
In this review, we will address evidence of the involvement of mitochondria and neuroinflammation in PD pathology. Furthermore, we will discuss the different genes identified as causing genetic forms of PD, and how they affect mitochondrial activity and inflammation in the CNS. Finally, we will describe links between mitochondrial activity and neuroinflammation, as a result of gene mutation, in the pathology of PD.
Mitochondria and reactive oxygen species in PD
Mitochondria are the organelles that constitute a cell’s major source of adenosine triphosphate (ATP) production, the chemical energy of the cell. Within mitochondria, ATP is produced via the action of four respiratory complexes (I, II, III, IV) in the matrix, and ATP synthase in the mitochondrial inner membrane (Haelterman et al. 2014). Mitochondrial mechanisms have been implicated in a variety of human disorders (Vafai and Mootha 2012) including PD (Haelterman et al. 2014).
Animal models for PD are usuaslly based on adminstration of toxins to produce PD-related pathology and symptomatology. A common feature of all toxin-induced models is their ability to induce mitochondrial damage and to cause cell death in dopaminergic neuronal cells that reflect what is seen in PD (Blesa et al. 2012). This first led to focusing the research on mitochondrial dysfunction in PD.
Defects in mitochondrial complex-I lead to increased free radical stress and reactive oxygen species (ROS), and increased neuronal vulnerability to glutamate excitotoxicity (Sherer et al. 2002). Studies of human post-mortem brains indicate that ROS and abnormal complex-I function play an important role in the pathogenesis of PD (Orth and Schapira 2002; Schapira et al. 1998). The activity of complex-I, a major component of the electron transport chain, is decreased in the substantia nigra (Schapira et al. 1990) and frontal cortex (Parker et al. 2008) in patients with PD.
Oxidative damage to lipids, proteins and DNA (Dexter et al. 1989; Zhang et al. 1999) as a result of mitochondrial dysfunction, as well as a decrease in the levels of antioxidant glutathione (Perry and Yong 1986), has been detected in autopsy tissue from the brains of individuals with PD. This abnormality is predicted to render cells more vulnerable to apoptosis, and possibly contributes to the dysfunction and eventual death of cells during the PD disease process (Henchcliffe and Beal 2008).
There are also findings that provide a link between oxidative damage and formation of the Lewy body protein aggregates that are characteristic of PD, as oxidative damage induces αSyn aggregation and impairs proteasomal ubiquitination and degradation of proteins (Jenner 2003). Oxidative stress may cause the oxidation of dopamine to the quinone products, which are believed to be damaging to brain mitochondria (Jenner 2003; Koutsilieri et al. 2002; McGeer and McGeer 2008), suggesting positive feedback of damaged mitochondria, oxidative stress and quinone production, resulting in PD pathology and dopaminergic cell death.
Inflammation and Parkinson’s disease
Neuroinflammatory mechanisms might contribute to the cascade of events leading to neuronal degeneration (Hirsch and Hunot 2009). Several reports suggest the important role of neuroinflammation in triggering dopaminergic neuronal death in PD (Hirsch 2000; Jenner 2003; Koutsilieri et al. 2002).
There is evidence of elevated levels of proinflammatory cytokines, such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNFα) and IL-6, which leads to increased production of inducible nitric oxide synthase (iNOS), secretion of nitric oxide (NO), oxidative sterss, neuronal stress, and further neuronal dysfunction and death in postmortem PD brains (McGeer et al. 1988; Mogi et al. 1994a, b) and PD models (Hirsch and Hunot 2009). As key components of the innate immune response in the brain, glial cells, and in particular microglia and astrocytes, can function as a double-edged sword, with both neurotoxic and neurotrophic effects (Block et al. 2007).
Most proposed etiologies of PD, including environmental toxins (Sherer et al. 2003), and bacterial and viral exposure (Carvey et al. 2006), induce inflammatory activation. For example, lipopolysaccharide (LPS), endotoxin from gram-negative bacteria, is a potent inductor of inflammation and presents diverse effects on microglia and astrocytes (Benveniste 1992). Interestingly, inflammatory activation by intracerebral LPS administration into the cortex, hippocampus, striatum or SN of mice and rats enhances the death of only dopaminergic neurons in the SN (Castano et al. 1998; Herrera et al. 2000), which is in accordance with the inflammatory response reported in PD. In addition to infectious responses, non-infectious inflammatory response were also linked to PD. High mobility group box 1 (HMGB1), a type of damage-associated molecular pattern molecule (DAMP), is increased following exposure of microglia to several PD-inducing toxins, such as rotenone and toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), (Gao et al. 2011b). HMGB1 was also shown to create a non-infectious inflammatory response, through secretion and paracrine activation of TLRs.
It was recently shown that human microglia express dopamine receptors (Mastroeni et al. 2009) and exposure to dopamine may increase secretion of proinflammatory cytokines (Rogers et al. 2007; Trudler et al. 2014). Indeed, increase sensitivity of microglia to dopamine was previously suggested to trigger neurotoxicity to dopaminergic neurons (Trudler et al. 2014). Activated microglia can produce large amounts of superoxide radicals, which may be the major source of the oxidative stress responsible for dopaminergic cell death in PD (McGeer and McGeer 2008).
Astrocytes maintain brain plasticity and protect the brain from injuries (Sofroniew 2005) by secreting protective neurotrophic factors and toxic inflammatory mediators (Segev-Amzaleg et al. 2013). In most cases of PD, there is an elevation in glial fibrillary acidic protein (GFAP) expression that marked astrocytes activation (Cabezas et al. 2013). In experimental models of the toxin such as MPTP, there is marked increase in proliferation and activation of astrocytes in the striatum and substantia nigra (Kohutnicka et al. 1998). Astrocytes activation may mediate neuroinflammatory response towards dopaminergic neurons by secretion of proinflammatory cytokines, generation of ROS and lipid peroxidation (Cabezas et al. 2013). This astrocytic activation was also shown to be accompanied by increased expression of nuclear factor erythroid 2 [NF-E2]-related factor 2 (NRF2) and Wnt1, which exerted neuroprotective effects on dopaminergic neurons (Chen et al. 2009; L’Episcopo et al. 2011). This may suggest that astrocytes activation may also play a neuroprotective role in PD.
