
Abstract
Aim
Developmental and epileptic encephalopathies (DEEs) are a group of devastating disorders caused by epileptic activity, resulting in deterioration in developmental, cognitive, and motor functions. The number of genes identified as being responsible for DEEs has been increasing rapidly. However, despite a comprehensive molecular analysis, a molecular diagnosis can only be established in 50% of cases. The aim of this project is to use whole exome sequencing (WES) to determine the molecular etiology of DEEs in undiagnosed patients with a pedigree suggestive of an autosomal recessive single gene disease.
Methods
Three DEE families, having either consanguineous parents of an affected individual and/or having more than one affected offspring, were enrolled in the project. Prior to this project, the families had been evaluated using a next-generation sequencing panel including 16 DEE genes in a previous study; however, no molecular diagnosis could be established. In five cases from the three selected DEEs families in our study, the genetic etiology was investigated using WES.
Results
All patients in the study group had infantile onset epileptic seizures; however, semiologies varied. All patients presented with severe developmental delay. WES revealed biallelic disease causing mutations in DENDD5A, GRN, and TBCD genes in family 1, family 2, and family 3, respectively. In each family, the identified variants associated with the disease were segregated. Reverse phenotyping supported the molecular analysis.
Conclusion
This study provided a valuable contribution to the genotype-phenotype relationship by determining rare epilepsy syndromes in undiagnosed patients previously. WES is a useful diagnostic alternative, particularly in consanguineous families.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Developmental and epileptic encephalopathies (DEEs) are a group of devastating disorders caused by epileptic activities, resulting in deterioration in developmental, cognitive, and motor functions [1, 2]. Early-onset epileptic encephalopathies appear during neonatal and infantile period. DEEs are a heterogeneous group of disorders resulted by congenital or acquired cerebral damage. The number of genes which been identified as being responsible for DEEs has been increasing rapidly, and over 50 genes have been identified during the last 3 years [3,4,5]. The most common genetic anomalies causing DEEs are single nucleotide variations responsible for 30–40% of genetic etiology. Copy number variations are seen rarely and responsible for 5–10% of cases [5]. Molecular diagnosis of DEEs is not only important for confirming the clinical diagnosis but also for the selection of appropriate treatment options, giving an accurate prognosis as well as the provision of appropriate genetic counseling. There are several molecular analysis methods including gene panels, whole exome sequencing, or genome sequencing able to detect molecular pathology underlying DEEs. These methods have some advantages and disadvantages compared with each other. Hebbar et al. [5] had recommended selecting most appropriate test considering several factors such as age at seizure onset, severity of disease, other associated features, and patient insurance. However, despite a comprehensive molecular analysis, a molecular diagnosis can only be established in 20–40% of cases [2, 4, 6].
In a previous study, we evaluated the molecular etiology of early-onset DEEs using a targeted next-generation sequencing (NGS) panel [7]. This panel included 16 genes responsible for DEEs: ARX, CDKL5, CNTNAP2, FOLR1, FOXG1, LAMC3, MBD5, MECP2, NTNG1, PCDH19, PNKP, SCN1A, SCN1B, SCN2A, STXBP1, and KCNQ2. In that study group, a molecular diagnosis could only be made in 40% of cases. However, the diagnostic success rate was found to be higher in patients born to non-consanguineous parents (55.5%) than in patients born to consanguineous parents (16%). In consanguineous families, due to the panel containing mainly de novo and channel-encoding genes, a sufficient diagnostic success rate could not be achieved.
In this study, we aimed to evaluate the diagnostic utility of whole exome sequencing (WES) in previously molecularly undiagnosed DEE patients with a pedigree suggestive of an autosomal recessive single gene disease.
Material and methods
Study group
Five patients from 3 unrelated families having early-onset refractory seizures with global developmental delay and cognitive dysfunction were enrolled into the study. Prior to this project, we evaluated the molecular etiology of early-onset DEEs using a targeted NGS panel in 30 patients [7]. NGS panel which included 16 DEE genes (ARX, CDKL5, CNTNAP2, FOLR1, FOXG1, LAMC3, MBD5, MECP2, NTNG1, PCDH19, PNKP, SCN1A, SCN1B, SCN2A, STXBP1, KCNQ2) failed to identify a molecular etiology in 60% of these families. Among these patients who could not be molecularly diagnosed, patients those pedigrees suggestive of an autosomal recessive inheritance (having either consanguineous parents and/or more than one affected sibling) were evaluated. Demographic data, family history, and laboratory and imaging test results were all obtained from hospital records. Clinical, laboratory, and electrophysiological findings of the patients were evaluated by experienced pediatric neurologist and geneticist, and three families with no specific clinical diagnosis were selected.
