
Abstract
Idiopathic pulmonary hemosiderosis (IPH) is a rare disease of unknown etiology. Due to the frequent findings of autoimmune antibodies — autoantibodies, immunologic causation of the diffuse alveolar hemorrhage in IPH has been proposed, to assess the prevalence/frequency and type of autoantibodies in pediatric patients with IPH. In addition, the patient demographics, diagnostic modalities used to diagnose IPH, treatment, and outcomes were also evaluated. Scoping review: The PubMed, Medline, and Embase databases were searched with appropriate MeSH terms to identify relevant papers consistent with the defined inclusion criteria. Thirteen observational studies comprising a total of 352 pediatric patients were included in this review. The majority of subjects were girls 217 out of 352 (61.6%). The mean and median ages of patients ranged from 3.1–6.5 years to 2.3–7 years, respectively. In the 10 studies that specified the number of patients in their cohorts with either at least one positive autoantibody or no antibody, the overall prevalence of autoantibodies was 76 out of 288 patients (26.4%). The prevalence of specific antibodies was as follows: ANA, 20.3%; ANCA, 17%; anti-dsDNA, 9.1%; RF, 12%; anti-SMA, 23.2%; and celiac antibodies, 25.9%. Cow’s milk protein allergy was present in 16.2% of the children. The significance of an association between IPH and the presence of autoantibodies has not been clarified. The autoantibodies could be suggestive of an overall immune dysregulation rather than causation. However, limited evidence based on a single study suggests that the presence of ANA may be associated with a higher risk of recurrence and worse outcomes. Further research, including prospective studies, will be crucial to explore a possible genetic linkage between vasculitides, systemic rheumatologic diseases, and IPH.
Key Points • Approximately one in four pediatric patients with IPH demonstrate autoantibodies. • Antibodies suggestive of celiac disease are the most prevalent autoantibody. • The presence of antinuclear autoantibody may be associated with unfavorable outcomes. • All patients with a positive ANCA demonstrated anti-myeloperoxidase (MPO-ANCA) antibody. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary hemosiderosis (IPH) is a rare disease of unknown etiology [1,2,3]. Based on the reported cases in the literature, IPH is more prevalent in children than adults [2]. Patients with IPH suffer from recurrent episodes of diffuse alveolar hemorrhage (DAH), causing a variable degree of respiratory symptoms. The classic clinical triad in IPH includes hemoptysis, radiologic chest infiltrate, and anemia [4]. This constellation is, however, uncommon, and patients may present with nonspecific symptoms, such as cough, chest pain, shortness of breath, fever, and fatigue [5]. The symptoms can be chronic, relapsing, and remitting, but massive pulmonary hemorrhage precipitating acute respiratory failure can also occur [5,6,7]. Recurrent episodes of DAH may result in pulmonary fibrosis and end-stage lung disease [8]. Additionally, pediatric patients may also present with unexplained anemia. IPH is a diagnosis of exclusion, and all competing diagnoses therefore need to be carefully evaluated and excluded [1, 2, 9,10,11].
The pathobiology of DAH in IPH remains uncertain. Although multiple pathophysiologic models have been proposed, the immunological hypothesis is strongly favored by clinicians [12,13,14]. One of the rationales that have promoted the immunologic hypothesis is the occurrence of autoimmune antibodies — autoantibodies in patients with IPH. Sometimes, these autoantibodies are found concurrently at the time of diagnosis, and at other times, they develop during the subsequent course of the disease, occasionally after years. The exact prevalence of autoantibodies in pediatric patients with IPH is unknown but has been reported as high as 92% in an observational study [13].
Previous reports have described several autoantibodies in patients with IPH. These include antinuclear antibody (ANA), antineutrophil cytoplasmic antibody (ANCA), rheumatoid factor (RF), anti-cyclic citrullinated peptide (anti-CCP), anti-smooth muscle antibody (anti-SMA), anti-Sjogren syndrome antibody (anti-SSA), and antibodies associated with celiac disease [12]. However, the significance of these autoantibodies in the pathogenesis of DAH is unknown. In addition, DAH mimicking IPH (bland pulmonary hemorrhage on lung biopsy) has been described in a number of autoimmune and rheumatologic diseases [15], raising the question that IPH may be an attenuated presentation of a systemic disease, and if the patient survives long enough, other organ involvements may be observed. Indeed, there are a few reports of patients who were diagnosed with IPH by lung biopsy but subsequently developed a vasculitis syndrome [16,17,18]. We, therefore, undertook this project to review the literature regarding the occurrence of autoantibodies in pediatric patients with IPH to elucidate the overall prevalence and identify any potential clinical association with causation and outcomes. We only included pediatric patients for this study as no prospective or retrospective studies exist in adult patients with IPH.
Methods
This scoping review was performed and reported in accordance with Preferred Reporting Items for Systematic Review and Meta-Analyses extension for Scoping Reviews (PRISMA-ScR) guidelines. Approval from Institutional Review Board (IRB) and informed patient consent were not obtained for this study as it was a scoping review of previously published literature. The study was not registered in PROSPERO as this is not a systematic review.
Study objectives
The primary objectives of the study were to determine the prevalence/frequency and type of autoantibodies in pediatric patients with IPH. The secondary objectives were to assess patient demographics, diagnostic modalities used to diagnose IPH, treatment, and outcomes in this patient population.