Peripheral immune cells such as T-cells also appear to play a role in the pathology of PD. In PD patients’ brains, there is an increase in CD4+ and CD8+ T-cells, which appears to be specific to the substantia nigra (Brochard et al. 2009). MPTP model in CD4 knockout resulted in higher survival of dopaminergic neurons and markedly reduced activation of microglia (Brochard et al. 2009). In this review, however, we will focus on the role of CNS resident cells in mediating inflammatory processes in PD.
Genetic links between mitochondrial activity and neuroinflammation in PD
Several genes have been identified to cause monogenetic forms of PD. Most of these genes are involved in mitochondrial activity, some of which also play a part in the inflammatory process in the CNS. In this section, we will review these genes and discuss the involvement in those processes (see Fig. 1).
Alpha synuclein
Alpha synuclein (αSyn) is the major component of PD Lewy bodies and clinically, patients with mutated αSyn have relatively young age of onset, rapid progression, and high prevalence of dementia, psychiatric, and autonomic disturbances (Xiromerisiou et al. 2010). The normal function of αSyn is poorly understood, but it is implicated in the regulation of vesicle dynamics at the presynaptic membrane, and is important in learning and neuronal plasticity (Sidhu et al. 2004).
αSyn can localize to the mitochondria via an N-terminal targeting motif (Shavali et al. 2008). αSyn can also impair the normal dynamics of mitochondria and this effect is more substantial in A53T mutant (Xie and Chung 2012). αSyn overexpression in neurons results in abnormal mitochondrial function and impaired complex-I activity, specifically in the dopaminergic neurons of the nigrostriatal pathways (Subramaniam et al. 2014). Overexpression of the A53T mutant of αSyn resulted in accumulation of αSyn in the mitochondria, in correlation with complex-I inhibition and aging (Chinta et al. 2010). Elevated expression levels of αSyn in the brain can lead to the development of PD (Miller et al. 2004; Singleton et al. 2003). This enhanced expression increases the deposition of soluble αSyn into insoluble aggregates (Miller et al. 2004). Pathogenic mutations in αSyn (notably A53T) have increased rates of self-assembly and fibrillization, and are considered gain of function mutations, resulting in autosomal dominant inheritance (Conway et al. 2000). Furthermore, it was recently suggested that cell–cell transmission of αSyn pathology is the basis for disease spreading between interconnected areas in the brain. However, while several brain regions, such as the thalamus and cortical regions, exhibit formation of Lewy bodies after injection of αSyn, the neurodegeneration appears to be selective to dopaminergic neurons in the substantia nigra (Luk et al. 2012).
Watson et al. (2012) have demonstrated that in αSyn overexpressing mice, despite the presence of high levels of αSyn in other brain regions, there was a selective early inflammatory response in regions of the nigrostriatal pathway. They suggested that specific factors, that may involve an increase in the expression of Toll-like receptors (TLRs), mediate αSyn-induced inflammatory responses in the SN, which could explain the selective vulnerability of dopaminergic neurons in PD (Watson et al. 2012). Transgenic mice overexpressing of human αSyn exhibited increased vulnerability of dopaminergic neurons to LPS-induced inflammation, and this inflammatory response led to the accumulation of insoluble αSyn aggregates and the formation of cytoplasmatic αSyn inclusions bodies within dopaminergic neurons (Gao et al. 2008). LPS-challenged transgenic mice expressing mutant αSyn (A53T) showed nitration and aggregation of αSyn and degeneration of dopaminergic neurons. Furthermore, those Tg mice have prolonged microglial activation, expressing iNOS and NADPH oxidase enzymes which generate nitric oxide and superoxide, respectively (Gao et al. 2011a). This finding suggests that microglia-dependent oxidative stress is a causative factor in αSyn accumulation and toxicity. It has been suggested that αSyn has a role in neuroinflammation and microglial activation in PD (Meulener et al. 2005). Microglial cells that were exposed to extracellular αSyn show increased proinflammatory phenotype, with increased IL1α, IL1β, TNFα and IL6 secretion (Alvarez-Erviti et al. 2011; Roodveldt et al. 2010). In addition, nitrated αSyn was shown to increase the expression of HMGB1 in microglia (Reynolds et al. 2008) suggesting that the microglial activation pattern following exposure to aggregated αSyn shows similarities to other non-infectious inflammatory responses. Finally, when microglia was treated with the A53T mutation of αSyn, this effect was exacerbated (Alvarez-Erviti et al. 2011; Roodveldt et al. 2010) (see Fig. 1).
αSyn stimulates human astrocytes as well as human U-373 MG astrocytoma cells to up-regulate IL-6 and inflammatory mediator intercellular adhesion molecule-1 (ICAM-1). The mutated forms of αSyn are more potent stimulators than the WT forms (Klegeris et al. 2006). Moreover, mice that selectively express mutated αSyn in astrocytes developed an early onset movement disability, and demonstrated dysfunctional astrocytes. These astrocytes were able to activate microglia and induce neurodegeneration (Gu et al. 2010).
A possible mechanism underlying the effect of αSyn on immune activation is Nurr1, a transcription factor belonging to the orphan nuclear receptor family. The expression of Nurr1 is reduced in αSyn expressing cells, as demonstrated in mice (Lagace-Wiens et al. 2008), and PD patients (Frohman et al. 2005). Nurr1 is a potential regulator of cytokine and growth factor action through regulating the inflammatory response (Lev et al. 2009). Moreover, Nurr1 has a neuroprotective effect because of its capability to inhibit the production of inflammatory mediators in microglia by interaction with CoREST, an NF-κB repressor, and by the promotion of the bound formation with NF-kB subunit p65 resulting in reduction of TNFa secretion (Saijo et al. 2009).