The study was approved by the Ethical Committee of the Ege University Medical Faculty (Date: March 9, 2015; number: 15-9/49) and financially supported by Ege University Scientific Research Projects Coordination (Grant Number 17-TIP-006). Samples from the patients were collected in accordance with the Helsinki Declarations. Written informed consent for genetic testing was obtained from all cases or their parents/guardians.
Whole exome sequencing
Whole exome sequencing was performed on affected siblings in families 1 and 3 and affected offspring and healthy parents in family 2. Genomic DNA samples were extracted from 1 ml of peripheral blood leukocytes obtained from the parents and probands using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany). DNA quality and quantity were assessed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Approximately 2 μg of high-quality genomic DNA from each sample was prepared as the starting material for generating the sequencing library using the SeqCap EZ Human Exome Library v3.0 (F. Hoffmann-La Roche Ltd., Basel, Switzerland), in accordance with the manufacturer’s instructions.
SeqCap EZ Exome v3.0 Kit (F. Hoffmann-La Roche Ltd., Basel, Switzerland) was used for target enrichment. Paired-end sequencing was performed in all samples using the Illumina HiSeq 2000 platform (Illumina Inc., San Diego, CA).
Burrows–Wheeler Alignment (BWA) tool was used for mapping to the reference genome (hg19) and alignment [8]. Variant calling was performed using SAMtools and GATK best practices pipeline [9, 10].
Data analysis
Sequencing data was analyzed using Illumina VariantStudio 3.0 software and Integrative Genomics Viewer (IGV). Variants with a frequency of less than 0.5% were selected based on NCBI dbSNP build141 (http://www.ncbi.nlm.nih.gov/SNP/), 1000 Genomes Project (http://www.1000genomes.org/), Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org/), and Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/). Any variants which had a read depth below 10X were excluded. The impact of the variants on the protein structure was identified using several in silico prediction tools such as MutationTaster, SIFT, REVEL, DANN, dbscSNV, and CADD [11,12,13,14,15,16]. Conservation of residues across species was evaluated using PhyloP algorithm and GERP [17, 18]. Variant pathogenicity was classified in accordance with American Collage of Medical Genetics (ACMG) recommendations [19].
Confirmation and segregation analysis
The most likely disease-causing variants, identified by data analysis, were then confirmed using direct Sanger sequencing on ABI PRISM 3130 DNA analyzer (Applied Biosystems). Following this segregation analysis was performed.
Results
Study group
Family 1
In case 1, a 14-year-old boy was presented with a focal seizure at the age of 2.5 months. He was born at the 39th week of gestation via spontaneous vaginal delivery following an uneventful pregnancy. He had intellectual disability and motor retardation. There was no consanguinity between the parents; however, both were coming from the same small village.
On physical examination, weight, height, and head circumference were measured as 32 kg (< 3 p, − 2.9 SD), 141 cm (< 3 p, − 3.1 SD), and 48.5 cm (< 3 p, −4.9 SD), respectively. Intellectual disability was considered severe. Poor eye contact and repetitive stereotypic movements were observed. He also had microcephaly, wide nasal tip, short philtrum, open mouth, thick and everted lips, and spasticity on lower extremities (Fig. 1).

Dysmorphic facial features of the study group: case 1 (a–b), case 2 (c), case 3 (d), case 4 (e), and case 5 (f)
His biochemical and metabolic screening tests and hormonal profile, hearing, and ophthalmologic examination were all normal. Cranial MRI revealed periventricular nodular heterotopia, and EEG findings were compatible with multifocal epileptic activity.
In case 2, a 9-year-old boy is the second affected offspring of this family. Firstly, he displayed a generalized tonic-clonic seizure at the age of 3 months. He displayed refractory focal and generalized motor seizures in spite of antiepileptic treatment. In conjunction with his older sibling, intellectual disability, stereotypic movements, microcephaly, and dysmorphic facial features were also present.
