
Abstract
Idiopathic pulmonary hemosiderosis (IPH) is a rare disease without a known incidence or prevalence in adults. Our knowledge of this entity is limited as there is no prospective or retrospective study with a reasonable number of patients. The objective is to describe the demographics, clinical manifestations, diagnosis, treatment, and prognosis of adult patients with IPH. The Medline and Embase databases were searched from inception to 2021 with appropriate search formulas to identify relevant articles following strict inclusion and exclusion criteria. Statistical analyses were performed for the entire cohort and prespecified subgroups. A total of 84 patients were identified. The majority of patients were males 54/84 (64.3%). The median age was 27 years. The manifesting symptoms were present in the following frequencies: anemia 76/83 (91.6%), dyspnea 71/83 (85.5%), hemoptysis 70/84 (83.3%), cough 22/84 (26.2%), and chest pain 9/84 (10.7%). The classic triad was present in 61/84 (79%) patients. The mean hemoglobin during the initial presentation was 8.4 gm/dL. A total of 16/57 (19.5%) tested positive for autoantibodies. The median delay in the diagnosis of IPH was 1.02 years. Immunosuppressive therapy was prescribed in 49/79 (62%) patients, and recurrence occurred in more than half of the patients 36/66 (54.5%). A total of 63/79 (79.7%) patients were alive during the final follow-up. IPH is more common in young adults with a male predominance. A high index of suspicion is necessary to attain an early diagnosis and possibly reduce the short-term mortality of nearly 20% and long-term complications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary hemosiderosis (IPH) is characterized by diffuse alveolar hemorrhage (DAH) by an unknown mechanism. Our knowledge of adult IPH is limited, primarily due to the lack of prospective or retrospective studies with a sizeable number of patients. As a result, clinical information, often inadequate, has been obtained mostly from case reports and small case series. Given the rarity of IPH, the feasibility of a large prospective study in the future is also questionable.
A recent study published in 2017 attempted to elucidate several aspects of adult IPH by analyzing the cases reported between 2000 and 2015 in Medline and Chinese databases [1]. The authors identified a total of 37 patients and reported their demographics, symptomatology, delay in diagnosis, treatment, and prognosis. However, the study had two critical limitations. First, the number of patients was modest at best, as only patients reported in the past 15 years were included. Second, several patients included in this study were either diagnosed with IPH when they were in the pediatric age group [2] or had provided inadequate information about individual patients [3, 4].
We undertook this project to overcome these challenges and shed light on IPH in the adult population. To do so, we have created an “Adult IPH Database” from all reported adult cases in the Medline and Embase databases since their inception. We have followed strict criteria for inclusion of patients into our database. We aim to inform the reader regarding the demographics, clinical manifestations, diagnosis, treatment, and prognosis of adult patients with IPH.
Materials and methods
Creation of IPH database
This is a retrospective study of adult IPH cases reported in the literature. We searched the Medline and Embase databases from their inception to December 31, 2021, with the appropriate MeSH terms and formula to identify adult patients with IPH. The following search formula was used: “idiopathic pulmonary hemosiderosis OR IPH OR pulmonary hemosiderosis AND adult”; and “idiopathic pulmonary hemosiderosis OR IPH OR pulmonary hemosiderosis.” The identified citations were then screened to exclude duplicates, abstracts, and pediatric patients. The full texts of the remaining manuscripts were examined by two independent reviewers (BKS and AB). The reviewers also meticulously hand searched the bibliography of the included manuscripts to identify additional papers. An exhaustive effort was implemented to obtain all available manuscripts. However, we could not find some manuscripts that were primarily reported in the 1950s and 1960s.
Patient population
The patient population consisted of adult patients with IPH reported in the literature. Patients diagnosed with IPH as a child but subsequently reported as adults were excluded from this study. Similarly, if the diagnosis of IPH was made based on clinical presentation and radiologic appearance without demonstration of hemosiderin-laden macrophages (HLM) from respiratory tract samples, they were not considered to have IPH. Patients with pulmonary hemorrhage (without lung biopsy) who had clinical manifestations suggestive of established autoimmune diseases were also excluded. For the purpose of subgroup analysis, the entire cohort was divided into two, patients reported between 1950 and 1990 (cohort A) and from 1990 to 2021 (cohort B). We arbitrarily chose this cut-off as testing for autoantibodies, including antineutrophil cytoplasmic antibody (ANCA), became more common in clinical practice in the early 1990s.
Study objectives
The primary objectives of the study were as follows: (1) determination of demographics, delay in diagnosis, clinical and radiologic manifestations, the prevalence of autoantibodies, treatment, and prognosis among adult patients with IPH. The secondary objectives were to assess any potential differences between cohorts A and B.
Inclusion criteria
Reported manuscripts fulfilling the following criteria were included in the study: (1) adult patients who were diagnosed with IPH at the age of 18 and above, (2) the diagnosis of IPH was made by histopathologic analysis of lung tissue obtained either by biopsy (bronchoscopic or surgical) or on autopsy studies, (3) in the absence of a tissue sample, demonstration of HLM from bronchoalveolar lavage (BAL) and sputum were considered adequate, (4) patients reported from the inception of the databases to December 31, 2021.
Exclusion criteria
The exclusion criteria were as follows: (1) Patients with adult-onset IPH for whom adequate information could not be obtained from the published manuscript, (2) studies that primarily reported long-term complications of a previously diagnosed case of IPH, (3) adult IPH cases reported as meeting abstracts, (4) manuscripts reported in a non-English language.
Definitions
“Delay in diagnosis” denoted the duration between the first reported respiratory symptoms and the diagnosis of IPH. “Definite IPH” was defined as patients for whom the diagnosis was secured by histopathologic examination of the lung, either by lung biopsy or on autopsy. “Probable IPH” was characterized by consistent clinical and radiologic findings and demonstration of HLM from a sample obtained from the respiratory tract without a histopathologic proof. “Recurrence of disease” represented any deterioration of the patient’s condition with signs or symptoms consistent with IPH, new or worsening radiologic infiltrate, or requirement of reinitiation of increased immunosuppressive medications. Regarding clinical manifestations, systemic symptoms included any of the following: fever, night sweats, chills, weight loss, loss of appetite, fatigue, and malaise.