Search criteria
The Medline, PubMed, and Embase databases were searched using the following formula:
“(Idiopathic pulmonary hemosiderosis OR IPH) AND (Pediatric patients OR children)” and “(Idiopathic pulmonary hemosiderosis OR IPH) AND (Pediatric patients OR children) AND Antibody.” The bibliography of the identified literature was then carefully searched to identify additional articles that met the inclusion criteria for our study.
Inclusion criteria
Articles fulfilling the following criteria were included: (1) prospective or retrospective case control or cohort studies that reported the incidence of autoantibodies in pediatric patients with a diagnosis of IPH; (2) patients initially diagnosed with IPH either clinically (consistent clinical presentation, radiology and demonstration of hemosiderin-laden macrophages from sputum, bronchoalveolar lavage [BAL], or gastric aspirate) or by lung biopsy, who subsequently developed autoantibodies and/or vasculitis were included; 3) and articles published in English language in peer-reviewed journals between January 1, 1980 and October 4, 2021.
Exclusion criteria
The exclusion criteria were as follows: (1) articles that reported simultaneous findings of autoantibodies and vasculitis from lung and/or kidney biopsies during the initial diagnosis of IPH (we considered that patient to be suffering from vasculitis and not IPH); (2) articles on patients with biopsy proven “bland pulmonary hemorrhage” but with clinical and laboratory findings suggestive of connective tissue disease; and (3) articles reported as case reports or abstracts.
Data collection process
Two researchers (BKS and AB) independently performed eligibility assessments of the data in a standardized manner to identify articles that reported autoantibodies in patients with IPH. The reviewers were blinded to each other’s assessment. Any disagreement between the researchers was resolved by discussion and input from a third investigator, PC. After the citations were identified, duplicate records were removed. The abstract of each citation was then screened for relevance to our study. Citations that were deemed unrelated to our research after independent evaluation of the abstract by the reviewers were excluded. The full texts of the remaining citations were then reviewed, along with careful examination of the bibliography of the published articles.
Data items
Included studies were coded, and the extracted data from the studies were then tabulated in a standardized Excel sheet (Microsoft Corporation). The following data were gathered from full-text articles: study design, year of reporting, country of the study, patient demographics, prevalence of antibodies, type of autoantibodies, temporal relationship of autoantibody determination with the diagnosis of IPH, initial presentation, as well as course and clinical outcome of disease.
Study risk and bias assessment
The quality of the studies and risks of bias were not assessed as this is a scoping review of the literature.
Results
Study characteristics
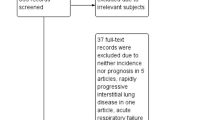
Thirteen manuscripts that fulfilled the eligibility criteria were included in this scoping review [13, 14, 19,20,21,22,23,24,25,26,27,28,29]. Figure 1 shows the selection process of the articles.

Flow chart showing selection process of the included studies
All studies were retrospective and included a cohort of pediatric patients diagnosed with IPH. The oldest study was published in 1999 and the latest study in 2021 (Table 1). Two of the French studies were reviews [13, 22] from a French database on rare lung diseases established in 2008 comprising 12 pediatric centers. The included studies were heterogeneous and assessed a wide range of outcomes (Table 1).
Subject demographics
A total of 352 children (61.6% girls) were included in this study. The mean and/or median age during the diagnosis of IPH was specified in all but one study [29]; the mean age ranged from 3.1 to 6.5 years and the median age from 2.3 to 7 years (observed range 0.16–18.2 years). Kiper et al. reported a retrospective cohort of 23 children whose age ranged from 11 months to 13 years with a calculated median of 6 years [29].
A delay in the diagnosis of IPH was reported in 6/13 studies [14, 19,20,21, 26, 27, 29]. The interval between the initial presentation and the final diagnosis varied widely, ranging from 2 months to 12 years (Table 2). In two studies, approximately 75% of patients were initially misdiagnosed [19, 21].
A high rate of consanguinity (48%) was reported in two Turkish studies [19, 29]. Down syndrome was present in 9/34 (26.4%) and 5/25 (20%) of cohorts reported by Alimi et al. [22] and Taytard et al. [13], respectively. Among the studies, 5/13 (38.5%) included patients being diagnosed with IPH before 1990.
Diagnosis of IPH
The diagnosis of IPH was made primarily by the clinical presentation, radiologic abnormalities on chest X-ray, and the demonstration of hemosiderin-laden macrophages in the sputum, gastric aspirate, or BAL. Rarely, IPH was diagnosed from clinical and radiologic appearance alone without documentation of hemosiderin-laden macrophages [20, 28].
Among the studies specifying the number of patients who underwent BAL or gastric aspiration, BAL was performed in 95/185 (51.3%) and gastric aspiration in 70/129 (54.2%) of the patients. Lung biopsies were performed in 39/209 (18.7%) of patients.
Prevalence of autoantibodies
The overall frequency of autoantibodies varied widely, ranging from 0 to 92% among the reported cohorts (Table 2) [13, 21]. A thorough evaluation for antibodies was not performed in all studies. Studies that included patients who were diagnosed with IPH prior to 1990 (approximately when testing for ANCA became standard practice) typically had less consistent evaluation for autoantibodies (less number of patients and type of antibodies tested) [14, 28, 29]. Although reported as part of the diagnostic workup, no study assessed the incidence of autoantibodies as the primary outcome measure (Table 1).