Microglial activation following phagocytosis of aggregated αSyn also enhanced dopaminergic neurodegeneration through production of ROS, in a NADPH oxidase dependent manner (Zhang et al. 2005b). These results suggest that damage to neurons in the substantia nigra may release aggregated αSyn to the substantia nigra, promoting microglia to produce proinflammatory mediators, thereby further inducing nigral neurodegeneration in PD (Zhang et al. 2005b). The extent of microglial activation was correlated with the degree of dopaminergic neurotoxicity induced by both wild-type and mutant αSyn. Exposure to mutated αSyn also induced greater production of reactive oxygen species (ROS) than WT αSyn. These results demonstrated that microglia have an important role in mediating the enhanced neurotoxicity induced by mutant forms of αSyn (Zhang et al. 2007). Interestingly, recent results suggest that passive immunization with monoclonal antibodies against αSyn may be of therapeutic relevance in patients with PD (Masliah et al. 2011), providing further evidence for the involvement of immune activity in disease progression (see Fig. 1).
Leucine-rich repeat kinase 2 (LRRK2)
LRRK2 is a large gene that consists of 51 exons. It encodes the 2527 amino acid cytoplasmic protein leucine-rich repeat kinase 2 (LRRK2) that consists of a leucine-rich repeat towards the amino terminus of the protein, and a kinase domain toward the carboxyl terminus with various conserved domains in between (Klein and Westenberger 2012). There are more than 50 different missense and nonsense mutations reported in LRRK2 to date (Nuytemans et al. 2010), and at least 16 of them are pathogenic, with gain of function attributes, and the G2019S mutation has been extensively researched (Klein and Westenberger 2012; West et al. 2005). However, the pathogenic mechanism leading to PD caused by LRRK2 mutations is still uncertain. LRRK2 is a large protein with many domains capable of protein–protein interactions, and thus it is plausible that changes in these domains would influence the LRRK2′s relationship with other proteins (Berger et al. 2010; Klein and Westenberger 2012; Venderova et al. 2009).
LRRK2 can localize to the outer membrane of the mitochondria (Biskup et al. 2006). LRRK2 mediated mitochondrial fragmentation and dysfunction, which is exacerbated in the G2019S mutation, leading to neurotoxicity (Wang et al. 2012). Fibroblasts acquired from LRRK2 mutated patient (G2019S), showed decreased mitochondrial membrane potential and intracellular ATP levels (Mortiboys et al. 2010). This was also demonstrated recently with induced pluripotent stem cells (iPSC) from PD patients carrying LRRK2 mutations. Mutant LRRK2 iPSCs demonstrated mitochondrial dysfunction and mitochondrial DNA damage (Cooper et al. 2012; Sanders et al. 2014), further supporting the role of LRRK2 in mitochondrial function.
High LRRK2 expression has been discovered in macrophages and monocytes (Thevenet et al. 2011), as well as T-cells (Hakimi et al. 2011), leading to the speculation of a functional role for LRRK2 in the immune system (see Fig. 1).
Moehle et al. (2012) have shown that microglial activation triggers LRRK2 expression, and that LRRK2 inhibition, by either small-molecule kinase inhibitors or shRNA, attenuates microglial proinflammatory response and reduces TNFα and NO levels following LPS activation (Moehle et al. 2012). This suggests that LRRK2 plays an important role in mediating proinflammatory responses in microglia cells. Recently, it was demonstrated that LRRK2 acts through regulation of p38 MAPK and NF-κB signaling pathways to stimulate microglial inflammatory responses. p38 MAPK plays a crucial role in regulating oxidative stress-induced cell death and survival (Runchel et al. 2011). LRRK2 knockdown attenuated the inflammatory response to LPS and reduces cytokine and NO secretion, and LRRK2 overexpression increased the response (Kim et al. 2012). Furthermore, mutated LRRK2 microglia showed a decrease in IL-10 production in response to LPS stimuli, as compared to WT (Gillardon et al. 2012).
In a small cohort of patients carrying LRRK2 mutations, patients showed neuronal degeneration in substantia nigra, and some of the patients exhibited Lewy body pathology (Zimprich et al. 2004). Similarly, double mutant mice carrying mutated αSyn and mutated LRRK2 showed increased striatal neurodegeneration, compared to mice carrying only mutated αSyn (Lin et al. 2009), suggesting a possible connection between LRRK2 and αSyn pathologies.
Taken together, these results suggest that LRRK2 mutations, which are gain of function mutations, could change the microglia towards a proinflammatory phenotype, which changes the microenvironment of brain, and thereby triggers and/or enhances the pathogenesis of PD.
Parkin
Parkin is a 465 amino acid protein with a molecular mass of 52 kDa, which is highly expressed in heart, testis, brain and skeletal muscle (Kitada et al. 1998). Parkin was identified to have E3 ubiquitin ligase activity (Shimura et al. 2000), and parkin mutations in patients resulted in the loss of E3 activity (Imai et al. 2000). Parkin also has a role in the mitochondrial activity and integrity (Abou-Sleiman et al. 2006), and in mammalian cells, parkin was shown to be recruited to the dysfunctional mitochondria, where it mediates the engulfment and degradation of deficient mitochondria, a process known as mitophagy (Narendra et al. 2008). Parkin-null Drosophila demonstrate disordered mitochondria (Greene et al. 2003), parkin−/− mice showed decreased mitochondrial respiratory capacity and increased oxidative stress (Palacino et al. 2004). Mitochondrial quality control is important for vital energy production processes, and was also linked to apoptosis-related processes such as the release of cytochrome c (Youle and van der Bliek 2012), suggesting that impairments in mitophagy could have detrimental effects on cells. In similar experiments using parkin−/− mice, however, there was no significant loss of dopaminergic neurons in the substantia nigra and inclusion bodies (Goldberg et al. 2003) Interestingly, Parkin−/− mice bearing mutated αSyn, did not show a more severe phenotype compared to mice carrying only mutated αSyn, regarding Lewy bodies deposition and neurodegeneration (von Coelln et al. 2006). These findings suggest that parkin-mediated toxicity is not necessarily synuclein-dependent, and may require additional factors to induce marked neurodegeneration.
