
Abstract
Control of refractory bleeding in idiopathic pulmonary hemosiderosis (IPH) is challenging. Based on the effect of liposteroid (dexamethasone palmitate) for acute bleeding in two reported cases, the long-term utility was assessed in all nine IPH children (including the first two cases) treated in a tertiary center for 20 years. The median at disease onset was 2.3 years (range, 1.2 to 8.6). All had life-threatening and/or repetitive bleeding on prednisolone (PSL) therapy. Liposteroid was intravenously infused at 0.8 mg/kg/day for three consecutive days at the time of acute bleeding. Single infusion was followed by a longer interval from weekly to monthly accompanied by low-dose PSL (less than 0.3 mg/kg/day). Monthly infusion as maintenance therapy was continued for prophylaxis of bleeding. Treatment outcomes were retrospectively analyzed. During the observation period of a median of 11.0 years (range 2.4–16.9 years), no one died. Five patients were weaned and the other one was being weaned from liposteroid for the cure or long remission (median, 5.5 years). Three others were on liposteroid therapy because of active disease. Neither patient had respiratory symptoms, although three showed subnormal %vital capacity. Serum levels of KL-6 and ferritin were normal in all and all but one patient(s), respectively. Four patients (three on liposteroid therapy) showed low bone mineral density. There were no obese patients. Height SD score did not significantly decrease except for one patient. Conclusion: The liposteroid therapy might improve the survival of IPH patients with reducing the adverse effects of steroids, although prospective control studies are needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic pulmonary hemosiderosis (IPH) is a rare disorder causing recurrent bleeding and subsequent hemosiderin accumulation in the lung [20, 21]. It occurs mostly in children, characterized by a triad of iron deficiency anemia, diffuse parenchymal infiltrates on chest radiographs, and chronic and/or repetitive pulmonary symptoms. Affected children have dyspnea, cough, wheezes, and cyanosis, along with occasional fever and inflammatory responses. Absent hemoptysis by swallowing bloody sputa might mislead to the diagnosis of common diseases such as pneumonia and gastrointestinal bleeding [30]. The incidence of IPH is estimated to be low at 0.24–1.23 cases per million [15, 23]. Patients die from acute bleeding and/or progressive cardiopulmonary failure due to hemosiderin deposition and fibrosis in the lung. The etiology of IPH remains unclear, although immune-mediated mechanisms are postulated. Local epidemics might suggest that pulmonary hemosiderosis occurs in the association with triggering factors [2, 5, 10].
More than 60 % of IPH patients died with a median of survival period of 2.5–5 years in earlier reports [4, 27]. However, the 5-year overall survival rate increased to 67–86 % at the end of last century [14, 23]. The improved survival might be explained by the early corticosteroid (CS) therapy for acute bleeding and the CS prophylaxis for recurrent bleeding [14, 17, 25, 31]. Nevertheless, persistent bleeding and hemorrhagic bouts occur unexpectedly even after the long remission [11]. The steady decline in survival beyond 5 years after diagnosis implied that patients continued to be at risk of death from massive bleeding after CS therapy was discontinued [21]. Treatment effects of immunosuppressant are varied on either acute or chronic bleeding. Prolonged administration of CS and anti-metabolites such as 6-mercaptopurine (6MP) and azathioprine (AZA) renders detrimental effects on the growth and raises the risk of developing malignancy [6]. No maintenance therapy has been established for children with refractory IPH.
Liposteroid is the dexamethasone palmitate incorporated in lipid microspheres. Because liposome is easily taken up by and retained in phagocytes, it is used for the treatment of rheumatic diseases in Japan [19]. We reported that intravenous administration of liposteroid effectively controlled acute pulmonary bleeding in two cases with refractory IPH [22]. The drastic effect was confirmed in other institutions [26]. In the present study, clinical utility of liposteroid was assessed in terms of the pulmonary function and adverse effects during and after the long-term therapy.