Family 2
In case 3, a 10-year-old girl had a normal neurocognitive development until the age of 7 months. Loss of cognitive and motor functions was observed following this period. At the age of 12 months, she had a seizure in the form of infantile spasm for the first time. These seizures repeated 10–12 times a day and were resistant to antiepileptic drugs. She was born to consanguineous parents at the 38th week via cesarean section. Family history revealed that an older sister with neurodegeneration, severe developmental delay, and refractory epilepsy died due to pneumonia at the age of 7 years. She was a 10-year-old girl, and her parents described that she had normal development until 7 months of age. She exhibited severe loss of cognitive and motor functions within time. At the age of 12 months, she presented with epileptic spasms refractory to multiple antiepileptic drugs.
On physical examination, weight, height, and head circumference were 41 kg (10–25 p, 0.87 SD), 120 cm (< 3 p, − 3.07 SD), and 56.5 cm (> 97 p, 2.26 SD), respectively. She had severe developmental delay. Generalized hypotonia, macrocephaly, proptosis, and strabismus were observed (Fig. 1). Deep tendon reflexes were hypoactive.
Biochemical and metabolic screening tests were found to be normal. A cranial MRI revealed severe diffuse cerebral, cerebellar, and brain stem atrophy.
Family 3
In case 4, an 18-year-old boy was presented with focal motor seizures at the age of 12 months. Despite multidrug regimen, his seizures had not been fully controlled. He was born at the 39th week of gestation via a spontaneous vaginal delivery. There was no consanguinity between the parents; however, both were coming from the same small village.
On physical examination, weight, height, and head circumference were 47 kg (10–25 p, − 1.08 SD), 156 cm (10–25 p, − 0.81 SD), and 53 cm (− 1.72 SD), respectively. He had poor eye contact, and he was not able to speak. Severe global developmental delay was noted. Deep tendon reflexes were diminished.
His biochemical and metabolic screening tests and hormonal profile, auditory, and ophthalmologic examinations were all normal. EMG revealed sensorimotor axonal polyneuropathy. Cranial MRI demonstrated bilateral periventricular leukomalacia and thin corpus callosum.
In case 5, the second affected child of family 3 had refractory epileptic seizures since 7-months-old. She had also severe developmental delay, periventricular leukomalacia, and sensorimotor axonal polyneuropathy.
Clinical features, biochemical analysis, and electrophysiological and imaging test results of all patients are given in Table 1.
WES and segregation analysis
Sequencing depth for each sample was more than 95.16% of target regions having at least a 10-fold coverage. Detailed coverage metric values for each sample within the target region are given in Table 2.
Family-based variant filtering was performed (two affected siblings in families 1 and 3, proband and parents in family 2). After selection variants with a minor allele frequency of < 0.5% in public databases, a read depth > 10X, and having a predicted deleterious effect (missense, nonsense, frameshift, and splice site variants), 47, 140, and 152 variants remained in families 1, 2, and, 3, respectively. To evaluate their putative pathogenic impact, a series of prediction tools were used and a comprehensive literature search was performed for each variant.
In family 1, a homozygous c.110-3T>G variant in DENND5A gene was identified in both affected siblings. This variant had not been reported in any database to date. It was predicted as being deleterious based on its DANN, GERP, and dbscSNV (v1.1) scores. The variant classified as “variant of unknown significance (VUS)” in accordance with ACMG recommendations. Via segregation analysis, both parents were found to be heterozygous carriers. The mutation analysis of a healthy brother was negative.
In family 2, a homozygous missense variant c.480G>A (p.Asp144Asn) was detected in GRN gene. This variant (rs200591137) had been reported in gnomAD exomes database with a minor allele frequency of 1/125.729. It was considered to be deleterious based on prediction tools and classified as “VUS” in accordance with ACMG recommendations. Both parents were found to be heterozygous carriers for the same variant.