Parkin is abundantly expressed in microglia, and parkin-null mice have increased number of microglia, as measured by immunocytochemical analysis (Casarejos et al. 2006). Additionally, parkin loss of function in microglia resulted in enhanced toxicity to dopaminergic neurons after rotenone treatment (Casarejos et al. 2006).
A possible mediator for the effect of parkin on inflammation is TNF-α receptor-associated factor (TRAF) 2/6 signaling pathway. TRAF2 and TRAF6 are essential mediators of cytokine signaling by regulating c-Jun N-terminal kinase (JNK) and nuclear factor-κB (NF-κB) signaling. Parkin expression is inversely related to TRAF2/6 expression, and it promotes the proteasomal degradation of TRAF2/6 and protects inflammatory signaling in response to cytokine activation. These results suggest that TRAF2/6 may be a physiological target of parkin, and they provide an explanation for why loss of parkin is detected in PD (Chung et al. 2013), and how it relates to the neuroinflammation observed in PD. This indicates that the loss of parkin may induce an inflammatory response in PD (see Fig. 1).
Parkin levels are reduced in microglia in response to LPS of TNFα activation, in an NF-κB dependent manner. In addition, activated macrophages from Parkin-null mice expressed increased levels of TNFα, IL-1β, and iNOS mRNA compared to WT macrophages (Tran et al. 2011). Moreover, loss of parkin function increases the vulnerability of dopaminergic neurons in the SN to inflammation-related degeneration, that was triggered by i.p. injection of low dose LPS (Frank-Cannon et al. 2008).
These reports indicate that parkin serves as an anti-inflammatory factor, suggesting that reduced parkin levels, or mutated parkin which has a loss of function phenotype, may be proinflammatory, and increase the neuronal death attributed to inflammation.
PTEN induced putative kinase 1 (PINK1)
PINK1 gene encodes a 581 amino acid protein with an N-terminal mitochondrial targeting motif and a highly conserved kinase domain homologous to the serine/threonine kinases of the Ca2+/calmodulin family (Gandhi et al. 2006). PINK1 is crucial for the intact function of cells, and is involved in cell respiration (Gandhi et al. 2009), protein folding and degradation (Dagda et al. 2009; Moriwaki et al. 2008), and in several mitochondrial functions, such as fission/fusion dynamics (Deng et al. 2005; Poole et al. 2008), trafficking (Weihofen et al. 2009) and calcium signaling (Marongiu et al. 2009). In addition, PINK1 is required for recruitment of parkin to the mitochondria with impaired membrane potential (Narendra et al. 2010; Vives-Bauza et al. 2010). PINK1 loss of function impairs the mitochondrial respiratory chain and ATP production, and induces neuronal aggregation of αSyn (Liu et al. 2009). Conversely, PINK1 overexpression can rescue αSyn-induced neuronal degeneration in a fly model (Todd and Staveley 2008), as well as αSyn-induced mitochondrial fragmentation in a Caenorhabditis elegans model (Kamp et al. 2010). These reports emphasize the important role of PINK1 in modulation of mitochondrial activity under normal conditions, as well under disease conditions.
PINK1 can interact with several chaperone proteins and such interaction may affect both its stability and its neuroprotective effects (Deas et al. 2009). PINK1 has been shown to protect cells against oxidative stress (Deas et al. 2009) by phosphorylating the mitochondrial protein TRAP1 (chaperone tumor necrosis factor receptor-associated protein 1) (Pridgeon et al. 2007).
Mutations in PINK1 gene account for 1–8 % of the early onset autosomal recessive PD cases (Klein et al. 2007; Xiromerisiou et al. 2010). PINK1 deficiency, results in calcium overload within mitochondria, causing ROS production in the mitochondria and cytosol (Gandhi et al. 2009). Moreover, PINK1 knockdown induced mitophagy, i.e., lysosomal degradation of mitochondria, a decrease in mitochondria levels within cells (Dagda et al. 2009).
PINK−/− embryonic fibroblasts showed decreased basal and inflammatory cytokine-induced NF-κB activity. In addition, PINK−/− mice had increased levels of IL-1β, IL-12 and IL-10 in the striatum after peripheral stimulation with LPS (Akundi et al. 2011) (see Fig. 1). Kim and colleages (2013) have shown that in organotypic cultures, PINK1−/− slices expressed higher mRNA levels of TNFα, IL1β and IL-6 compared to those of WT slices. These results indicate that PINK1 deficiency increases the production of proinflammatory cytokines, leading to a neurotoxic effect. The observed effect was mediated by reduced serine/theronine kinase Akt activation and enhanced IκB degradation in response to brain injury (Kim et al. 2013). Another possible mechanism for the effect is that PINK1 directly interacts with two members of the IL-1β-mediated downstream signaling pathway, TRAF6 and transforming growth factor β activated kinase-1 (TAK1), and positively regulates their activation leading to enhanced IL-1β-mediated cytokine production (Lee et al. 2012). These reports demonstrate that PINK1 plays a role in the proinflammatory response in PD, possibly through interactions with the mitochondria.