Methods
Patients
Eleven Japanese patients less than 15 years of age were diagnosed as having IPH in Kyushu University Hospital between 1992 and 2008. Two patients were excluded from the study because of less than 1 year of therapeutic course; one 2-month-old female (Heiner syndrome: positive precipitating antibodies against cow’s milk) died with no receiving liposteroid 16 h after the first bleeding. The other 4-year-old female obtained remission 2 months after liposteroid therapy; the duration of which was too short to evaluate the long-term effects. Nine fulfilled the diagnostic criteria of IPH; the classical triad of iron deficiency anemia, diffuse infiltrates on chest X-ray, and respiratory signs, along with detection of siderophages in the gastric lavage fluids or sputa, and no underlying causes of bleeding. Bleeding and hemosiderosis in the lung were assessed by high-resolution computed tomography, magnetic resonance imaging, and in part histopathology. Liposteroid therapy was started after the informed consent was obtained from parents.
Liposteroid therapy
Liposteroid (limethasone®, 2.5 mg/ml, Mitsubishi Tanabe Pharma Co., Osaka, Japan) therapy was introduced when patients had life-threatening and/or recurrent hemorrhagic bouts during the treatment course. It was intravenously infused at 0.8 mg/kg/day for three consecutive days at the time of acute bleeding [22, 26], after the obtainment of informed consent from parents. The single infusion was followed by the step-up widening interval from weekly to monthly accompanied by low-dose oral prednisolone (PSL) (less than 0.3 mg/kg). When bleeding recurred, the 3-day liposteroid therapy was followed by intermittent infusion in the same manner. Monthly infusion was given for at least 4 years as long as subclinical, persistent, and/or abrupt bleeding continued. Immunosuppressants (MZB) were limitedly added to the patients who did not attain the maintenance monthly infusion.
Assessment on the late effects
During the treatment course, complete blood counts, liver and kidney functions, iron profiles, and diabetic and ophthalmologic screenings were examined. Chest X-ray, and/or computed tomography of the lung were assessed at the bleeding. Height and weight were measured by a Harpenden stadiometer and an electronic scale, respectively. Body mass index (BMI) was calculated to assess the degree of obesity [9, 16]. Height was transformed into a standard deviation (SD) score for chronological age and sex, based on the standard growth charts for Japanese children [28], or those with Down syndrome [13]. Bone mineral density (BMD) was measured with a Hologic densitometer. Target heights were calculated by the heights of parents. Pulmonary function was assessed by the values of vital capacity as percent of predicted (%VC) and forced expiratory volume in 1 s as percent of forced vital capacity (FEV1.0 %) measured on the spirometry. Serum KL-6 levels were monitored as an active and sensitive marker of pulmonary fibrosis [32]. The present quality of life was assessed by the activity in school and preschool life (the Karnofsky Performance Status Scale).
Results
Treatment courses
Demographics of IPH patients are shown in Table 1. Male to female ratio was 4 to 5. Median age at the disease onset was 2.3 years (range, 1.2 to 8.6). Patient (Pt) 1 and Pt 4 were reported previously [22]. Pt 1, Pt 2, and Pt 8 were born as low-birth-weight infants. Pt 9 had trisomy 21. However, they had no any cause of pulmonary bleeding at diagnosis. Pt 5 might be sensitive to milk, but had no specific antibodies against cow’s milk. No patient was positive for anti-neutrophil cytoplasmic autoantibody. Two had very low titers of anti-nuclear antibody but did not develop autoimmune diseases. Despite the favorable control of bleeding, Pt 9 suffered from thyroiditis 6 years after the onset of IPH.
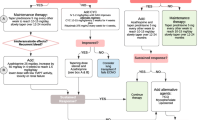
Seven received blood transfusions at the first presentation, when a median of hemoglobin level was 5.0 g/dl (range, 2.7 to 7.4). Liposteroid therapy was started in six patients for uncontrollable bleeding by high-dose PSL with or without high-dose methyl-PSL (mPSL) pulse therapy (30 mg/kg/day for 3 days), and one (Pt 8) for the most severe first bleeding with shock (Fig. 1). The liposteroid therapy was early introduced to Pts 2, 3, and 9. Prior to liposteroid therapy, three patients received immunosuppressants of AZA, cyclosporine-A (CSA). No patient died during the observation period (median, 11.0 years; range, 2.4–16.9 years).