In family 3, two heterozygous variants in TBCD gene were identified in both affected siblings. The variant c.202C>T (p.Gln68Ter) had not previously been reported in the databases and was classified as “pathogenic.” The second variant c.880C>T (p.Arg294Trp) (rs200591137) had been reported in gnomAD genomes and gnomAD exomes databases with a minor allele frequency of 1/31.182 and 1/123.543, respectively. It was classified as “VUS” in accordance with ACMG recommendations. The amino acid Arg294 is highly conserved across species, and additionally, a different missense variant affecting the same amino acid has been registered on ClinVar as “likely pathogenic.” Segregation analysis revealed that the mother was a heterozygous carrier for c.202C> T variant, with the father a heterozygous carrier for c.880C>T variant.
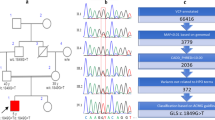
Confirmation and segregation analysis results of all families are given in Fig. 2.

Sequencing electropherograms of the mutations identified in family 1 (a), family 2 (b), and family 3 (c)
Reverse phenotyping
Following WES results, some additional tests were performed in families to evaluate genotype-phenotype correlation. In family 1, a cranial computed tomography was performed to investigate intracranial calcifications reported in biallelic DENND5A mutation carriers. Intracranial calcifications on deep white matter, basal ganglia, and thalamus were detected in both siblings. In family 2, a detailed family history revealed that the grandmother of the father showed clinical features of dementia and cognitive impairment at the age of 60 years. On peripheral blood smear using light microscopy, vacuolated lymphocytes were observed (Fig. 3). However, further electron microscopic evaluation for lipopigment accumulation or any biochemical tests to determine blood progranulin dosage were not available at the time of study. An ophthalmological examination revealed no signs for retinal degeneration. In family 3, a cranial MRI was considered necessary for the confirmation of cerebral atrophy. However, due to adverse behavioral characteristics of both siblings, it was an impossibility.

Peripheral blood smear of case 3 revealing vacuolated lymphocytes
Discussion
In this study, the diagnostic success of WES in three DEE families with pedigrees suggestive of autosomal recessive single gene disorders was evaluated. Prior to WES, the families had been analyzed using a next-generation sequencing panel which included 16 known DEE genes; unfortunately, no molecular diagnosis could be established [7]. WES was able to provide a diagnosis of rare Mendelian disorders in each family.
DENND5A, located at 11p15.4, plays a role in the regulation of membrane traffic between Golgi and endosomal complex. In 2016, Han et al. showed that biallelic DENND5A mutations were responsible for DEEs [20]. They reported two different homozygous null variants in three epileptic encephalopathy cases from two unrelated families. Dysmorphic facial features, microcephaly, and global growth retardation were described in all three cases. Seizures started in the newborn period, with tonic, myoclonic, or generalized tonic-clonic seizures all been observed. Intracranial calcifications had been noted in all three cases. They also showed the loss of function of DENND5A alternating neurite and dendrite outgrowth during neuronal differentiation via functional studies. In 2017, Anazi et al. also reported homozygous variants in the DENND5A gene in 2 different cases presenting with epilepsy, global developmental delay, and microcephaly [21]. Detailed clinical features of biallelic DENND5A mutations are given in Table 3. In our study, we identified a novel splice site DENND5A variant in family 1. The clinical findings of both siblings were consistent with DENND5A-related epileptic encephalopathy. As reported in the literature, seizures are early onset and have been observed in different semiologies. Dysmorphic facial features of our patients (triangular face, thick eyebrows, wide nasal tip, prominent nostrils, short and deep philtrum, open mouth, and thick lower and upper vermilion) have been found to be similar to the patients described in the literature.
GRN gene is located at 17q21.31 and encodes the progranulin glycoprotein. It is known that monoallelic loss of function GRN mutations are responsible for frontotemporal lobar degeneration (FTLD) [22]. FTLD is the most common cause of dementia after Alzheimer’s disease. In 2012, Smith et al. described a homozygous mutation in the GRN gene in two siblings presenting with adult onset progressive vision loss, retinal dystrophy, recurrent seizures, ataxia, and cerebellar atrophy [23]. Supporting molecular findings, they also showed that plasma progranulin levels were low in heterozygous carriers and undetectable in homozygous in related family. The authors considered that biallelic GRN mutations were responsible for neuronal ceroid lipofuscinosis (NCL) type 11. In a further study conducted by the same group, fingerprint storage and the absence of progranulin protein were observed in the skin biopsy and peripheral blood leukocytes of those two siblings [24]. Following this, three further NCL cases carrying homozygous GRN mutations were reported (Table 4) [25, 26].