DJ-1
DJ-1 encodes a small 189 amino acid protein that is ubiquitously expressed and highly conserved throughout diverse species (Bandopadhyay et al. 2004; Nagakubo et al. 1997). DJ-1 is localized to the cytoplasm, nucleus (Bader et al. 2005; Nagakubo et al. 1997) and mitochondria (Zhang et al. 2005a) of the cells, and it has been suggested that oxidation promotes the mitochondrial localization of DJ-1 (Blackinton et al. 2005). It was shown that the loss of functional DJ-1 causes 1–2 % of autosomal recessive early onset PD cases (Abou-Sleiman et al. 2003; Bonifati et al. 2003).
Several hypotheses have been introduced to explain the mechanism of DJ-1 involvement in PD pathogenesis. It has been suggested that DJ-1 plays a role in maintenance of mitochondrial complex-I activity (Hayashi et al. 2009) as well as in maintenance of mitochondrial morphology through fission/fusion dynamics (Irrcher et al. 2010), and that loss of DJ-1 function impairs nigrostriatal dopaminergic function (Chen et al. 2005; Goldberg et al. 2005; Kim et al. 2005). Of note, similarly to PINK1, DJ-1 overexpression can also rescue αSyn-induced mitochondrial fragmentation in a C. elegans model (Kamp et al. 2010). Neuronal cells that carry mutant forms of DJ-1, become more susceptible to death in parallel with the loss of oxidized forms of DJ-1 (Taira et al. 2004; Yokota et al. 2003), and are more sensitive to toxins (Lev et al. 2008). Moreover, DJ-1−/− mice are more vulnerable to several toxicity models of PD, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treatment (Kim et al. 2005), 6-hydroxydopamine (6-OHDA), and rotenone toxicity without αSyn pathology (Lev et al. 2008, 2013). In all of the above models, DJ-1−/− induced a greater loss of dopaminergic neurons in these mice. It was demonstrated that ROS production, induced by bisphenol A (BPA) compromised mitochondrial function and elevated the expression and oxidization of DJ-1. DJ-1 was found to maintain the complex-I activity against BPA-induced oxidative stress after the localization in mitochondria. Furthermore, it was suggested that DJ-1 plays a role in the prevention of mitochondrial injury-induced cell death (Ooe et al. 2005).
It has been previously reported that DJ-1 is important for the proinflammatory response in astrocytes, and that DJ-1−/− astrocytes have neurotoxic properties, with increased NO production and enhanced induction of COX2 and IL-6 (Waak et al. 2009). Moreover, recent research demonstrated that DJ knockout attenuates astrocytes neuroprotection against 6-OHDA toxicity (Lev et al. 2013), suggesting a protective role for astrocytes in the 6-OHDA model for PD, which depends on DJ-1 activation. When DJ-1 is knocked out, the protective effect is diminished, suggesting that DJ-1 is important for the neuroprotective ability of astrocytes, and that mutated DJ-1 reduces the neuroprotection.
Stimulation of macrophages with LPS induced an elevation in the DJ-1 protein levels (Mitsumoto and Nakagawa 2001). We have recently demonstrated that DJ-1-deficient microglia have increased monoamine oxidase (MAO) activity which resulted in elevation of intracellular ROS, NO, and proinflammatory cytokines, leading to increased dopaminergic neurotoxicity. This phenotype was ameliorated by rasagaline, a MAO inhibitor which is approved for treatment in PD patients. Those results demonstrate the important role of DJ-1 in immune activation in Parkinson’s disease (Trudler et al. 2014).
Conclusion
There is increasing evidence that connects neuroinflammation and PD progression. PD gene mutations were previously suggested to lead to neuronal dysfunction leading to neuronal death. Here, we described the potential role of PD genes in mediating brain inflammation, as well as their effect on mitochondrial structure and function. Furthermore, we suggest that PD genes may trigger neuroinflammation to dopaminergic neurons either as a result of neuronal stress or by direct activation of glia cells in the microenvironment towards the dopaminergic neurons. This phenomenon supports the notion of a non-cell-autonomous mechanism in PD. Further understanding of the mechanisms underlying impairment of microglia or astrocyte activity, which are linked to PD genes, may provide new avenues for the etiology of PD and new therapeutic approaches.