Data items
The included studies were coded, and the extracted data were then tabulated in a standardized Excel sheet (Microsoft Corporation). The following data were gathered: author name, year of reporting, country of the study, patient demographics, presenting symptoms, delay between the symptom onset and the diagnosis of IPH, testing for autoantibodies, autoantibody positivity, type of positive autoantibodies, temporal relationship of autoantibody determination with the diagnosis of IPH, clinical and radiologic manifestations, lung biopsy results when available, treatment and clinical outcome of the disease.
Statistical analysis
Descriptive and inferential statistical analyses were performed using the IBM SPSS statistics software package 28. Normally and non-normally distributed data were reported as mean (standard deviation, SD) and median (interquartile range, IQR), respectively. In addition, independent t-test was used to compare continuous variables, and chi-square test was used to compare categorical variables. Univariate and multivariate analyses of continuous variables were performed to identify risk factors for poor outcomes, namely risks of recurrence and survival.
Results
Study characteristics
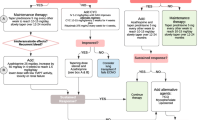
Seventy-four manuscripts fulfilling the inclusion criteria were included in this study [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79]. The study selection process is shown in Fig. 1. No prospective studies were identified. A retrospective study by Miwa et al. that included nine patients was not included in this review as the information regarding individual patients was limited [3]. Similarly, patients from 2 additional retrospective studies were excluded due to inadequate information, inaccurate diagnosis, and being previously reported by other authors [80, 81]. Of the 74 studies, 67 papers were a single case report, whereas the other 7 were small case series. The patients were reported from all continents (Table 1).

Flow chart showing the selection of studies
Subject demographics
A total of 84 patients were included in this review. A total of 31/84 (36.9%) patients were reported between 1950 and 1990 and the remaining 53/84 (63.1%) between 1991 and 2021. The median age of the entire cohort at the time of IPH diagnosis was 27 (IQR 22–39.7 years). The majority of patients were males 54/84 (64.3%). The demographic data among the subgroup of patients are detailed in Table 2. The age at diagnosis of IPH did not vary statistically depending on the gender of the patient in the entire cohort, t(82) = 0.44, p = 0.329. The reported cases with age distribution are shown in Fig. 2.

Scatterplot showing reported cases over the years and age at the time of IPH diagnosis. The majority of patients were less than 30 years old at the time of diagnosis
Clinical, laboratory, and radiologic findings
The manifesting symptoms were present in the following frequencies: anemia 76/83 (91.6%), dyspnea 71/83 (85.5%), hemoptysis 70/84 (83.3%), cough 22/84 (26.2%), and chest pain 9/84 (10.7%). Systemic symptoms were present in nearly half, 38/84 (45.2%) patients. The classic triad consisting of hemoptysis, anemia, and radiologic chest abnormalities were present in 61/84 (79%) patients. Respiratory failure during the acute phase of the disease was reported in 13/84 (15.5%).
The mean hemoglobin during initial presentation was 8.4 gm/dL (SD, 2.59) (n = 63) and the median serum ferritin concentration was 72.3 ng/mL (IQR, 12.67–130.1) (n = 13). A total of 54.2% (45/83) of all patients underwent chest computed tomography (CT), and the rest were imaged by chest radiography. No patient in the older cohort was evaluated by a CT scan, whereas 86.5% (45/52) of patients reported between 1991 and 2021 were. Bilateral alveolar infiltrate on chest imaging was present in 73/81 (90.1%). Other abnormalities included interstitial opacity in 30/81 (37%) and cystic or emphysematous changes in 12/81 (14.8%). Rare radiologic presentations were thoracic lymphadenopathy and pleural effusion.
Autoantibody testing
One or more autoantibodies were tested in 57/82 (69.5%) patients. Among these patients, 16 (19.5%) tested positive. A total of 12/16 (62.5%) of these patients had antibodies suggestive of celiac disease (CD). The other antibodies were antithyroid antibodies (2), rheumatoid factor (1), and anti-GM-CSF (1). Only 8/31 (25.8%) patients underwent antibody testing in cohort A. In contrast, 49/51 (96.1%) patients were tested for autoantibodies in cohort B.
Diagnosis of IPH
A diagnosis of “definite IPH” by histopathologic analysis was obtained in 65/84 (77.4%) of patients. Lung tissue was obtained by lung biopsy in 56 patients and during autopsy in 9. Among these patients, surgical lung biopsy (SLB) was performed in 29, video-assisted thoracoscopic surgery (VATS) in 11, and bronchoscopic transbronchial lung biopsy (TBLB) in 16. The diagnosis of “probable IPH” was made in the remaining 19 patients by demonstration of HLM from BAL and sputum in 10 and 7 patients, respectively. In the other two patients, sequential BAL demonstrated progressively hemorrhagic fluid return.
Delay in diagnosis
The median delay in the diagnosis of IPH for the entire cohort was 53.5 (IQR 8–195) weeks or 1.02 years. Subgroup analysis revealed a longer delay in cohort A compared to cohort B. The detailed data are presented in Table 2. The diagnosis of IPH was also delayed for women compared to men (Table 2). However, the difference in the diagnostic delay between gender was not statistically significant in the entire cohort or previously specified subgroups by one-way independent t-test analysis, t(74) = 0.213, p = 0.416.
Treatment
Following the diagnosis of IPH, immunosuppressive therapy was initiated in 49/79 (62%) patients. A total of 47/49 (95.91%) patients received corticosteroid (CS) as the first-line therapy, and the other two were treated with azathioprine (AZA). A second agent was required in 15/47 (31.91%) of these patients. The most commonly used second-line medications were AZA (9), ACTH (3), antimalarials (2), and cyclophosphamide (CYC) in 1. Among 14 patients simultaneously diagnosed with CD (in 2, the diagnosis was made on small bowel biopsy without any antibody testing), 12 were treated with only gluten-free diet (GFD). The other two patients received immunosuppressive therapy in addition to GFD. Treatment with CS was not associated with a higher survival (chi-square test, p = 0.928).
Follow-up, recurrence, and survival
The median follow-up period for the entire cohort was 1 year (IQR 0.5–3) (n = 59). Recurrence of the disease was reported in more than half of the patients 36/66 (54.5%). The recurrence was reported in 16/25 (64%) patients in cohort A compared to that in 20/41 (48.8%) in cohort B. There was no statistical difference in the risk of recurrence between males and females, chi-square test, p = 0.777. A total of 63/79 (79.7%) patients were alive during the final follow-up. The survival was similar between the two cohorts, 75.9% and 82% in cohorts A and B, respectively. No statistical difference was identified in survival between men and women (chi-square test, p = 0.118). Univariate and multivariate analyses of several variables, such as age at diagnosis, delay in diagnosis, and admission hemoglobin, did not predict the risk of future recurrence or survival.