Neuronal ceroid lipofuscinosis is clinically and genetically heterogeneous neurodegenerative disease [27, 28]. To date, 14 different types have been identified. The disease is characterized by progressive loss of cognitive and motor functions, retinal degeneration, cerebellar atrophy, and seizures. It is classified as infantile, late-infantile, juvenile, or adult onset based on the age of onset of clinical features. The clinical features of the patient in family 2 including infantile onset progressive neurodegeneration, epilepsy, and cerebellar atrophy were considered to be compatible with NCL. Vacuolated lymphocytes shown in peripheral blood smear of case 3 supported the diagnosis. To our knowledge, our patient carrying biallelic GRN gene variant is the first described infantile onset NCL type 11 case in the literature. However, while visual loss and retinal degeneration are major clinical features of NCL type 11 in adults, no retinal involvement was observed in our patient or her sister during their clinical follow-up. Kamate et al. reported two siblings whose symptoms appeared at the age of 8 and 13, respectively [26]. No retinal involvement was observed in their study either. Retinal degeneration may be specific to NCL type 11 in adulthood.
Biallelic TBCD gene variants are responsible for early-onset and progressive encephalopathy [29]. To date, approximately 34 cases have been described in the literature [29,30,31,32,33]. The disease is characterized by progressive encephalopathy which usually begins during infantile period. Epilepsy has been described in approximately 90% of cases, and seizure types vary [33]. The clinical findings of two affected siblings in family 3 were considered to be compatible with the clinical picture caused by biallelic TBCD gene mutations.
The majority of DEEs are resulted by de novo variants in responsible genes [4]. However, in a recent study conducted by Papuc et al. [34], autosomal recessive inheritance was found to be responsible for 38% of molecularly diagnosed cases. In a highly consanguineous study group, Nashabat et al. [35] also identified autosomal recessive etiology in 50% of 72 molecularly characterized early infantile epileptic encephalopathy cases. In our previous study, a targeted gene panel failed to succeed a high molecular diagnostic rate in consanguineous families. In three of these undiagnosed families, WES revealed molecular diagnosis of rare autosomal recessive genetic disorders. WES can be considered as a first step diagnostic test in the DEE families with consanguineous marriage and/or two or more affected siblings.
The limitation of the present study is that we could not perform functional studies to determine the disease-causing effects of the identified variants in the patients. Therefore, the molecular diagnosis could only be supported by segregation analysis and reverse phenotyping.
To conclude, in this study, three rare syndromes associated with epilepsy were identified, contributing to genotype-phenotype relationship. It was thought that WES might provide a more appropriate diagnostic method in DEE cases with suspected autosomal recessive inheritance than a gene targeted panel.
References
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshe SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM (2017) ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58(4):512–521. https://doi.org/10.1111/epi.13709
Noh GJ, Jane Tavyev Asher Y, Graham JM Jr (2012) Clinical review of genetic epileptic encephalopathies. Eur J Med Genet 55(5):281–298. https://doi.org/10.1016/j.ejmg.2011.12.010
Milh M, Riccardi F, Denis J (2019) Genetics of neonatal onset epilepsies: an overview. Rev Neurol 176:2–9. https://doi.org/10.1016/j.neurol.2019.01.396
Moller RS, Dahl HA, Helbig I (2015) The contribution of next generation sequencing to epilepsy genetics. Expert Rev Mol Diagn 15(12):1531–1538. https://doi.org/10.1586/14737159.2015.1113132