Therapeutic course and cumulative dose of steroids and immunosuppressant. The dosages of liposteroid and methylprednisolone (mPSL) (mg/kg of body weight) were calculated to the equivalent doses of prednisolone (PSL) based on the anti-inflammatory potency. In therapeutic course, open marks mean initiation of each drug, and closed marks mean the discontinuation of each drug. The circles mean PSL and the squares mean liposteroid. The star signs mean the hospitalization due to bleeding attacks. The open star signs indicate the bleedings that were treated with oral PSL/3-day liposteroid therapy. The closed star signs indicate the bleedings which required additional high-dose mPSL pulse therapy. The solid lines indicate the period of steroid therapy and the dashed lines indicate the observation period (off therapy). The arrowed lines under the solid lines indicate the period of immunosuppressant administration. AZA: azathioprine, CSA: cyclosporine-A, mPSL: methylprednisolone, MZB: mizoribine, PSL: prednisolone, Pt: patient
Treatment effects on the pulmonary function
Three patients (Pts 1, 2, and 3) attained cure based on more than 1.4 years of medication-free remission (Fig. 1 and Table 2). Pt 4 weaned liposteroid on tapering dose of PSL. Pt 5 was being weaned from liposteroid therapy for more than 2 years of remission. Three others (Pts 7, 8, and 9) were on liposteroid therapy less than 2 years after the last bleeding. Of the three patients, Pt 7 had poor adherence to the therapy. Because his parents refused the hospitalization at bleeding attacks, Pt 7 required the increased doses of PSL and liposteroid as an outpatient (Fig. 1). At the time of study, no one had anemia or respiratory problem at rest. Pts 1, 5, and 7 showed less than 80 % of predicted VC, and Pt 7 only showed mild dyspnea at hard exercise (Table 3). Spirometry was not completed in two patients for the lack of skill. Serum levels of KL-6 and ferritin were normal in 9 and 8 patients, respectively.
Late effects on the growth and quality of life
Anthropometric measurements are shown in Table 3. No patients showed cushingoid face, hypertension, striae cutis, pathological bone fracture, obesity (all BMI z score <2), and steroid-induced visual impairment at the time of study. Four patients (Pts 5, 7–9) showed low BMD (z score < −2). All four evaluable patients attained more than 85 % of target height. The height of Pt 9 was normal for Japanese Down syndrome (±0.5SD), although the SD score was less than −2.5. The height SD scores of seven patients were within −2.5, but the score of Pt 7 who was only less adherent to the therapy was −3.9. The SD scores of height (p = 0.154) and weight (p = 0.308) did not significantly differ between at the disease onset and at present (Fig. 2). All six patients with cure or long remission had complete performance scores and no restriction of exercise in school life at the time of study. Three others on active disease attended school or preschool without infection proneness during the maintenance liposteroid therapy.

The standard deviation (SD) score of height and weight in IPH patients. The mean SD score of height (a) and weight (b) of IPH patients did not significantly differ between at the onset of disease and at present, respectively. The statistical comparison was assessed by paired t test (a: p = 0.154, b: p = 0.308). The square represents the value of Pt 7 who was not closely adherent to the therapy
Discussion
The notable findings in the observational study were (1) no death during and after the long-term liposteroid therapy, (2) complete cure or long remission in more than 65 % patients, and (3) favorable pulmonary functions and tolerable effects of CS accumulation. The declining pattern of height SD score might be associated with lower BMD on active disease. The liposteroid therapy could improve the survival and quality of life in CS-dependent IPH children. Further prospective studies are needed to optimize the therapy in large number of patients.