References
Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW (2003) The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann Neurol 54:283–286. doi:10.1002/ana.10675
Abou-Sleiman PM, Muqit MM, Wood NW (2006) Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci 7:207–219. doi:10.1038/nrn1868
Akundi RS et al (2011) Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One 6:e16038. doi:10.1371/journal.pone.0016038
Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ (2011) Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res 69:337–342. doi:10.1016/j.neures.2010.12.020
Bader V, Ran Zhu X, Lubbert H, Stichel CC (2005) Expression of DJ-1 in the adult mouse CNS. Brain Res 1041:102–111. doi:10.1016/j.brainres.2005.02.006
Bandopadhyay R et al (2004) The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 127:420–430. doi:10.1093/brain/awh054
Benveniste EN (1992) Inflammatory cytokines within the central nervous system: sources, function, and mechanism of action. Am J Physiol 263:C1–C16
Berger Z, Smith KA, Lavoie MJ (2010) Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49:5511–5523. doi:10.1021/bi100157u
Biskup S et al (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol 60:557–569. doi:10.1002/ana.21019
Blackinton J et al (2005) Effects of DJ-1 mutations and polymorphisms on protein stability and subcellular localization. Brain Res Mol Brain Res 134:76–83. doi:10.1016/j.molbrainres.2004.09.004
Blesa J, Phani S, Jackson-Lewis V, Przedborski S (2012) Classic and new animal models of Parkinson’s disease. J Biomed Biotechnol 2012:845618. doi:10.1155/2012/845618
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69. doi:10.1038/nrn2038
Bonifati V et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259. doi:10.1126/science.1077209
Brochard V et al (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Investig 119:182–192. doi:10.1172/JCI36470
Cabezas R, Avila MF, Torrente D, El-Bachá RS, Morales L, Gonzalez J, Barreto GE (2013) Astrocytes role in Parkinson: a double-edged sword. In: Uday Kishore (ed) Neurodegenerative disease. doi:10.5772/54305
Carvey PM, Punati A, Newman MB (2006) Progressive dopamine neuron loss in Parkinson’s disease: the multiple hit hypothesis. Cell Transplant 15:239–250
Casarejos MJ, Menendez J, Solano RM, Rodriguez-Navarro JA, Garcia de Yebenes J, Mena MA (2006) Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J Neurochem 97:934–946. doi:10.1111/j.1471-4159.2006.03777.x
Castano A, Herrera AJ, Cano J, Machado A (1998) Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. J Neurochem 70:1584–1592
Chen L et al (2005) Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem 280:21418–21426. doi:10.1074/jbc.M413955200
Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA (2009) Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proc Natl Acad Sci USA 106:2933–2938. doi:10.1073/pnas.0813361106
Chinta SJ, Mallajosyula JK, Rane A, Andersen JK (2010) Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett 486:235–239. doi:10.1016/j.neulet.2010.09.061
Chung JY et al (2013) Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity laboratory investigation. J Tech Methods Pathol. doi:10.1038/labinvest.2013.60
Conway KA, Harper JD, Lansbury PT Jr (2000) Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 39:2552–2563
Cooper O et al (2012) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Trans Med 4:141ra190. doi:10.1126/scitranslmed.3003985
Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB (2005) Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflamm 2:14. doi:10.1186/1742-2094-2-14
Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284:13843–13855. doi:10.1021/bi991447r
Deas E, Plun-Favreau H, Wood NW (2009) PINK1 function in health and disease. EMBO Mol Med 1:152–165. doi:10.1002/emmm.200900024
Deng H, Jankovic J, Guo Y, Xie W, Le W (2005) Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem Biophys Res Commun 337:1133–1138. doi:10.1016/j.bbrc.2005.09.178
Dexter DT et al (1989) Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem 52:381–389
Fahn S, Clarence-Smith KE, Chase TN (1998) Parkinson’s disease: neurodegenerative mechanisms and neuroprotective interventions—report of a workshop. Mov Disord 13:759–767. doi:10.1002/mds.870130502
Frank-Cannon TC et al (2008) Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci 28:10825–10834. doi:10.1523/JNEUROSCI.3001-08.2008
Frohman EM et al (2005) Therapeutic considerations for disease progression in multiple sclerosis: evidence, experience, and future expectations. Arch Neurol 62:1519–1530. doi:10.1001/archneur.62.10.1519
Gandhi S et al (2006) PINK1 protein in normal human brain and Parkinson’s disease. Brain 129:1720–1731. doi:10.1093/brain/awl114
Gandhi S et al (2009) PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33:627–638. doi:10.1016/j.molcel.2009.02.013
Gao HM, Kotzbauer PT, Uryu K, Leight S, Trojanowski JQ, Lee VM (2008) Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J Neurosci 28:7687–7698. doi:10.1523/JNEUROSCI.0143-07.2008
Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS (2011a) Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ Health Perspect 119:807–814. doi:10.1289/ehp.1003013
Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS (2011b) HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci 31:1081–1092. doi:10.1523/JNEUROSCI.3732-10.2011
Gillardon F, Schmid R, Draheim H (2012) Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 208:41–48. doi:10.1016/j.neuroscience.2012.02.001
Goldberg MS et al (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635. doi:10.1074/jbc.M308947200
Goldberg MS et al (2005) Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45:489–496. doi:10.1016/j.neuron.2005.01.041
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100:4078–4083. doi:10.1073/pnas.0737556100
Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H (2010) Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain 3:12. doi:10.1186/1756-6606-3-12
Haelterman NA, Yoon WH, Sandoval H, Jaiswal M, Shulman JM, Bellen HJ (2014) A mitocentric view of Parkinson’s disease. Annu Rev Neurosci 37:137–159. doi:10.1146/annurev-neuro-071013-014317
Hakimi M et al (2011) Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm 118:795–808. doi:10.1007/s00702-011-0653-2
Hayashi T et al (2009) DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem Biophys Res Commun 390:667–672. doi:10.1016/j.bbrc.2009.10.025
Henchcliffe C, Beal MF (2008) Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis Nature clinical practice. Neurology 4:600–609. doi:10.1038/ncpneuro0924
Herrera AJ, Castano A, Venero JL, Cano J, Machado A (2000) The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis 7:429–447. doi:10.1006/nbdi.2000.0289