Discussion
Waldenström reported the first antemortem case of IPH in 1944 [82]. Due to the rarity of IPH in adults, most aspects of this illness have remained somewhat speculative even after seven decades. In this manuscript, we have provided a thorough analysis of adult patients with IPH. To date, this is the most comprehensive review of adult IPH cases. Our paper has confirmed several findings reported earlier by Chen et al. [1] while refuting some previous assumptions and elucidating less well-known aspects of the disease.
IPH is a rare disease that affects both adults and children. The incidence and prevalence of IPH in adults are currently unknown. However, based on the number of reported cases, IPH appears to be more common in children [40]. The incidence ranges between 0.24 and 1.23 cases per million per year in select group of pediatric patients [83, 84]. IPH is also considered more aggressive in children with a higher recurrence rate and poorer prognosis [40]. Most children are diagnosed before ten, and IPH has a distinct female predominance in this population [85,86,87]. In contrast, we found that among adult patients, IPH is more common in males. Nearly two-thirds of the patients were males in our study. Similar findings were also reported in previous studies [1, 3, 88, 89]. Although previous studies reported most adult IPH patients to be younger than 30 years [40, 41], this was contradicted by Chen et al., who reported 57% of patients to be older than 30 years in their paper [1]. On the contrary, we have found that most patients are, in fact, younger than 30 years (Fig. 2). This trend persisted even when prespecified subgroups were analyzed (Table 2). Approximately 40% of patients were over 30 years at the time of diagnosis.
Regarding presenting signs and symptoms, our study is in stark contrast with Chen et al. [1]. The proportion of patients with anemia (91.6% vs. 54%), dyspnea (85.5% vs. 62%), hemoptysis (83.3% vs. 81%), productive or non-productive cough without hemoptysis (26.2% vs. 5%), and chest pain (10.7% vs. 5%) were higher in our study. The classic triad, comprising hemoptysis, anemia, and radiologic chest abnormalities, were present in almost 80% of our cohort (not reported by Chen et al.). Systemic symptoms, such as fever, night sweats, chills, weight loss, or loss of appetite, were seen in nearly half of the patients (45%). This is a crucial finding, as the presence of fever and other systemic symptoms may incorrectly dissuade clinicians from strongly considering the diagnosis of IPH and lean toward an infectious etiology for the respiratory symptoms. Due to the nonspecificity of symptoms and variable recognition of IPH among clinicians, a delay and misdiagnosis have often been reported in pediatric and adult patients [1, 87]. Like Chen et al., we also found the mean delay to be 2.3 years from symptom onset to the diagnosis of IPH. It is crucial to emphasize that using an average value could be misleading, as the data was skewed due to outliers. The median delay of 1.02 years is likely more representative of the real-world scenario. Subgroup analysis revealed a trend toward an earlier diagnosis of IPH in the 1991–2021 cohort compared to the older cohort (52 vs. 92 weeks).
The kinetics of iron metabolism in patients with IPH requires special mention. These patients develop functional iron deficiency manifested by the microcytic or normocytic and hypochromic RBCs in peripheral blood smear, low serum iron, normal or high total iron-binding capacity, and bone marrow depletion of iron storage. However, the serum ferritin level may be normal or even elevated, thus mimicking anemia of chronic disease as seen in patients with a systemic inflammatory condition. After an episode of DAH, the RBCs are phagocytosed by alveolar macrophages (AMs). The AMs produce free iron after oxidation of hemoglobin, which subsequently saturates the intracellular apo-ferritin pool and gets stored in the lung storage in the lung as ferritin [90]. Once the ferritin storage reaches maximal capacity, with ongoing hemorrhage, the iron is deposited as hemosiderin—the unusable form of iron storage—leading to the development of low serum iron and reduced storage in the bone marrow. The ferritin escapes the AMs by an unknown mechanism maintaining or even raising the serum ferritin level [40]. We found an elevated mean serum ferritin level (177.34 ng/mL) despite a mean hemoglobin level of 8.4 gm/dL in our cohort. On the other hand, patients with coexisting CD may have blood work more consistent with iron deficiency anemia [65, 66]. As these patients often suffer from undiagnosed malabsorption for a prolonged duration before medical contact, the elevated ferritin may not be present. In the absence of any systemic inflammatory condition, elevated or normal serum ferritin level in the face of microcytic hypochromic anemia and hemoptysis should prompt the consideration of IPH.
The pathogenesis of IPH is currently unknown. An immunologic mechanism appears to be most likely. We have previously proposed renaming IPH to immune-mediated pulmonary hemosiderosis (ImPH) to signify the immunologic association/causation [91, 92]. The identification of autoantibodies and response to CS therapy have propagated a positive outlook regarding the immune hypothesis among clinicians [85, 86, 93,94,95]. The other hypotheses proposed by authors in the past are genetic, environmental, and allergic causation of IPH [96, 97]. Identification of autoantibodies has been reported in both pediatric and adult patients with IPH [95]. Approximately one in every five patients in our cohort tested positive for an autoantibody, with antibodies specific for CD being the most common (Table 1). In the 1991–2021 cohort, when antibody testing for CD became more common, 12/47 (25.5%) patients tested positive. The coexistence of IPH and CD has been described in the past and is known as Lane Hamilton syndrome (LHS) [98]. Interestingly, most patients with LHS do not suffer from GI symptoms and should be tested from antibodies for CD when diagnosed with IPH [99]. The other commonly reported autoantibodies in adults include rheumatoid factor and antithyroid antibodies [30, 50, 52]. A more diverse group of antibodies, such as ANA, ANCA, and smooth muscle antibody, have been reported in pediatric patients but not in adults [85, 91]. All patients with IPH who subsequently developed ANCA were diagnosed with IPH as a child [93, 94].
The radiologic appearance of IPH is nonspecific. During the acute phase of the disease, a chest X-ray may demonstrate bilateral infiltrate primarily in the middle and lower lung zones [100]. As a general rule, computed tomography (CT) is more sensitive than conventional chest radiology. Unsurprisingly, no patient in the older cohort were evaluated by chest CT as the technology only became available in 1970s and was not widely available until the 1990s [101]. As a result, most of the patients in the recent cohort received CT imaging as a part of their evaluation. CT scan of the chest shows ground glass opacity (GGO) and, sometimes, consolidative changes in the same distribution. If the hemorrhage is severe, all lung lobes could be involved. In the absence of ongoing bleeding, the alveolar opacity morphs into an interstitial opacity after 48–72 h [102]. Reticular and reticulonodular shadow may predominate at this time. Long-standing disease may result in pulmonary fibrosis involving the posterobasilar area [48, 103, 104]. Less common radiologic features are emphysema or cystic changes [4, 23, 35, 46, 48, 49, 77, 78]. There were rare cases of intrathoracic lymphadenopathy and pleural effusion [77].