The treatment practice of IPH has not changed over the last decade. Lung-specific autoimmunity has been implicated for the pathogenesis. Matrix metalloproteinases and the inhibitors might be associated with the progressive damage and remodeling of the lung tissue [29]. Clinical application of high-dose γ-globulin or rituximab remains elusive, since no pathognomonic factors for IPH have yet been identified. The rare disease is hard to diagnose for the infrequent hemoptysis [24]. Lung biopsy has less diagnostic importance, because the intervention raises a risk of bleeding and only excludes secondary causes of pulmonary bleeding. In our series of patients, not all patients underwent lung biopsy because of the severity at diagnosis. If poor prognostic diseases such as pulmonary capillaritis were included, the outcomes could be more impressive. Treatment effects of high-dose CS therapy on acute bleeding were well-recognized [15]. Kiper et al. suggested that long-term CS treatment could lead to a milder course and prevent crises, based on the prolonged survival of 22 IPH children between 1979 and 1994 [14]. Saeed et al. reported that extended courses of immunosuppressive therapy yielded a better outcome, based on the analysis of 17 IPH children treated between 1972 and 1998 [25]. Low-dose CS prophylaxis against chronic bleeding has come to the front by improving the overall survival of patients. Recent reports supported the benefit of prolonged CS therapy for the outcome of patients [3, 12]. In this study, liposteroid was at first planned to employ in limited patients who had repetitive bleeding during the oral PSL therapy. However, in our tertiary center, all IPH patients were recruited as a consequence during the study period. The primary goal of IPH management is the prophylaxis of fatal bleeding as experienced in an infant with Heiner syndrome. None of nine patients with liposteroid therapy died, in contrast to the poor outcome in historical control of 36 patients without liposteroid therapy in Japan [23] (p < 0.05, Supplementary figure). Historical comparison tends to overestimate the impact of the present study; however, clinical outcomes of our patients appeared to be successful compared with any reported controls without liposteroid therapy [4, 23, 27].
Liposteroid is the dexamethasone palmitate ester incorporated in liposome, which is developed as a drug carrier vehicle for appropriate distribution to the inflamed site and decreasing systemic side effects [1, 19, 33]. Although anti-inflammatory effect depends on the concentration of steroid in inflamed tissue, this agent is easily taken up and can inhibit the activation of pulmonary macrophages to accumulate the hemosiderin. Furthermore, dexamethasone has higher affinity for the glucocorticoid receptor than PSL or mPSL and has relatively stronger anti-inflammatory effect and longer biological half-life. Bonanomi et al. [1] showed no suppression of the endogenous cortisol levels in liposteroid administrated rabbit arthritis models. Hoshi et al. [7] revealed that this agent had a higher improvement rate of rheumatoid arthritis and lower frequency of side effects than dexamethasone. Even in the varied and prolonged courses (Fig. 1), systemically low-dose liposteroid therapy might accumulate effectively in the hemorrhagic inflamed site of the lung. It could, therefore, reduce the chance of high-dose mPSL therapy and prevent the dose escalation of oral PSL therapy.
There is no standard to control the refractory IPH bleeding. Treatment choice of additional immunosuppressant remains elusive [8, 11, 34]. Oral CSA and AZA were administrated as a prior therapy in three patients, but the treatment effects were all equivocal. In Pts 1 and 5, liposteroid therapy instead of CSA effectively changed the course of repetitive bleeding. Consecutive 3-day infusion of liposteroid succeeded in the prophylaxis of bleeding during surgical intervention for acute abdomen in Pt 2. The 3-day infusion in outpatient clinic could also control bleeding triggered by upper respiratory infection. Recently, Luo et al. reported the utility of 6MP combined with oral PSL therapy [18]. 6MP and AZA could reduce the maintenance dose of PSL for the control of refractory bleeding. However, the optimal dose should be individualized to avoid the adverse event of myelosuppression. FDA has reported the risk of developing malignancy in patients with juvenile idiopathic arthritis or inflammatory bowel disease, who continued to receive immunosuppressant such as 6MP/AZA [6]. In this setting, liposteroid therapy has an advantage in the long-term therapy for children.