In the 10 studies that specified the number of patients with either at least one positive or no autoantibody in their cohort, the overall incidence of autoantibodies was 76/288 (26.4%), and some patients had more than one antibody. Taytard et al. reported that 23/25 (92%) of their patients had a positive antibody screening; among these patients, 17/23 (74%) presented antibodies at the time of diagnosis of IPH, whereas 6/24 (26%) developed antibodies during follow-up [13]. Three other studies specified the development of autoantibodies during the follow-up period (Table 2) [14, 19, 24]. A few patients subsequently developed autoimmune diseases [14]. Clainche et al. reported the development of rheumatoid arthritis-like illness in 3/15 patients [14].
Type of autoantibodies
Patients reported in the past decade [13, 19,20,21,22] had more complete antibody profile than in studies reported in the late 1990s and early 2000s [14, 27,28,29]. The most commonly evaluated antibodies included ANA, ANCA, anti-double-stranded DNA (anti-dsDNA), RF, anti-SMA, antibodies for celiac disease, and allergy to cow’s milk protein (Table 2). The occurrence of particular antibodies in the individual studies is summarized in Table 2.
The exact incidence for specific autoantibodies was difficult to ascertain as this was not reported in all studies. Based on the studies reporting specific autoantibodies, the pooled incidences were ANA 26/128 (20.3%) [7 studies], ANCA 21/123 (17%) [7 studies], anti-dsDNA 4/44 (9.1%) [3 studies], RF 6/50 (12%) [3 studies], anti-SMA 10/43 (23.2%) [3 studies], celiac antibodies 23/89 (25.9%) [4 studies], and cow’s milk protein allergy 25/154 (16.2%) [n = 9]. The presence of autoantibodies was generally not associated with the development of autoimmune diseases except in a few patients [13]. Le Clainche et al. reported three patients in their cohort who developed rheumatoid arthritis like illness during follow-up [14]. Furthermore, some studies reported a reduction in antibody titer and disappearance of autoantibodies in some patients during subsequent follow-up [13, 19].
Treatment and outcomes
The majority of patients received immunosuppressive therapy. The first-line therapy included systemic corticosteroid with or without additional immunomodulators. Recurrence of disease occurred frequently in all studies. Experimental treatment included liposteroid, mesenchymal stem cell transplant in addition to corticosteroid, and leflunomide [23, 24]. Common second-line agents included hydroxychloroquine, methotrexate, azathioprine, and cyclophosphamide.
Only one study specifically addressed clinical outcomes in patients with IPH with and without the presence of autoantibodies [19]. Hizal et al. reported 7/17 and 1/25 patients to be ANA positive in the respective cohorts with “unfavorable” and “favorable” outcomes. This association was found to be statistically significant, p = 0.008. The authors, therefore, concluded that ANA positivity was associated with an unfavorable response to therapy, including recurrence of bleeding [19].
Discussion
Based on the reported cases in the literature, IPH affects pediatric patients more often than adults. Approximately two-thirds of all reported cases have included pediatric patients [2]. The estimated incidence of IPH in selective pediatric groups ranges from 0.24 to 1.23 cases per million children per year [30, 31]. A review of all reported adult cases between 2000 and 2015 identified only 37 cases [5]. Among pediatric patients, IPH is more prevalent in girls [13], which was also the case in this review. More than 60% of subjects included in our study were girls. In contrast, males may be affected more frequently in adult onset disease [5]. The onset of disease predominantly occurs before the age of 10 years [14], although patients of all ages have been reported in the literature [5]. Initial misdiagnosis and a delay in diagnosis are common in IPH patients. Most of our included studies reported a significant delay in the diagnosis. Chen et al. reported a mean diagnostic delay of 2.3 years in adult patients [5]. The delay in diagnosis is likely due to nonspecificity of clinical presentation and lack of awareness among clinicians. Additionally, hemoptysis, which is the most prevalent symptom among adults, may be completely absent in children, making it even more challenging [15].
Due to their retrospective nature, antibody profiles were not available for all patients in the majority of studies. Even when evaluated, not all relevant antibodies were checked routinely. The ANA became available for clinical use in 1957 [32]. However, the existence and significance of ANCA antibodies were unknown until the mid-1980s. Therefore, descriptions of IPH before that point in time (especially when the diagnosis was made without lung biopsy) may have mistakenly included patients with ANCA-associated vasculitis (AAV) [33,34,35].
The data regarding pediatric AAV are less robust in the literature. The incidence of AAV in adults can vary based on geographic location. For example, in Europe, the annual incidence of AAV ranges from 13 to 20 cases per million [36]. In contrast, the estimated incidence of AAV in children approximates 0.45 cases per million per year (nearly 30 times lower than adults) [37]. Unlike the adult population, girls are affected more frequently than boys. The median age for disease onset is 11.5 years [38]. Alveolar hemorrhage is present in approximately 30% of patients and can be life threatening [37, 39]. The clinical manifestations in AAV may vary depending on the type of autoantibody. Although early research suggested otherwise [40], the incidence of DAH appears to be more frequent in patients with anti-proteinase 3 (PR3-ANCA) than with anti-myeloperoxidase (MPO-ANCA) [41, 42]. Patients with PR3-ANCA are also more likely to suffer from relapse of their disease [43]. In patients with AAV, the presence of DAH is associated with an increased mortality [44].