IPH is a diagnosis of exclusion. All competing diagnoses need to be excluded before a confident diagnosis of IPH can be made [40, 105,106,107]. In this manuscript, we have specified the diagnosis as “definite” and “probable.” Patients with a definite IPH diagnosis either underwent lung biopsy or autopsy studies, and the histopathologic analysis was consistent with bland pulmonary hemorrhage without any evidence of inflammatory cellular infiltration, necrosis, vasculitis, or granulomatous inflammation. A “probable” diagnosis of IPH is made without histopathologic examination of the lung but with evidence of hemosiderin-laden macrophages from respiratory tract samples. Although we included the cases of “probable IPH” for analysis in this review as IPH, it is important to remember that even with a negative autoantibody screening, patients may still suffer from seronegative ANCA-associated vasculitis (AAV) or isolated pauci-immune pulmonary capillaritis [108, 109]. A biopsy, either transbronchial or surgical, is therefore preferable. The SLB is more invasive but allows for more tissue allowing the pathologist to do a thorough analysis. Transbronchial lung biopsy is less invasive but may not provide an adequate sample. Transbronchial cryobiopsy provides more tissue with a higher risk of bleeding than conventional TBLB [73]. The classic histopathologic findings in IPH include evidence of bland pulmonary hemorrhage (BH) and change associated with recurrent bleeding. BH refers to the occurrence of alveolar hemorrhage without any evidence of vasculitis, necrosis, granulomatous inflammation, and the absence of immunocomplex deposition or inflammatory cellular infiltration of the pulmonary parenchyma [92, 103]. Hemosiderin deposition can take place intracellularly (in the alveolar macrophages), extracellularly and along the interalveolar septum. Collagen deposition and emphysematous changes can be present in advanced disease [103].
One of the limitations that we faced during this study was the absence of long-term follow-up in most patients. The median follow-up was 1 year, and more than half of the patients suffered from recurrence during this period. Corticosteroid (CS) represents the first line of therapy for patients with IPH [110]. A multinational survey among pediatricians showed CS to be the most commonly used medication for both induction and maintenance therapy [111]. The use of corticosteroid (CS) was associated with clinical improvement even in patients with respiratory failure requiring mechanical ventilation. Interestingly, the survival with CS treatment did not reach statistical significance. This is likely due to the very short overall follow-up period. One-third of patients required a second-line agent. Azathioprine appears to be the most commonly used agent in adults, whereas, in pediatric patients, antimalarials are the second most commonly used medication [111]. Experimental treatment includes mesenchymal stem cell transplant and liposteroid therapy [112, 113]. An overwhelming majority of patients with LHS appear to do well on GFD. However, in case of recurrent bleeding, even with GFD, additional immunosuppressants may be necessary.
Since no prospective or retrospective studies are available in adult patients with IPH, we attempted to identify risk factors for poorer prognosis in terms of “recurrence of disease” and “survival.” Unfortunately, univariate and multivariate analysis of variables, such as age at diagnosis of IPH, duration of symptoms before the diagnosis, and admission hemoglobin, failed to identify any such potential risk factor. This is likely due to the modest number of patients in our study and inadequate information and follow-up time.
Strength and limitations of the study
Our study has several limitations. First, we had excluded manuscripts published in the non-English literature, which would have increased the number of subjects in our cohort. Second, some manuscripts were excluded as we could not obtain the full texts. The lack of availability was primarily the case for older manuscripts, which led to a fewer number of patients in the 1950–1990 cohort. Third, since most manuscripts were retrospective reports of single cases or small case series, the reported variables were not uniform. Similarly, the risk of publication bias was likely high. Fourth, the follow-up period was limited to assess prognosis with confidence as many patients could have suffered from recurrence of their disease later. Despite the limitations, to our knowledge, this is the most complete review of adult patients with IPH ever reported in the literature. Our study has consolidated the knowledge regarding the demographics, clinical manifestations, modalities used for diagnosing IPH, treatment, and prognosis for these patients.
Conclusion
IPH is a rare cause of DAH in adult patients and is associated with a significant delay in diagnosis. In this manuscript, we have provided a detailed account of all reported cases of IPH in adults. IPH is more common in males, and the majority of patients are diagnosed before the age of 30 years. Anemia, dyspnea, and hemoptysis are the most frequent symptoms. The classic triad is present in nearly 80% of patients. LHS affects 25% of patients with IPH. Therefore, screening for CD should be performed routinely at diagnosis of IPH. Recurrence is common, and nearly half the patients experience recurrence even with short-term follow-up. CS represents the first line of therapy. Treatment with CS appears to be efficacious but was not statistically associated with improved survival. Prospective studies are required to evaluate the long-term efficacy of CS therapy.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AAV:
-
ANCA-associated vasculitis
- AM:
-
Alveolar macrophage
- ANCA:
-
Antineutrophil cytoplasmic antibody
- AZA:
-
Azathioprine
- BAL:
-
Bronchoalveolar lavage
- CD:
-
Celiac disease
- CS:
-
Corticosteroid
- DAH:
-
Diffuse alveolar hemorrhage
- GFD:
-
Gluten-free diet
- HLM:
-
Hemosiderin-laden macrophages
- IPH:
-
Idiopathic pulmonary hemosiderosis
- SLB:
-
Surgical lung biopsy
- TBLB:
-
Transbronchial lung biopsy
- VATS:
-
Video-assisted thoracoscopic surgery
References
Chen X-Y, Sun J-M, Huang X-J (2017) Idiopathic pulmonary hemosiderosis in adults: review of cases reported in the latest 15 years. Clin Respir J 11:677–681. https://doi.org/10.1111/crj.12440
Calabrese F, Giacometti C, Rea F et al (2002) Recurrence of idiopathic pulmonary hemosiderosis in a young adult patient after bilateral single-lung transplantation. Transplantation 74:1643–1645. https://doi.org/10.1097/00007890-200212150-00027
Miwa S, Imokawa S, Kato M et al (2011) Prognosis in adult patients with idiopathic pulmonary hemosiderosis. Intern Med 50:1803–1808. https://doi.org/10.2169/internalmedicine.50.4715
Ioachimescu OC, Jennings C (2006) intercostal lung cyst hernia in idiopathic pulmonary hemosiderosis (cyst necessitans). Mayo Clin Proc 81:692. https://doi.org/10.4065/81.5.692
Tait GB, Corridan M (1952) Idiopathic pulmonary haemosiderosis. Thorax 7:302–304. https://doi.org/10.1136/thx.7.4.302
Manderson WC (1954) Idiopathic pulmonary haemosiderosis: with report of a case in an adult. Glasgow Med J 35:19–24
Hamer NA (1955) Idiopathic pulmonary haemosiderosis in a young adult. Br Med J 1:1008–1009. https://doi.org/10.1136/bmj.1.4920.1008
Browning JR, Houghton JD (1956) Idiopathic pulmonary hemosiderosis. Am J Med 20:374–382. https://doi.org/10.1016/0002-9343(56)90122-x
Wynn-Williams N, Young RD (1956) Idiopathic pulmonary haemosiderosis in an adult. Thorax 11:101–104
Boyd DH (1959) Idiopathic pulmonary haemosiderosis in adults and adolescents. Br J Dis Chest 53:41–51. https://doi.org/10.1016/s0007-0971(59)80108-x
Gurewich V, Thomas MA (1959) Idiopathic pulmonary hemorrhage in pregnancy. Report of a case suggesting early pulmonary hemosiderosis with clinical recovery after steroid therapy. N Engl J Med 261:1154–1159. https://doi.org/10.1056/NEJM195912032612303
Nickol KH (1960) Idiopathic pulmonary haemosiderosis presenting with spontaneous pneumothorax. Tubercle 41:216–218. https://doi.org/10.1016/s0041-3879(60)80082-7
Yettra M, Goldenberg E, Weiner H (1960) Idiopathic pulmonary hemosiderosis. Calif Med 93:330–336
Sprecace GA (1963) Idiopathic pulmonary hemosiderosis. Personal experience with six adults treated within a ten-month period, and a review of the literature. Am Rev Respir Dis 88:330–341. https://doi.org/10.1164/arrd.1963.88.3P1.330
Murphy KJ (1965) Pulmonary haemosiderosis (apparently idiopathic) associated with myocarditis, with bilateral penetrating corneal ulceration, and with diabetes mellitus. Thorax 20:341–347. https://doi.org/10.1136/thx.20.4.341
Samuels ML, Howe CD, Butler J Idiopathic pulmonary hemosiderosis. 5
Sarkar TK, Sarkar (1965) Idiopathic pulmonary hemosiderosis associated with renal changes and abnormal serum proteins: report of a case. Dis Chest 48:211–213. https://doi.org/10.1378/chest.48.2.211
Aledort LM, Lord GP (1967) Idiopathic pulmonary hemosiderosis: severe anemia without hemoptysis— one year follow-up of pulmonary function. Arch Intern Med 120:220–223. https://doi.org/10.1001/archinte.1967.00300020092012
Degowin RL, Sorensen LB, Charleston DB et al (1968) Retention of radioiron in the lungs of a woman with idiopathic pulmonary hemosiderosis. Ann Intern Med 69:1213–1220. https://doi.org/10.7326/0003-4819-69-6-1213
Elliott ML, Kuhn C (1970) Idiopathic pulmonary hemosiderosis. Am Rev Respir Dis 102:895–904. https://doi.org/10.1164/arrd.1970.102.6.895
Byrd RB, Trunk G (1973) Systemic lupus erythematosus presenting as pulmonary hemosiderosis. Chest 64:128–129. https://doi.org/10.1378/chest.64.1.128
Irwin RS, Cottrell TS, Hsu KC et al (1974) Idiopathic Pulmonary hemosiderosis: an electron microscopic and immunofluorescen. Chest 65:41–45. https://doi.org/10.1378/chest.65.1.41
Jiji V, Hofkin GA (1974) Idiopathic pulmonary hemosiderosis. South Med J 67:488–491. https://doi.org/10.1097/00007611-197404000-00025
Donlan CJ, Srodes CH, Duffy FD (1975) Idiopathic pulmonary hemosiderosis: electron microscopic, immunofluorescent, and iron kinetic studies. Chest 68:577–580. https://doi.org/10.1378/chest.68.4.577
Donald KJ, Edwards RL, McEvoy JD (1975) Alveolar capillary basement membrane lesions in Goodpasture’s syndrome and idiopathic pulmonary hemosiderosis. Am J Med 59:642–649. https://doi.org/10.1016/0002-9343(75)90225-9
Pozo-Rodriguez F, Freire-Campo JM, Gutierrez-Millet V et al (1980) Idiopathic pulmonary haemosiderosis treated by plasmapheresis. Thorax 35:399–400
Moses HW, Schreiner BF, Hyde RW, Kallay MC (1982) Heart block and idiopathic pulmonary hemosiderosis. Pacing Clin Electrophysiol 5:826–828. https://doi.org/10.1111/j.1540-8159.1982.tb06563.x
(1986) Hemoptysis, pulmonary infiltrates, and diarrhea in a 36-year-old man. Am J Med 80:930–938. https://doi.org/10.1016/0002-9343(86)90640-6
Nomura S, Kanoh T (1987) Association of idiopathic pulmonary haemosiderosis with IgA monoclonal gammopathy. Thorax 42:696–697. https://doi.org/10.1136/thx.42.9.696