Hirsch EC (2000) Glial cells and Parkinson’s disease. J Neurol 247(Suppl 2):II58–II62
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol 8:382–397. doi:10.1016/S1474-4422(09)70062-6
Imai Y, Soda M, Takahashi R (2000) Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem 275:35661–35664. doi:10.1074/jbc.C000447200
Irrcher I et al (2010) Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet 19:3734–3746. doi:10.1093/hmg/ddq288
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26–S36. doi:10.1002/ana.10483 (discussion S36–S28)
Kamp F et al (2010) Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 29:3571–3589. doi:10.1038/emboj.2010.223
Kim RH et al (2005) Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and oxidative stress. Proc Natl Acad Sci USA 102:5215–5220. doi:10.1073/pnas.0501282102
Kim B et al (2012) Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One 7:e34693. doi:10.1371/journal.pone.0034693
Kim J, Byun JW, Choi I, Kim B, Jeong HK, Jou I, Joe E (2013) PINK1 deficiency enhances inflammatory cytokine release from acutely prepared brain slices. Exp Neurobiol 22:38–44. doi:10.5607/en.2013.22.1.38
Kitada T et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. doi:10.1038/33416
Klegeris A, Giasson BI, Zhang H, Maguire J, Pelech S, McGeer PL (2006) Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J 20:2000–2008. doi:10.1096/fj.06-6183com
Klein C, Westenberger A (2012) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2:a008888. doi:10.1101/cshperspect.a008888
Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE (2007) Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol 6:652–662. doi:10.1016/S1474-4422(07)70174-6
Kohutnicka M, Lewandowska E, Kurkowska-Jastrzebska I, Czlonkowski A, Czlonkowska A (1998) Microglial and astrocytic involvement in a murine model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Immunopharmacology 39:167–180
Koutsilieri E, Scheller C, Grunblatt E, Nara K, Li J, Riederer P (2002) Free radicals in Parkinson’s disease. J Neurol 249(Suppl 2):II1–II5. doi:10.1007/s00415-002-1201-7
Lagace-Wiens PR, Decorby MR, Baudry PJ, Hoban DJ, Karlowsky JA, Zhanel GG (2008) Differences in antimicrobial susceptibility in Escherichia coli from Canadian intensive care units based on regional and demographic variables. Can J Infect Dis Med Microbiol 19:282–286
Lee HJ, Jang SH, Kim H, Yoon JH, Chung KC (2012) PINK1 stimulates interleukin-1beta-mediated inflammatory signaling via the positive regulation of TRAF6 and TAK1. Cell Mol Life Sci 69:3301–3315. doi:10.1007/s00018-012-1004-7
L’Episcopo F et al (2011) Reactive astrocytes and Wnt/beta-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Neurobiol Dis 41:508–527. doi:10.1016/j.nbd.2010.10.023
Lesage S, Brice A (2009) Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 18:R48–R59. doi:10.1093/hmg/ddp012
Lev N, Ickowicz D, Melamed E, Offen D (2008) Oxidative insults induce DJ-1 upregulation and redistribution: implications for neuroprotection. Neurotoxicology 29:397–405. doi:10.1016/j.neuro.2008.01.007
Lev N, Ickowicz D, Barhum Y, Melamed E, Offen D (2009) DJ-1 changes in G93A-SOD1 transgenic mice: implications for oxidative stress in ALS. J Mol Neurosci 38:94–102. doi:10.1007/s12031-008-9138-7
Lev N, Barhum Y, Ben-Zur T, Melamed E, Steiner I, Offen D (2013) Knocking out DJ-1 attenuates astrocytes neuroprotection against 6-hydroxydopamine toxicity. J Mol Neurosci. doi:10.1007/s12031-013-9984-9
Lin X et al (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64:807–827. doi:10.1016/j.neuron.2009.11.006
Liu W et al (2009) PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS One 4:e4597. doi:10.1371/journal.pone.0004597
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. doi:10.1126/science.1227157
Marongiu R et al (2009) Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J Neurochem 108:1561–1574. doi:10.1111/j.1471-4159.2009.05932.x
Masliah E et al (2011) Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 6:e19338. doi:10.1371/journal.pone.0019338
Mastroeni D et al (2009) Microglial responses to dopamine in a cell culture model of Parkinson’s disease. Neurobiol Aging 30:1805–1817. doi:10.1016/j.neurobiolaging.2008.01.001
Mayeux R (2003) Epidemiology of neurodegeneration. Annu Rev Neurosci 26:81–104
McGeer PL, McGeer EG (2008) Glial reactions in Parkinson’s disease. Mov Disord 23:474–483. doi:10.1002/mds.21751
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291
Meulener M et al (2005) Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson’s disease. Curr Biol 15:1572–1577. doi:10.1016/j.cub.2005.07.064
Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62:1835–1838
Mitsumoto A, Nakagawa Y (2001) DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radical Res 35:885–893
Moehle MS et al (2012) LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 32:1602–1611. doi:10.1523/JNEUROSCI.5601-11.2012
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994a) Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 180:147–150
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T (1994b) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett 165:208–210
Moriwaki Y, Kim YJ, Ido Y, Misawa H, Kawashima K, Endo S, Takahashi R (2008) L347P PINK1 mutant that fails to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a proteasome-dependent manner. Neurosci Res 61:43–48. doi:10.1016/j.neures.2008.01.006
Mortiboys H, Johansen KK, Aasly JO, Bandmann O (2010) Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 75:2017–2020. doi:10.1212/WNL.0b013e3181ff9685
Nagakubo D, Taira T, Kitaura H, Ikeda M, Tamai K, Iguchi-Ariga SM, Ariga H (1997) DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun 231:509–513. doi:10.1006/bbrc.1997.6132
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. doi:10.1083/jcb.200809125
Narendra DP et al (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8:e1000298. doi:10.1371/journal.pbio.1000298
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31:763–780. doi:10.1002/humu.21277
Ooe H, Taira T, Iguchi-Ariga SM, Ariga H (2005) Induction of reactive oxygen species by bisphenol A and abrogation of bisphenol A-induced cell injury by DJ-1. Toxicol Sci 88:114–126. doi:10.1093/toxsci/kfi278
Orth M, Schapira AH (2002) Mitochondrial involvement in Parkinson’s disease. Neurochem Int 40:533–541
Palacino JJ et al (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279:18614–18622. doi:10.1074/jbc.M401135200
Parker WD Jr, Parks JK, Swerdlow RH (2008) Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res 1189:215–218. doi:10.1016/j.brainres.2007.10.061
Perry TL, Yong VW (1986) Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett 67:269–274
Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA 105:1638–1643. doi:10.1073/pnas.0709336105