A complete assessment of ANCA requires both indirect immunofluorescence assay (IIF) and enzyme-linked immunosorbent assay (ELISA) testing [15]. The IIF is performed by incubating the serum from a patient suspected of AAV to the ethanol-fixed human neutrophils. In the presence of ANCA in the serum, two distinctive patterns can be observed on IIF. These patterns are perinuclear (p-ANCA) and cytoplasmic (c-ANCA). p-ANCA pattern signifies staining around the nucleus, while a c-ANCA pattern is characterized by diffuse cytoplasmic fluorescence. Sometimes a nonspecific pattern, known as atypical (a-ANCA), can be seen in patients with non-vasculitic autoimmune disorders. Although the majority of p-ANCA and c-ANCA antibodies are directed against anti-MPO and anti-PR3, respectively, this is not the case for all patients. Definitive identification of the antigenic target requires further testing by ELISA. The ELISA can easily differentiate among antibodies directed against anti-MPO, anti-PR3, and antibodies that are not directed to these antigens (so-called a-ANCA). Although more sensitive, an accurate estimation of the immunofluorescence pattern may be challenging, as this depends on the expertise of the operator. For example, a positive ANA pattern may be reported as a false-positive “p-ANCA.” Therefore, simultaneous testing with IIF and ELISA provides greater sensitivity and specificity.
It is crucial to emphasize that the presence of ANCA does not always imply a diagnosis of AAV. A positive ANCA can be present in virtually all systemic rheumatologic diseases [45]. However, most of these ANCA autoantibodies are directed towards non-MPO and non-PR3 antigens, designating “atypical” ANCA. PR3-ANCA and MPO-ANCA are extremely rare in CTDs [45]. Patients with inflammatory bowel disease frequently demonstrate atypical p-ANCA pattern with non-MPO-ANCA antibodies [46]. ANCA, including PR3- and MPO-ANCA, can also be present in patients with infective bacterial endocarditis, tuberculosis, as well as in parasitic and fungal infections [47,48,49].
The specific antigenic target of ANCA antibodies was not reported in the observational studies included in this review. Taytard et al. [13] and Clainche et al. [14] did not specify the occurrence of PR3-ANCA or MPO-ANCA in their patients. Yang et al. reported one patient each with a positive p-ANCA and c-ANCA by immunofluorescence, but the authors did not further specify the target further with ELISA [20]. Positive ANCA tests have also been reported in case reports and case series of patients with IPH. The ELISA primarily demonstrated MPO-ANCA positivity [16, 17, 50]. A total of 3/5 and 2/5 patients demonstrated perinuclear and cytoplasmic distribution of MPO-ANCA, respectively [16, 17, 50]. In the literature search, we did not identify any patient with IPH who had PR3-ANCA.
The role of ANCA in the causation of DAH in IPH is unclear. AAV is caused by pathologic ANCA, which histologically presents as necrotizing glomerulonephritis on kidney biopsy. In case of pulmonary involvement, pauci-immune small vessel vasculitis with parenchymal necrosis and granuloma formation (except in MPA) can be seen [15, 51]. Based on reported studies, the incidence of DAH in AAV varies between 8 and 36% [52]. As histopathologic analysis of the lung in IPH reveals bland pulmonary hemorrhage (BH), the presence of ANCA may not signify causation, instead a manifestation of overall immunologic dysregulation in IPH patients. However, it is also possible that some cases of IPH present as an attenuated form of AAV, and that with prolonged survival, a more systemic involvement of AAV becomes evident. The literature is limited in establishing any definitive association between IPH and AAV. However, given the yearly incidence of IPH and AAV being 1.2 and 0.45 cases per million in children, co-occurrence of these two rare diseases by chance alone would be 0.00054 cases per year per million or 0.54 cases per trillion children. Our literature review identified four pediatric patients with IPH who subsequently developed AAV [16, 17, 50]. This may suggest that a concurrent diagnosis of IPH and AAV may not be solely by chance but that a yet unidentified association may exist.
ANA has high sensitivity but low specificity for the diagnosis of a number of autoimmune diseases. A significantly elevated ANA titer, defined as 1:80 or higher, is present in approximately 2.5% of the apparently healthy population [53]. Similarly, the prevalence of a positive ANA titer (1:80 or higher dilution) in the pediatric population was 3% in a study from Thailand [54]. The prevalence increased to 15% with a cutoff value of 1:40. A positive ANA titer was present in 20.3% of the tested patients in our study, which appears to be higher than reported among apparently healthy pediatric and adult population, especially with a cutoff value of 1:80. However, since the specific ANA titers were not reported, the positive ANA in patients with IPH may have been coincidental.
Rheumatoid factor is also a nonspecific antibody, and a high titer can be present in many autoimmune diseases other than rheumatoid arthritis, and RF is also found in healthy individuals [55]. Additionally, chronic infections and malignancy can also be associated with positive RF [56, 57]. A high antibody titer increases the specificity of the test in the evaluation of autoimmune diseases. Although more specific than RF for the diagnosis of RA, anti-CCP can also be positive in patients with other rheumatologic conditions, such as SLE, Sjogren syndrome, and psoriatic arthritis [58, 59]. Moreover, patients with tuberculosis (TB) and alpha-1-antitrypsin deficiency may also have a positive assay for anti-CCP. The presence of anti-CCP is associated with a more aggressive disease course in RA [60].
In the appropriate setting, anti-SMA in high titers can be suggestive of autoimmune hepatitis (AIH). Interestingly, patients with AIH may also have other concomitant autoantibodies, such as ANA and atypical P-ANCA. Only three studies reported the presence of anti-SMA in their cohort of patients [13, 14, 22]. Two of these studies used the same French database, which would explain the fairly high incidence of anti-SMA (23.2%) [13, 22]. As with most autoimmune diseases, multiple autoantibodies can be present in a single patient with IPH [13, 14]. This finding strengthens the hypothesis that the autoantibodies in IPH are indicative of some sort of immune dysregulation.