Bain SC, Bryan RL, Hawkins JB (1989) Idiopathic pulmonary haemosiderosis and autoimmune thyrotoxicosis. Respir Med 83:447–450. https://doi.org/10.1016/s0954-6111(89)80082-4
Chaudhry AA, Dobson CM, Simpson FG (1991) Pulmonary haemosiderosis associated with left atrial myxoma. Thorax 46:539–540
Pacheco A, Casanova C, Fogue L, Sueiro A (1991) Long-term clinical follow-up of adult idiopathic pulmonary hemosiderosis and celiac disease. Chest 99:1525–1526. https://doi.org/10.1378/chest.99.6.1525
Bouros D, Panagou P, Rokkas T, Siafakas N (1994) Bronchoalveolar lavage findings in a young adult with idiopathic pulmonary haemosiderosis and coeliac disease. Eur Respir J
Bavry AA, Knoper S, Alpert JS (2000) Segmental wall motion abnormalities in an individual with idiopathic pulmonary hemosiderosis. CRD 93:201–204. https://doi.org/10.1159/000007027
Godoy I, Leite RM, Yoo HH et al (2000) Idiopathic pulmonary hemosiderosis with cystic lesions: a rare presentation. Am J Med Sci 319:411–413. https://doi.org/10.1097/00000441-200006000-00012
Tedeschi A, Lorini M, Giannini S et al (2001) Serum histamine-releasing activity in a patient with idiopathic pulmonary haemosiderosis. Allergol Immunopathol (Madr) 29:281–283. https://doi.org/10.1016/s0301-0546(01)79072-6
Helman DL, Sullivan A, Kariya ST et al (2003) Management of idiopathic pulmonary haemosiderosis in pregnancy: report of two cases. Respirology 8:398–400. https://doi.org/10.1046/j.1440-1843.2003.00477.x
Malhotra P, Aggarwal R, Aggarwal AN et al (2005) Coeliac disease as a cause of unusually severe anaemia in a young man with idiopathic pulmonary haemosiderosis. Respir Med 99:451–453. https://doi.org/10.1016/j.rmed.2004.09.007
Turay UY, Ergün P, Erdoğan Y et al (2004) Idiopathic pulmonary haemosiderosis. Tuberk Toraks 52:382–385
Ioachimescu OC, Sieber S, Kotch A (2004) Idiopathic pulmonary haemosiderosis revisited. Eur Respir J 24:162–169. https://doi.org/10.1183/09031936.04.00116302
Soto RG, Soares MM (2005) Idiopathic pulmonary hemosiderosis in pregnancy: anesthetic implications. J Clin Anesth 17:482–484. https://doi.org/10.1016/j.jclinane.2005.06.002
Corte, T., Tattersall, S. Iron deficiency anaemia: a presentation of idiopathic pulmonary haemosiderosis. Intern Med J 36:207–9
Deniz O, Ongürü O, Ors F et al (2007) Idiopathic pulmonary hemosiderosis in an adult patient responded well to corticosteroid therapy. Tuberk Toraks 55:77–82
Gencer M, Ceylan E, Bitiren M, Koc A (2007) Two sisters with idiopathic pulmonary hemosiderosis. Can Respir J 14:490–493. https://doi.org/10.1155/2007/150926
Thachil JV, Watson A, Williams S, O’Hea AM (2007) A mystery unravels. Am J Med 120:669–670. https://doi.org/10.1016/j.amjmed.2007.05.004
Bal A, Bhalla A, Joshi K (2008) Idiopathic pulmonary haemosiderosis with mineralizing pulmonary elastosis: a case report. J Med Case Rep 2:65. https://doi.org/10.1186/1752-1947-2-65
Chen C-H, Yang H-B, Chiang S-R, Wang P-C (2008) Idiopathic pulmonary hemosiderosis: favorable response to corticosteroids. J Chin Med Assoc 71:421–424. https://doi.org/10.1016/S1726-4901(08)70094-7
Harte S, Mcnicholas WT, Donnelly SC, Dodd JD (2008) Honeycomb cysts in idiopathic pulmonary haemosiderosis: high-resolution CT appearances in two adults. BJR 81:e295–e298. https://doi.org/10.1259/bjr/66050546
Mayes DH, Guerrero ML (2008) A few good men: a Marine with hemoptysis and diarrhea. Idiopathic pulmonary hemosiderosis and celiac sprue. Chest 134:644–647. https://doi.org/10.1378/chest.07-2834
Dutkiewicz R. A 74 Year‐Old male with intermittent hemoptysis. 6
Abbdallah Fatma CB, Amel C, Ridha M et al (2010) Idiopathic pulmonary hemosiderosis in adult. Respir Med CME 3:238–240. https://doi.org/10.1016/j.rmedc.2009.10.004
Fujii M, Miyahara N, Tanimoto Y et al (2010) Diffuse alveolar hemorrhage with chronic thyroiditis in an advanced-age adult. Respir Med CME 3:90–92. https://doi.org/10.1016/j.rmedc.2009.04.006
Man M, Alexandrescu D, Ra B. A rare cause of hemoptisys: idiopathic pulmonary hemosiderosys (IPH). CLINICAL ASPECTS 4
Nishino M, Patrick JL, Connors JM (2010) Case 155: Lane-Hamilton syndrome. Radiology 254:985–988. https://doi.org/10.1148/radiol.09082062
Oviedo Ramírez MI, Hop K, Carrera E, Soriano Rosas J (2010) Idiopathic pulmonary haemosiderosis in a young adult. Autopsy findings. Arch Bronconeumol 46:565–567. https://doi.org/10.1016/j.arbres.2010.06.006
Schroers R, Bonella F, Tötsch M, Costabel U (2010) A female soccer player with recurrent haemoptysis and iron deficiency anaemia: idiopathic pulmonary haemosiderosis (IPH)-case report and literature review. BMJ Case Rep 2010:bcr06.2009.1969. https://doi.org/10.1136/bcr.06.2009.1969
dos Santos JWA, de Mello Neto AB, Marchiori RC et al (2012) Pulmonary hemosiderosis associated with celiac disease: improvement after a gluten-free diet. J Bras Pneumol 38:412–414. https://doi.org/10.1590/s1806-37132012000300020
Kahraman H, Köksal N, Özkan F (2012) Eight years follow-up of a case with idiopathic pulmonary hemosiderosis after corticosteroid therapy. N Am J Med Sci 4:49–51. https://doi.org/10.4103/1947-2714.92907
Tzouvelekis A, Ntolios P, Oikonomou A et al (2012) Idiopathic pulmonary hemosiderosis in adults: a case report and review of the literature. Case Rep Med 2012:e267857. https://doi.org/10.1155/2012/267857
Cambruzzi E, Pêgas KL, Vedana T (2013) Idiopathic pulmonary hemorrhage: morphology and differential diagnosis. Case report. J Bras Patol Med Lab 49:216–221. https://doi.org/10.1590/S1676-24442013000300010