Hebbar M, Mefford HC (2020) Recent advances in epilepsy genomics and genetic testing. F1000Research 9. https://doi.org/10.12688/f1000research.21366.1
McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE (2016) The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol 15(3):304–316. https://doi.org/10.1016/S1474-4422(15)00250-1
Gokben S, Onay H, Yilmaz S, Atik T, Serdaroglu G, Tekin H, Ozkinay F (2017) Targeted next generation sequencing: the diagnostic value in early-onset epileptic encephalopathy. Acta Neurol Belg 117(1):131–138. https://doi.org/10.1007/s13760-016-0709-z
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Koboldt DC, Larson DE, Wilson RK (2013) Using VarScan 2 for Germline variant calling and somatic mutation detection. Current protocols in bioinformatics 44:15 14 11–17. https://doi.org/10.1002/0471250953.bi1504s44
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303. https://doi.org/10.1101/gr.107524.110
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, Cannon-Albright LA, Teerlink CC, Stanford JL, Isaacs WB, Xu J, Cooney KA, Lange EM, Schleutker J, Carpten JD, Powell IJ, Cussenot O, Cancel-Tassin G, Giles GG, MacInnis RJ, Maier C, Hsieh CL, Wiklund F, Catalona WJ, Foulkes WD, Mandal D, Eeles RA, Kote-Jarai Z, Bustamante CD, Schaid DJ, Hastie T, Ostrander EA, Bailey-Wilson JE, Radivojac P, Thibodeau SN, Whittemore AS, Sieh W (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99(4):877–885. https://doi.org/10.1016/j.ajhg.2016.08.016
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46(3):310–315. https://doi.org/10.1038/ng.2892
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4(7):1073–1081. https://doi.org/10.1038/nprot.2009.86
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7(8):575–576. https://doi.org/10.1038/nmeth0810-575
Quang D, Chen Y, Xie X (2015) DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31(5):761–763. https://doi.org/10.1093/bioinformatics/btu703
Jian X, Boerwinkle E, Liu X (2014) In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res 42(22):13534–13544. https://doi.org/10.1093/nar/gku1206
Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20(1):110–121. https://doi.org/10.1101/gr.097857.109
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6(12):e1001025. https://doi.org/10.1371/journal.pcbi.1001025
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Han C, Alkhater R, Froukh T, Minassian AG, Galati M, Liu RH, Fotouhi M, Sommerfeld J, Alfrook AJ, Marshall C, Walker S, Bauer P, Scherer SW, Riess O, Buchert R, Minassian BA, McPherson PS (2016) Epileptic encephalopathy caused by mutations in the guanine nucleotide exchange factor DENND5A. Am J Hum Genet 99(6):1359–1367. https://doi.org/10.1016/j.ajhg.2016.10.006
Anazi S, Maddirevula S, Faqeih E, Alsedairy H, Alzahrani F, Shamseldin HE, Patel N, Hashem M, Ibrahim N, Abdulwahab F, Ewida N, Alsaif HS, Al Sharif H, Alamoudi W, Kentab A, Bashiri FA, Alnaser M, AlWadei AH, Alfadhel M, Eyaid W, Hashem A, Al Asmari A, Saleh MM, AlSaman A, Alhasan KA, Alsughayir M, Al Shammari M, Mahmoud A, Al-Hassnan ZN, Al-Husain M, Osama Khalil R, Abd El Meguid N, Masri A, Ali R, Ben-Omran T, El Fishway P, Hashish A, Ercan Sencicek A, State M, Alazami AM, Salih MA, Altassan N, Arold ST, Abouelhoda M, Wakil SM, Monies D, Shaheen R, Alkuraya FS (2017) Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry 22(4):615–624. https://doi.org/10.1038/mp.2016.113
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442(7105):916–919. https://doi.org/10.1038/nature05016
Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 90(6):1102–1107. https://doi.org/10.1016/j.ajhg.2012.04.021
Canafoglia L, Morbin M, Scaioli V, Pareyson D, D'Incerti L, Fugnanesi V, Tagliavini F, Berkovic SF, Franceschetti S (2014) Recurrent generalized seizures, visual loss, and palinopsia as phenotypic features of neuronal ceroid lipofuscinosis due to progranulin gene mutation. Epilepsia 55(6):e56–e59. https://doi.org/10.1111/epi.12632