The association between celiac disease and IPH (Lane-Hamilton syndrome) has been reported by many authors. In our study, celiac disease antibodies were positive with the highest frequency (25.9%). Since the incidence of seropositivity for celiac disease in adults and children is approximately 1% in North America and Europe, there may be an association between these two entities [61]. Several hypotheses have been proposed to unify the coexistence of these diseases, but it has never been proven [62].
Similarly, cow’s milk protein allergy was also positive in a significant (16.2%) number of patients. In total, 9/13 (69.2%) of studies assessed for the presence of cow’s milk protein allergy in their patients.
There is limited data in the literature that have evaluated the clinical aspects in IPH patients with or without autoantibodies. Whether there is a difference in the presentation, treatment response, or overall prognosis is currently uncertain. It has been reported that patients with IPH and Down syndrome typically have more severe disease and worse prognosis compared to others [22, 63]. Similarly, whether there is a difference between patients who have autoantibodies during initial evaluation versus patients who develop antibodies during the course of the disease is unclear. Current data suggests that the presence of ANA may portend a higher risk of recurrence and worse outcome [19]. Although one report suggested a worse clinical course in patients who were ANCA positive, it is unclear whether those patients actually suffered from AAV rather than IPH [64]. Only a well-planned prospective study with a sufficient number of subjects can answer these questions. Unfortunately, IPH being a rare disease with an inconsistent diagnostic approach throughout the world, such endeavor is likely to prove difficult.
Our study identified the pooled prevalence of autoantibodies to be 26.2% in pediatric IPH patients. However, only a few patients were eventually diagnosed with an autoimmune disease (Table 2). An implication of identifying autoantibodies in patients with IPH is its impact on the “diagnosis of IPH.” By definition, IPH is a diagnosis of exclusion, and all competing diagnosis needs to be excluded with a reasonable workup. Therefore, the question would remain “Does the presence of autoantibodies indicate that the patient doesn’t have IPH or autoantibodies should be considered an immunologic phenomenon in IPH without a causative association?” The answer may be more straightforward if the patient has or develops ANCA antibodies and histopathologic evidence of vasculitis. But in the absence of a histopathologic diagnosis or a diagnosis of a systemic connective tissue disease, this distinction may be challenging. BH, defined as alveolar hemorrhage without vasculitis or inflammation, is the hallmark of DAH in IPH. However, BH can also occur in patients with autoimmune diseases. For example, nearly two-thirds of patients with DAH in SLE have been reported to suffer from BH [65]. As a result, the presence of autoantibodies may be the earliest clue regarding the subclinical presence of a systemic autoimmune disease, and patients with IPH should be routinely and carefully evaluated for subtle clues of rheumatologic diseases. A retrospective study identified four criteria to be associated with the immunologic causation of DAH. These were (a) onset of respiratory symptoms more than 10 days, (b) incapacitating fatigue or weight loss (5% in the past month), (c) arthralgia or arthritis, and (d) proteinuria [66]. Future genetic correlation studies involving patients with IPH, AAV, and connective tissue diseases may point towards a commonality.
Strengths and limitations of the study
Ours is the first study that has systematically scrutinized the literature to identify the frequency and role of autoantibodies in patients with IPH. However, the study has several limitations. Firstly, no prospective trials were identified in the literature. Therefore, the risk of bias is high among the reported patients. Secondly, the data were incomplete in many of these studies. Thirdly, several studies included patients before the existence of ANCA antibodies was discovered. Fourthly, the lack of exact characterization of the ANCA antibodies in the observational trials makes the results less reliable. Despite these limitations, this study provides a thorough analysis of the present knowledge of autoantibodies in IPH and proposes directions for further research.
Conclusions
The exact association between IPH and the occurrence of autoantibodies is unclear. Although many IPH patients develop one or multiple antibodies, these autoantibodies may manifest immunological dysregulation rather than causation. Whether an attenuated form of AAV could be responsible for BH in IPH remains speculative. However, the occurrence of two rare diseases, IPH and AAV, in the same patient only by chance appears highly improbable. The presence of ANA and ANCA may be associated with disease recurrence and worse outcomes. Further research is necessary to establish a genetic linkage among vasculitides, systemic rheumatologic diseases, and IPH.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AAV:
-
ANCA-associated vasculitis
- ANA:
-
Antinuclear antibody
- ANCA:
-
Antineutrophil cytoplasmic antibody
- BAL:
-
Bronchoalveolar lavage
- CCP:
-
Cyclic citrullinated peptide
- DAH:
-
Diffuse alveolar hemorrhage
- dsDNA:
-
Double-stranded DNA
- ELISA:
-
Enzyme-linked immunosorbent assay
- IIF:
-
Indirect immunofluorescence assay
- IPH:
-
Idiopathic pulmonary hemosiderosis
- MPO:
-
Myeloperoxidase
- PR3:
-
Proteinase 3
- RF:
-
Rheumatoid factor
- SMS:
-
Smooth muscle antibody
References
Saha BK (2020) Idiopathic pulmonary hemosiderosis: a state of the art review. Respir Med 176:106234. https://doi.org/10.1016/j.rmed.2020.106234
Ioachimescu OC, Sieber S, Kotch A (2004) Idiopathic pulmonary haemosiderosis revisited. Eur Respir J 24:162–169. https://doi.org/10.1183/09031936.04.00116302
Milman N, Pedersen FM (1998) Idiopathic pulmonary haemosiderosis. Epidemiology, pathogenic aspects and diagnosis. Respir Med 92:902–907. https://doi.org/10.1016/s0954-6111(98)90188-3
Milman N. Idiopathic pulmonary hemosiderosis. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. http://www.uptodate.com. Accessed Oct 4, 2021.)