Gerhardy B, Simpson G (2013) Melioidosis and idiopathic pulmonary hemosiderosis: a cast-iron case. Respirol Case Rep 1:46–47. https://doi.org/10.1002/rcr2.25
Patrucco F, Sarcoli M, Boldorini R, Balbo PE (2013) A young man with anemia and recurrent tachyarrhythmic episodes. RES 86:149–154. https://doi.org/10.1159/000348723
Singhal KK, Janmeja AK, Sodhi R, Punia RS (2013) Hemoptysis in patients of celiac disease with disproportionately severe anemia: tip of the iceberg? Multidiscip Respir Med 8:25. https://doi.org/10.1186/2049-6958-8-25
JackinMoses R, Sinha N, Madhusmita M et al (2014) Idiopathic pulmonary hemosiderosis without hemoptysis in an adult: a rare presentation. https://doi.org/10.5348/IJCRI-2014-03-480-CR-11
Berger N, Nichols J, Datta D (2016) Idiopathic pulmonary haemosiderosis with celiac disease (Lane-Hamilton syndrome) in an adult - a case report. Clin Respir J 10:661–665. https://doi.org/10.1111/crj.12258
Khilnani GC, Jain N, Tiwari P et al (2015) A young man with hemoptysis: rare association of idiopathic pulmonary hemosiderosis, celiac disease and dilated cardiomyopathy. Lung India 32:70–72. https://doi.org/10.4103/0970-2113.148457
Sherani KM, Upadhyay HN, Sherani FK et al (2015) Idiopathic pulmonary hemosiderosis presenting in an adult: a case report and review of the literature. Lung India 32:395–397. https://doi.org/10.4103/0970-2113.159594
Karatas M, Ozyurt S, Gumus A et al (2017) Idiopathic pulmonary hemosiderosis with celiac disease; Lane-Hamilton syndrome. J Clin Anal Med 8.https://doi.org/10.4328/jcam.4847
Popp A, Jurcuţ C, Balaban DV et al (2016) Severe alveolar hemorrhage - what’s in it for the gastroenterologist? J Gastrointestin Liver Dis 25:555–558. https://doi.org/10.15403/jgld.2014.1121.254.cut
Prabhu S, Rai S, Acharya V et al (2016) Ceelen-Gellerstedt syndrome in an elderly Indian man: case report of an unusual case. https://doi.org/10.4103/0975-9727.174675
Agarwal Idiopathic pulmonary hemosiderosis in a young adult patient: a rare case. https://www.jacpjournal.org/article.asp?issn=2320-8775;year=2018;volume=6;issue=1;spage=38;epage=40;aulast=Agarwal. Accessed 12 Jan 2022
Silva P, Ferreira PG (2017) Idiopathic pulmonary hemosiderosis: hemorrhagic flare after 6 years of remission. Rev Port Pneumol (2006) 23:368–369. https://doi.org/10.1016/j.rppnen.2017.07.006
Kania A, Misiaszek M, Vašáková M et al (2019) Cryobiopsy in the diagnosis of idiopathic pulmonary hemosiderosis: a case report. J Thorac Dis 11:3195–3201. https://doi.org/10.21037/jtd.2019.07.17
Butt A, Ahmed R, Sheikh MDA et al (2020) Idiopathic pulmonary hemosiderosis - a rare cause of chronic anemia. Monaldi Arch Chest Dis 90. https://doi.org/10.4081/monaldi.2020.1267
Gocho K, Sato K, Imasaka K et al (2020) A case of idiopathic pulmonary hemosiderosis associated with emphysematous change in an adult who underwent lung transplantation. Intern Med. https://doi.org/10.2169/internalmedicine.5142-20
Poberezhets V, Poberezhets O (2020) A case of idiopathic pulmonary hemosiderosis in a 30-year-old man. Clin Pulm Med 27:64–66. https://doi.org/10.1097/CPM.0000000000000357
Austin S, Kobrin D, Villgran V et al (2021) Coincidence or connection? A patient with concurrent Lane Hamilton syndrome and idiopathic membranous nephropathy. Respir Med Case Rep 33:101446. https://doi.org/10.1016/j.rmcr.2021.101446
Iwasaki K, Matsuzawa Y, Wakabayashi H, Kumano K (2021) Diffuse alveolar haemorrhage with suspected idiopathic pulmonary hemosiderosis and decrease in lung diffusing capacity and chronic respiratory failure. BMJ Case Rep 14:e242901. https://doi.org/10.1136/bcr-2021-242901
Walsh L, McCarthy C, Henry M (2021) Autoimmune pulmonary alveolar proteinosis and idiopathic pulmonary haemosiderosis: a dual pathology. BMJ Case Rep 14:e241048. https://doi.org/10.1136/bcr-2020-241048
Wright PH, Menzies IS, Pounder RE, Keeling PW (1981) Adult idiopathic pulmonary haemosiderosis and coeliac disease. Q J Med 50:95–102
Wright PH, Buxton-Thomas M, Keeling PW, Kreel L (1983) Adult idiopathic pulmonary haemosiderosis: a comparison of lung function changes and the distribution of pulmonary disease in patients with and without coeliac disease. Br J Dis Chest 77:282–292
Waldenström J (1944) Relapsing, Diffuse, Pulmonary bleedings or hemosiderosis pulmonum — a New clinical diagnosis. Acta Radiol os-25:149–162. https://doi.org/10.1177/028418514402500207
Ohga S, Takahashi K, Miyazaki S et al (1995) Idiopathic pulmonary haemosiderosis in Japan: 39 possible cases from a survey questionnaire. Eur J Pediatr 154:994–995. https://doi.org/10.1007/BF01958645
Kjellman B, Elinder G, Garwicz S, Svan H (1984) Idiopathic pulmonary haemosiderosis in Swedish children. Acta Paediatr 73:584–588. https://doi.org/10.1111/j.1651-2227.1984.tb09978.x
Taytard J, Nathan N, de Blic J et al (2013) New insights into pediatric idiopathic pulmonary hemosiderosis: the French RespiRare(®) cohort. Orphanet J Rare Dis 8:161. https://doi.org/10.1186/1750-1172-8-161
Alimi A, Taytard J, Abou Taam R et al (2018) Pulmonary hemosiderosis in children with Down syndrome: a national experience. Orphanet J Rare Dis 13:60. https://doi.org/10.1186/s13023-018-0806-6
Hizal M, Polat SE, Gursoy TR et al (2021) Risk factors for recurrent pulmonary exacerbation in idiopathic pulmonary hemosiderosis. Pediatr Pulmonol 56:1060–1068. https://doi.org/10.1002/ppul.25189
Bronson SM (1960) Idiopathic pulmonary hemosiderosis in adults: report of a case and review of the literature. Am J Roentgenol Radium Ther Nucl Med 83:260–273
Soergel KH, Sommers SC (1962) Idiopathic pulmonary hemosiderosis and related syndromes. Am J Med 32:499–511. https://doi.org/10.1016/0002-9343(62)90051-7