Pridgeon JW, Olzmann JA, Chin LS, Li L (2007) PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol 5:e172. doi:10.1371/journal.pbio.0050172
Reynolds AD et al (2008) Nitrated alpha-synuclein-activated microglial profiling for Parkinson’s disease. J Neurochem 104:1504–1525. doi:10.1111/j.1471-4159.2007.05087.x
Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A (2007) Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol 82:235–246. doi:10.1016/S0074-7742(07)82012-5
Roodveldt C et al (2010) Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: differences between wild-type and Parkinson’s disease-linked mutants. PLoS One 5:e13481. doi:10.1371/journal.pone.0013481
Runchel C, Matsuzawa A, Ichijo H (2011) Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Signal 15:205–218. doi:10.1089/ars.2010.3733
Saijo K et al (2009) A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137:47–59. doi:10.1016/j.cell.2009.01.038
Sanders LH et al (2014) LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: reversal by gene correction. Neurobiol Dis 62:381–386. doi:10.1016/j.nbd.2013.10.013
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54:823–827
Schapira AH, Gu M, Taanman JW, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM (1998) Mitochondria in the etiology and pathogenesis of Parkinson’s disease. Ann Neurol 44:S89–S98
Segev-Amzaleg N, Trudler D, Frenkel D (2013) Preconditioning to mild oxidative stress mediates astroglial neuroprotection in an IL-10-dependent manner. Brain Behav Immun 30:176–185. doi:10.1016/j.bbi.2012.12.016
Shavali S, Brown-Borg HM, Ebadi M, Porter J (2008) Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett 439:125–128. doi:10.1016/j.neulet.2008.05.005
Sherer TB, Betarbet R, Greenamyre JT (2002) Environment, mitochondria, and Parkinson’s disease. Neuroscientist 8:192–197
Sherer TB, Betarbet R, Kim JH, Greenamyre JT (2003) Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci Lett 341:87–90
Shimura H et al (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25:302–305. doi:10.1038/77060
Sidhu A, Wersinger C, Moussa CE, Vernier P (2004) The role of alpha-synuclein in both neuroprotection and neurodegeneration. Ann N Y Acad Sci 1035:250–270. doi:10.1196/annals.1332.016
Singleton AB et al (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi:10.1126/science.1090278
Singleton AB, Farrer MJ, Bonifati V (2013) The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord 28:14–23. doi:10.1002/mds.25249
Sofroniew MV (2005) Reactive astrocytes in neural repair and protection. Neuroscientist 11:400–407. doi:10.1177/1073858405278321
Spillantini MG, Crowther RA, Jakes R, Hasegawa M (1998) Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Subramaniam SR, Vergnes L, Franich NR, Reue K, Chesselet MF (2014) Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol Dis 70:204–213. doi:10.1016/j.nbd.2014.06.017
Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H (2004) DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep 5:213–218. doi:10.1038/sj.embor.7400074
Thevenet J, Pescini Gobert R, Hooft van Huijsduijnen R, Wiessner C, Sagot YJ (2011) Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One 6:e21519. doi:10.1371/journal.pone.0021519
Thomas B (2009) Parkinson’s disease: from molecular pathways in disease to therapeutic approaches. Antioxid Redox Signal 11:2077
Todd AM, Staveley BE (2008) Pink1 suppresses alpha-synuclein-induced phenotypes in a Drosophila model of Parkinson’s disease. Genome 51:1040–1046. doi:10.1139/G08-085
Tran TA, Nguyen AD, Chang J, Goldberg MS, Lee JK, Tansey MG (2011) Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor-kappa B. PLoS One 6:e23660. doi:10.1371/journal.pone.0023660
Trudler D, Weinreb O, Mandel SA, Youdim MB, Frenkel D (2014) DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J Neurochem 129:434–447. doi:10.1111/jnc.12633
Vafai SB, Mootha VK (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491:374–383. doi:10.1038/nature11707
Venderova K et al (2009) Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum Mol Genet 18:4390–4404. doi:10.1093/hmg/ddp394
Vives-Bauza C et al (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107:378–383. doi:10.1073/pnas.0911187107
von Coelln R et al (2006) Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy. J Neurosci Off J Soc Neurosci 26:3685–3696. doi:10.1523/JNEUROSCI.0414-06.2006
Waak J et al (2009) Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J 23:2478–2489. doi:10.1096/fj.08-125153
Wang X et al (2012) LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 21:1931–1944. doi:10.1093/hmg/dds003
Watson MB et al (2012) Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol 237:318–334. doi:10.1016/j.expneurol.2012.06.025
Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ (2009) Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48:2045–2052. doi:10.1021/bi8019178
West AB et al (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 102:16842–16847. doi:10.1073/pnas.0507360102
Wood-Kaczmar A, Gandhi S, Wood NW (2006) Understanding the molecular causes of Parkinson’s disease. Trends Mol Med 12:521–528. doi:10.1016/j.molmed.2006.09.007
Xie W, Chung KK (2012) Alpha-synuclein impairs normal dynamics of mitochondria in cell and animal models of Parkinson’s disease. J Neurochem 122:404–414. doi:10.1111/j.1471-4159.2012.07769.x
Xiromerisiou G et al (2010) Genetic basis of Parkinson disease. Neurosurg Focus 28:E7. doi:10.3171/2009.10.FOCUS09220
Yokota T, Sugawara K, Ito K, Takahashi R, Ariga H, Mizusawa H (2003) Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem Biophys Res Commun 312:1342–1348
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337:1062–1065. doi:10.1126/science.1219855
Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ (1999) Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154:1423–1429. doi:10.1016/S0002-9440(10)65396-5
Zhang L et al (2005a) Mitochondrial localization of the Parkinson’s disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet 14:2063–2073. doi:10.1093/hmg/ddi211
Zhang W et al (2005b) Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 19:533–542. doi:10.1096/fj.04-2751com
Zhang W et al (2007) Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia 55:1178–1188. doi:10.1002/glia.20532
Zimprich A et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. doi:10.1016/j.neuron.2004.11.005
Acknowledgments
The work was supported by grant from the ISF to D.F.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Trudler, D., Nash, Y. & Frenkel, D. New insights on Parkinson’s disease genes: the link between mitochondria impairment and neuroinflammation. J Neural Transm 122, 1409–1419 (2015). https://doi.org/10.1007/s00702-015-1399-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-015-1399-z