Chen X-Y, Sun J-M, Huang X-J (2017) Idiopathic pulmonary hemosiderosis in adults: review of cases reported in the latest 15 years. Clin Respir J 11:677–681. https://doi.org/10.1111/crj.12440
de Klerk KD, Bau S, Günther G (2018) Diffuse pulmonary small nodular and patchy infiltrates on chest X-ray with hemoptysis: TB or not TB?-A call for scale up of respiratory medicine services in African TB high burden countries: a case of idiopathic pulmonary hemosiderosis. Pan Afr Med J 30:121. https://doi.org/10.11604/pamj.2018.30.121.12691
Gutierrez S, Shaw S, Huseni S et al (2014) Extracorporeal life support for a 5-week-old infant with idiopathic pulmonary hemosiderosis. Eur J Pediatr 173:1573–1576. https://doi.org/10.1007/s00431-013-2130-4
Saha BK, Chong WH (2021) Lung transplant to manage end-stage lung disease due to idiopathic pulmonary hemosiderosis: a review of the literature. Respir Investig S2212–5345(21):00118. https://doi.org/10.1016/j.resinv.2021.06.009
Saha BK, Chong WH (2021) Diffuse alveolar hemorrhage in cardiac diseases. Lung 199:103–112. https://doi.org/10.1007/s00408-021-00433-x
Imtiaz M, Saha B, Sana Ullah U, Saha A (2019) A case of acute life-threatening pulmonary hemorrhage from synthetic cannabinoid abuse. Case Rep Pulmonol 2019:8137648. https://doi.org/10.1155/2019/8137648
Saha S, Chong WH, Saha BK (2021) Unilateral diffuse alveolar hemorrhage due to selective directionality of mitral regurgitant jet in a patient with severe aortic stenosis. Cureus 13:e14714. https://doi.org/10.7759/cureus.14714
Saha BK (2021) Is it time to call idiopathic pulmonary hemosiderosis by the correct name: immune-mediated pulmonary hemosiderosis? Am J Med Sci 361:809–811. https://doi.org/10.1016/j.amjms.2021.01.006
Taytard J, Nathan N, de Blic J et al (2013) New insights into pediatric idiopathic pulmonary hemosiderosis: the French RespiRare(®) cohort. Orphanet J Rare Dis 8:161. https://doi.org/10.1186/1750-1172-8-161
Le Clainche L, Le Bourgeois M, Fauroux B et al (2000) Long-term outcome of idiopathic pulmonary hemosiderosis in children. Medicine (Baltimore) 79:318–326. https://doi.org/10.1097/00005792-200009000-00005
Saha BK, Chong WH, Milman NT (2021) Differentiation of idiopathic pulmonary hemosiderosis from rheumatologic and autoimmune diseases causing diffuse alveolar hemorrhage: establishing a diagnostic approach. Clin Rheumatol. https://doi.org/10.1007/s10067-021-05895-1
Freitas A, Senra V, Marinho A, Guedes M (2015) Chronic alveolar haemorrhage in a paediatric patient: a diagnostic and treatment challenge. BMJ Case Rep 2015. https://doi.org/10.1136/bcr-2014-206856
Stainer A, Rice A, Devaraj A et al (2019) Diffuse alveolar haemorrhage associated with subsequent development of ANCA positivity and emphysema in three young adults. BMC Pulm Med 19:185. https://doi.org/10.1186/s12890-019-0947-y
Buckley M, Van Mater H (2020) Idiopathic pulmonary hemosiderosis as a mimic of pulmonary vasculitis: a case report and review of the literature. Curr Allergy Asthma Rep 20:13. https://doi.org/10.1007/s11882-020-00907-7
Hizal M, Polat SE, Gursoy TR et al (2021) Risk factors for recurrent pulmonary exacerbation in idiopathic pulmonary hemosiderosis. Pediatr Pulmonol 56:1060–1068. https://doi.org/10.1002/ppul.25189
Yang C-T, Chiang B-L, Wang L-C (2021) Aggressive corticosteroid treatment in childhood idiopathic pulmonary hemosiderosis with better outcome. J Formos Med Assoc Taiwan Yi Zhi 120:838–846. https://doi.org/10.1016/j.jfma.2020.05.022
Zhang Y, Luo F, Wang N et al (2019) Clinical characteristics and prognosis of idiopathic pulmonary hemosiderosis in pediatric patients. J Int Med Res 47:293–302. https://doi.org/10.1177/0300060518800652
Alimi A, Taytard J, Abou Taam R et al (2018) Pulmonary hemosiderosis in children with Down syndrome: a national experience. Orphanet J Rare Dis 13:60. https://doi.org/10.1186/s13023-018-0806-6
Xu L-H, Ou R-Q, Wu B-J et al (2017) Corticosteroid in combination with leflunomide and mesenchymal stem cells for treatment of pediatric idiopathic pulmonary hemosiderosis. J Trop Pediatr 63:389–394. https://doi.org/10.1093/tropej/fmx002
Doi T, Ohga S, Ishimura M et al (2013) Long-term liposteroid therapy for idiopathic pulmonary hemosiderosis. Eur J Pediatr 172:1475–1481. https://doi.org/10.1007/s00431-013-2065-9
Luo X-Q, Ke Z-Y, Huang L-B et al (2008) Maintenance therapy with dose-adjusted 6-mercaptopurine in idiopathic pulmonary hemosiderosis. Pediatr Pulmonol 43:1067–1071. https://doi.org/10.1002/ppul.20894