Saha BK (2020) Idiopathic pulmonary hemosiderosis: a state of the art review. Respir Med 176:106234. https://doi.org/10.1016/j.rmed.2020.106234
Saha BK (2021) Is it time to call idiopathic pulmonary hemosiderosis by the correct name: immune-mediated pulmonary hemosiderosis? Am J Med Sci 361:809–811. https://doi.org/10.1016/j.amjms.2021.01.006
Saha BK, Chong WH, Milman NT (2021) Differentiation of idiopathic pulmonary hemosiderosis from rheumatologic and autoimmune diseases causing diffuse alveolar hemorrhage: establishing a diagnostic approach. Clin Rheumatol. https://doi.org/10.1007/s10067-021-05895-1
Stainer A, Rice A, Devaraj A et al (2019) Diffuse alveolar haemorrhage associated with subsequent development of ANCA positivity and emphysema in three young adults. BMC Pulm Med 19:185. https://doi.org/10.1186/s12890-019-0947-y
Freitas A, Senra V, Marinho A, Guedes M (2015) Chronic alveolar haemorrhage in a paediatric patient: a diagnostic and treatment challenge. BMJ Case Rep 2015.https://doi.org/10.1136/bcr-2014-206856
Saha BK, Bonnier A, Chenna P, Milman NT (2022) Prevalence of autoantibodies in pediatric patients with idiopathic pulmonary hemosiderosis: a scoping review of the literature in the period 1980–2021. Clin Rheumatol. https://doi.org/10.1007/s10067-021-06029-3
Watanabe H, Ayusawa M, Kato M et al (2015) Idiopathic pulmonary hemosiderosis complicated by Down syndrome. Pediatr Int 57:1009–1012. https://doi.org/10.1111/ped.12690
Dearborn DG, Yike I, Sorenson WG et al (1999) Overview of investigations into pulmonary hemorrhage among infants in Cleveland, Ohio. Environ Health Perspect 107:495–499
Lane DJ, Hamilton WS (1971) Idiopathic steatorrhoea and idiopathic pulmonary haemosiderosis. Br Med J 2:89–90. https://doi.org/10.1136/bmj.2.5753.89
Saha BK, Saha S, Bonnier A, Saha BN. Association between idiopathic pulmonary hemosiderosis and celiac disease in pediatric patients: a scoping review of the literature over the past 50 years. Pediatr Pulmonol. https://doi.org/10.1002/ppul.25847
Primack SL, Miller RR, Müller NL (1995) Diffuse pulmonary hemorrhage: clinical, pathologic, and imaging features. AJR Am J Roentgenol 164:295–300. https://doi.org/10.2214/ajr.164.2.7839958
Wesolowski JR, Lev MH (2005) CT: history, technology, and clinical aspects. Semin Ultrasound CT MR 26:376–379. https://doi.org/10.1053/j.sult.2005.07.007
Khorashadi L, Wu CC, Betancourt SL, Carter BW (2015) Idiopathic pulmonary haemosiderosis: spectrum of thoracic imaging findings in the adult patient. Clin Radiol 70:459–465. https://doi.org/10.1016/j.crad.2014.11.007
Saha BK, Chong WH (2021) Lung transplant to manage end-stage lung disease due to idiopathic pulmonary hemosiderosis: a review of the literature. Respir Investig S2212–5345(21):00118. https://doi.org/10.1016/j.resinv.2021.06.009
Cheah FK, Sheppard MN, Hansell DM (1993) Computed tomography of diffuse pulmonary haemorrhage with pathological correlation. Clin Radiol 48:89–93. https://doi.org/10.1016/S0009-9260(05)81078-5
Saha BK, Chong WH (2021) Diffuse alveolar hemorrhage in cardiac diseases. Lung 199:103–112. https://doi.org/10.1007/s00408-021-00433-x
Saha S, Chong WH, Saha BK (2021) Unilateral diffuse alveolar hemorrhage due to selective directionality of mitral regurgitant jet in a patient with severe aortic stenosis. Cureus 13:e14714. https://doi.org/10.7759/cureus.14714
Imtiaz M, Saha B, Sana Ullah U, Saha A (2019) A case of acute life-threatening pulmonary hemorrhage from synthetic cannabinoid abuse. Case Rep Pulmonol 2019:8137648. https://doi.org/10.1155/2019/8137648
Mollaeian A, Chan N, Aloor R et al (2021) ANCA-negative microscopic polyangiitis with diffuse alveolar hemorrhage masquerading as congestive heart failure. Autoimmunity Highlights 12:1. https://doi.org/10.1186/s13317-020-00143-z
Thompson G, Specks U, Cartin-Ceba R (2015) Isolated pauciimmune pulmonary capillaritis successfully treated with rituximab. Chest 147:e134–e136. https://doi.org/10.1378/chest.14-1884
Saha BK, Milman NT (2021) Idiopathic pulmonary hemosiderosis: a review of the treatments used during the past 30 years and future directions. Clin Rheumatol 40:2547–2557. https://doi.org/10.1007/s10067-020-05507-4
Chin CIC, Kohn SL, Keens TG et al (2015) A physician survey reveals differences in management of idiopathic pulmonary hemosiderosis. Orphanet J Rare Dis 10:98. https://doi.org/10.1186/s13023-015-0319-5
Xu L-H, Ou R-Q, Wu B-J et al (2017) Corticosteroid in combination with leflunomide and mesenchymal stem cells for treatment of pediatric idiopathic pulmonary hemosiderosis. J Trop Pediatr 63:389–394. https://doi.org/10.1093/tropej/fmx002
Saha BK, Milman NT (2021) Short review of liposteroid: a novel targeted glucocorticoid preparation for treatment of autoimmune and inflammatory diseases. Prague Med Rep 122:257–268. https://doi.org/10.14712/23362936.2021.23
Author information
Authors and Affiliations
Contributions
BKS, AB, and SS planned the study. BKS, AB, and SS collected the data. BKS, AB, SS, and BS performed data synthesis. BKS, AB, SS, BS, and BNS prepared the initial manuscript. All the authors contributed to the finalization of the manuscript.
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Saha, B.K., Bonnier, A., Saha, S. et al. Adult patients with idiopathic pulmonary hemosiderosis: a comprehensive review of the literature. Clin Rheumatol 41, 1627–1640 (2022). https://doi.org/10.1007/s10067-022-06104-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-022-06104-3