Almeida MR, Macario MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I (2016) Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging 41:200 e201–200 e205. https://doi.org/10.1016/j.neurobiolaging.2016.02.019
Kamate M, Detroja M, Hattiholi V (2019) Neuronal ceroid lipofuscinosis type-11 in an adolescent. Brain and Development 41(6):542–545. https://doi.org/10.1016/j.braindev.2019.03.004
Nita DA, Mole SE, Minassian BA (2016) Neuronal ceroid lipofuscinoses. Epileptic Disord 18(S2):73–88. https://doi.org/10.1684/epd.2016.0844
Anderson GW, Goebel HH, Simonati A (2013) Human pathology in NCL. Biochim Biophys Acta 1832(11):1807–1826. https://doi.org/10.1016/j.bbadis.2012.11.014
Miyake N, Fukai R, Ohba C, Chihara T, Miura M, Shimizu H, Kakita A, Imagawa E, Shiina M, Ogata K, Okuno-Yuguchi J, Fueki N, Ogiso Y, Suzumura H, Watabe Y, Imataka G, Leong HY, Fattal-Valevski A, Kramer U, Miyatake S, Kato M, Okamoto N, Sato Y, Mitsuhashi S, Nishino I, Kaneko N, Nishiyama A, Tamura T, Mizuguchi T, Nakashima M, Tanaka F, Saitsu H, Matsumoto N (2016) Biallelic TBCD mutations cause early-onset neurodegenerative encephalopathy. Am J Hum Genet 99(4):950–961. https://doi.org/10.1016/j.ajhg.2016.08.005
Stephen J, Nampoothiri S, Vinayan KP, Yesodharan D, Remesh P, Gahl WA, Malicdan MCV (2018) Cortical atrophy and hypofibrinogenemia due to FGG and TBCD mutations in a single family: a case report. BMC Med Genet 19(1):80. https://doi.org/10.1186/s12881-018-0597-6
Edvardson S, Tian G, Cullen H, Vanyai H, Ngo L, Bhat S, Aran A, Daana M, Da'amseh N, Abu-Libdeh B, Cowan NJ, Heng JI, Elpeleg O (2016) Infantile neurodegenerative disorder associated with mutations in TBCD, an essential gene in the tubulin heterodimer assembly pathway. Hum Mol Genet 25(21):4635–4648. https://doi.org/10.1093/hmg/ddw292
Pode-Shakked B, Barash H, Ziv L, Gripp KW, Flex E, Barel O, Carvalho KS, Scavina M, Chillemi G, Niceta M, Eyal E, Kol N, Ben-Zeev B, Bar-Yosef O, Marek-Yagel D, Bertini E, Duker AL, Anikster Y, Tartaglia M, Raas-Rothschild A (2017) Microcephaly, intractable seizures and developmental delay caused by biallelic variants in TBCD: further delineation of a new chaperone-mediated tubulinopathy. Clin Genet 91(5):725–738. https://doi.org/10.1111/cge.12914
Zhang Y, Zhang L, Zhou S (2019) Developmental regression and epilepsy of infancy with migrating focal seizures caused by TBCD mutation: a case report and review of the literature. Neuropediatrics. 51:068–071. https://doi.org/10.1055/s-0039-1698423
Papuc SM, Abela L, Steindl K, Begemann A, Simmons TL, Schmitt B, Zweier M, Oneda B, Socher E, Crowther LM, Wohlrab G, Gogoll L, Poms M, Seiler M, Papik M, Baldinger R, Baumer A, Asadollahi R, Kroell-Seger J, Schmid R, Iff T, Schmitt-Mechelke T, Otten K, Hackenberg A, Addor MC, Klein A, Azzarello-Burri S, Sticht H, Joset P, Plecko B, Rauch A (2019) The role of recessive inheritance in early-onset epileptic encephalopathies: a combined whole-exome sequencing and copy number study. Eur J Human Genet 27(3):408–421. https://doi.org/10.1038/s41431-018-0299-8
Nashabat M, Al Qahtani XS, Almakdob S, Altwaijri W, Ba-Armah DM, Hundallah K, Al Hashem A, Al Tala S, Maddirevula S, Alkuraya FS, Tabarki B, Alfadhel M (2019) The landscape of early infantile epileptic encephalopathy in a consanguineous population. Seizure 69:154–172. https://doi.org/10.1016/j.seizure.2019.04.018
Funding
This work was supported by the Ege University Scientific Research Projects Coordination (Grant Number 17-TIP-006).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study was approved by the Ethical Committee of the Ege University Medical Faculty (Date: March 9, 2015; number: 15-9/49). Written consent was obtained from all subjects.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Isik, E., Yilmaz, S., Atik, T. et al. The utility of whole exome sequencing for identification of the molecular etiology in autosomal recessive developmental and epileptic encephalopathies. Neurol Sci 41, 3729–3739 (2020). https://doi.org/10.1007/s10072-020-04619-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-020-04619-8