Kabra SK, Bhargava S, Lodha R et al (2007) Idiopathic pulmonary hemosiderosis: clinical profile and follow up of 26 children. Indian Pediatr 44:333–338
Yao T-C, Hung I-J, Wong K-S et al (2003) Idiopathic pulmonary haemosiderosis: an Oriental experience. J Paediatr Child Health 39:27–30. https://doi.org/10.1046/j.1440-1754.2003.00066.x
Saeed MM, Woo MS, MacLaughlin EF et al (1999) Prognosis in pediatric idiopathic pulmonary hemosiderosis. Chest 116:721–725. https://doi.org/10.1378/chest.116.3.721
Kiper N, Göçmen A, Ozçelik U, et al (1999) Long-term clinical course of patients with idiopathic pulmonary hemosiderosis (1979–1994): prolonged survival with low-dose corticosteroid therapy. Pediatr Pulmonol 27:180–184. https://doi.org/10.1002/(sici)1099-0496(199903)27:3<180::aid-ppul5>3.0.co;2-8
Kjellman B, Elinder G, Garwicz S, Svan H (1984) Idiopathic pulmonary haemosiderosis in Swedish children. Acta Paediatr 73:584–588. https://doi.org/10.1111/j.1651-2227.1984.tb09978.x
Ohga S, Takahashi K, Miyazaki S et al (1995) Idiopathic pulmonary haemosiderosis in Japan: 39 possible cases from a survey questionnaire. Eur J Pediatr 154:994–995. https://doi.org/10.1007/BF01958645
Holborow EJ, Weir DM, Johnson GD (1957) A serum factor in lupus erythematosus with affinity for tissue Nuclei. Br Med J 2:732–734
Davies DJ, Moran JE, Niall JF, Ryan GB (1982) Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? Br Med J Clin Res Ed 285:606. https://doi.org/10.1136/bmj.285.6342.606
Hall JB, Wadham BM, Wood CJ et al (1984) Vasculitis and glomerulonephritis: a subgroup with an antineutrophil cytoplasmic antibody. Aust N Z J Med 14:277–278. https://doi.org/10.1111/j.1445-5994.1984.tb03769.x
Falk RJ, Jennette JC (1988) Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med 318:1651–1657. https://doi.org/10.1056/NEJM198806233182504
Watts RA, Mahr A, Mohammad AJ et al (2015) Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrol Dial Transplant 30:i14–i22. https://doi.org/10.1093/ndt/gfv022
Sacri A-S, Chambaraud T, Ranchin B et al (2015) Clinical characteristics and outcomes of childhood-onset ANCA-associated vasculitis: a French nationwide study. Nephrol Dial Transplant 30:i104–i112. https://doi.org/10.1093/ndt/gfv011
Calatroni M, Oliva E, Gianfreda D et al (2017) ANCA-associated vasculitis in childhood: recent advances. Ital J Pediatr 43:46. https://doi.org/10.1186/s13052-017-0364-x
Plumb LA, Oni L, Marks SD, Tullus K (2018) Paediatric anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis: an update on renal management. Pediatr Nephrol Berl Ger 33:25–39. https://doi.org/10.1007/s00467-016-3559-2
Tervaert JW, Goldschmeding R, Elema JD et al (1990) Autoantibodies against myeloid lysosomal enzymes in crescentic glomerulonephritis. Kidney Int 37:799–806. https://doi.org/10.1038/ki.1990.48
Hilhorst M, van Paassen P, Tervaert JWC (2015) Proteinase 3-ANCA vasculitis versus myeloperoxidase-ANCA vasculitis. J Am Soc Nephrol JASN 26:2314–2327. https://doi.org/10.1681/ASN.2014090903
Hruskova Z, Casian AL, Konopasek P et al (2013) Long-term outcome of severe alveolar haemorrhage in ANCA-associated vasculitis: a retrospective cohort study. Scand J Rheumatol 42:211–214. https://doi.org/10.3109/03009742.2012.754939
Hilhorst M, Wilde B, van Paassen P et al (2013) Improved outcome in anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis: a 30-year follow-up study. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc 28:373–379. https://doi.org/10.1093/ndt/gfs428
de Joode AAE, Sanders JSF, Stegeman CA (2013) Renal survival in proteinase 3 and myeloperoxidase ANCA-associated systemic vasculitis. Clin J Am Soc Nephrol CJASN 8:1709–1717. https://doi.org/10.2215/CJN.01020113
Merkel PA, Polisson RP, Chang Y et al (1997) Prevalence of antineutrophil cytoplasmic antibodies in a large inception cohort of patients with connective tissue disease. Ann Intern Med 126:866–873. https://doi.org/10.7326/0003-4819-126-11-199706010-00003
Terjung B, Spengler U, Sauerbruch T, Worman HJ (2000) “Atypical p-ANCA” in IBD and hepatobiliary disorders react with a 50-kilodalton nuclear envelope protein of neutrophils and myeloid cell lines. Gastroenterology 119:310–322. https://doi.org/10.1053/gast.2000.9366
Mahr A, Batteux F, Tubiana S et al (2014) Brief report: prevalence of antineutrophil cytoplasmic antibodies in infective endocarditis. Arthritis Rheumatol Hoboken NJ 66:1672–1677. https://doi.org/10.1002/art.38389
Sherkat R, Mostafavizadeh K, Zeydabadi L, et al (2011) Antineutrophil cytoplasmic antibodies in patients with pulmonary tuberculosis. Iran J Immunol IJI 8:52–57. IJIv8i1A7
Vahid B, Wildemore B, Nguyen C, Marik P (2006) Positive C-ANCA and cavitary lung lesion: recurrence of Wegener granulomatosis or aspergillosis? South Med J 99:753–756. https://doi.org/10.1097/01.smj.0000217201.80408.8a
Leaker B, Cambridge G, du Bois RM, Neild GH (1992) Idiopathic pulmonary haemosiderosis: a form of microscopic polyarteritis? Thorax 47:988–990. https://doi.org/10.1136/thx.47.11.988
Fishbein GA, Fishbein MC (2011) Lung vasculitis and alveolar hemorrhage: pathology. Semin Respir Crit Care Med 32:254–263. https://doi.org/10.1055/s-0031-1279823
West S, Arulkumaran N, Ind PW, Pusey CD (2013) Diffuse alveolar haemorrhage in ANCA-associated vasculitis. Intern Med Tokyo Jpn 52:5–13. https://doi.org/10.2169/internalmedicine.52.8863
Grygiel-Górniak B, Rogacka N, Puszczewicz M (2018) Antinuclear antibodies in healthy people and non-rheumatic diseases – diagnostic and clinical implications. Reumatologia 56:243–248. https://doi.org/10.5114/reum.2018.77976
Wananukul S, Voramethkul W, Kaewopas Y, Hanvivatvong O (2005) Prevalence of positive antinuclear antibodies in healthy children. Asian Pac J Allergy Immunol 23:153–157
Shmerling RH, Delbanco TL (1991) The rheumatoid factor: an analysis of clinical utility. Am J Med 91:528–534. https://doi.org/10.1016/0002-9343(91)90190-9
Clifford BD, Donahue D, Smith L et al (1995) High prevalence of serological markers of autoimmunity in patients with chronic hepatitis C. Hepatol Baltim Md 21:613–619
Brickmann K, Brezinschek RI, Yazdani-Biuki B et al (2010) Superior specificity of anti-citrullinated peptide antibodies in patients with chronic lymphocytic leukemia and arthritis. Clin Exp Rheumatol 28:888–891
Qing Y-F, Zhang Q-B, Zhou J-G et al (2009) The detecting and clinical value of anti-cyclic citrullinated peptide antibodies in patients with systemic lupus erythematosus. Lupus 18:713–717. https://doi.org/10.1177/0961203309102817
Gottenberg J-E, Mignot S, Nicaise-Rolland P et al (2005) Prevalence of anti-cyclic citrullinated peptide and anti-keratin antibodies in patients with primary Sjögren’s syndrome. Ann Rheum Dis 64:114–117. https://doi.org/10.1136/ard.2003.019794
Meyer O, Labarre C, Dougados M et al (2003) Anticitrullinated protein/peptide antibody assays in early rheumatoid arthritis for predicting five year radiographic damage. Ann Rheum Dis 62:120–126. https://doi.org/10.1136/ard.62.2.120
Reilly NR, Green PHR (2012) Epidemiology and clinical presentations of celiac disease. Semin Immunopathol 34:473–478. https://doi.org/10.1007/s00281-012-0311-2
Nacaroglu HT, Sandal OS, Bag O et al (2015) Association of celiac disease with idiopathic pulmonary hemosiderosis. Lane Hamilton syndrome. Iran J Pediatr 25:e3312. https://doi.org/10.5812/ijp.3312
Watanabe H, Ayusawa M, Kato M et al (2015) Idiopathic pulmonary hemosiderosis complicated by Down syndrome. Pediatr Int 57:1009–1012. https://doi.org/10.1111/ped.12690
Blanco A, Solis P, Gomez S et al (1994) Antineutrophil cytoplasmic antibodies (ANCA) in idiopathic pulmonary hemosiderosis. Pediatr Allergy Immunol Off Publ Eur Soc Pediatr Allergy Immunol 5:235–239. https://doi.org/10.1111/j.1399-3038.1994.tb00246.x
Zamora MR, Warner ML, Tuder R, Schwarz MI (1997) Diffuse alveolar hemorrhage and systemic lupus erythematosus: clinical presentation, histology, survival, and outcome. Medicine (Baltimore) 76:192–202
Picard C, Cadranel J, Porcher R et al (2010) Alveolar haemorrhage in the immunocompetent host: a scale for early diagnosis of an immune cause. Respiration 80:313–320. https://doi.org/10.1159/000315144
Author information
Authors and Affiliations
Contributions
BKS and AB were involved in the planning and collection of data. BKS, AB, and PC analyzed the data and prepared the initial manuscript. NTM supervised the preparation of the initial manuscript. All authors contributed in the finalization of the manuscript.
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Saha, B.K., Bonnier, A., Chenna, P. et al. Prevalence of autoantibodies in pediatric patients with idiopathic pulmonary hemosiderosis: a scoping review of the literature in the period 1980–2021. Clin Rheumatol 41, 977–990 (2022). https://doi.org/10.1007/s10067-021-06029-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-021-06029-3