Final height was a major concern on liposteroid therapy, although intermittent infusion of liposteroid might reduce the accumulated CS effects. No patients became obese at the time of study. On the other hand, neither had plus SD scores of height nor exceeded the target height. Impaired bone mineralization on active phase would be normalized after the therapy. Pt 7 had often stopped any treatment by himself since parents divorced. Frequent bleeding might affect his height before the growth spurt.
In conclusion, our observation demonstrated that IPH children can be cured after appropriate maintenance therapy. The long-term liposteroid therapy could improve the outcome of IPH patients, regarding the mortality, pulmonary functions, and performance in school life. Further prospective studies are needed to establish the maintenance therapy for refractory IPH bleeding.
Abbreviations
- AZA:
-
Azathioprine
- BMD:
-
Bone mineral density
- CS:
-
Corticosteroid
- CSA:
-
Cyclosporine-A
- IPH:
-
Idiopathic pulmonary hemosiderosis
- 6MP:
-
6-Mercaptopurine
- mPSL:
-
Methylprednisolone
- MZB:
-
Mizoribine
- Pt:
-
Patient
- PSL:
-
Prednisolone
References
Bonanomi MH, Velvart M, Stimpel M, Roos KM, Fehr K, Weder HG (1987) Studies of pharmacokinetics and therapeutic effects of glucocorticoids entrapped in liposomes after intraarticular application in healthy rabbits and in rabbits with antigen-induced arthritis. Rheumatol Int 7(5):203–212
Cassimos CD, Chryssanthopoulos C, Panagiotidou C (1983) Epidemiologic observations in idiopathic pulmonary hemosiderosis. J Pediatr 102(5):698–702
Chen CH, Yang HB, Chiang SR, Wang PC (2008) Idiopathic pulmonary hemosiderosis: favorable response to corticosteroids. J Chin Med Assoc 71(8):421–424
Chryssanthopoulos C, Cassimos C, Panagiotidou C (1983) Prognostic criteria in idiopathic pulmonary hemosiderosis in children. Eur J Pediatr 140(2):123–125
Dearborn DG, Smith PG, Dahms BB, Allan TM, Sorenson WG, Montana E, Etzel RA (2002) Clinical profile of 30 infants with acute pulmonary hemorrhage in Cleveland. Pediatrics 110(3):627–637
Diak P, Siegel J, La Grenade L, Choi L, Lemery S, McMahon A (2010) Tumor necrosis factor alpha blockers and malignancy in children: forty-eight cases reported to the Food and Drug Administration. Arthritis Rheum 62(8):2517–2524
Hoshi K, Mizushima Y, Shiokawa Y, Kageyama T, Honma M, Kashiwazaki S, Shichikawa K, Tsunematsu T, Kaneko K (1985) Double-blind study with liposteroid in rheumatoid arthritis. Drugs Exp Clin Res 11(9):621–626
Huang SH, Lee PY, Niu CK (2003) Treatment of pediatric idiopathic pulmonary hemosiderosis with low-dose cyclophosphamide. Ann Pharmacother 37(11):1618–1621
Inokuchi M, Hasegawa T, Anzo M, Matsuo N (2006) Standardized centile curves of body mass index for Japanese children and adolescents based on the 1978–1981 national survey data. Ann Hum Biol 33(4):444–453
Investigation of acute idiopathic pulmonary hemorrhage among infants - Massachusetts, December 2002-June 2003 (2004). MMWR Morb Mortal Wkly Rep 53 (35):817-820
Ioachimescu OC, Sieber S, Kotch A (2004) Idiopathic pulmonary haemosiderosis revisited. Eur Respir J 24(1):162–170
Kabra SK, Bhargava S, Lodha R, Satyavani A, Walia M (2007) Idiopathic pulmonary hemosiderosis: clinical profile and follow up of 26 children. Indian Pediatr 44(5):333–338
Kimura J, Tachibana K, Imaizumi K, Kurosawa K, Kuroki Y (2003) Longitudinal growth and height velocity of Japanese children with Down’s syndrome. Acta Paediatr 92(9):1039–1042
Kiper N, Gocmen A, Ozcelik U, Dilber E, Anadol D (1999) Long-term clinical course of patients with idiopathic pulmonary hemosiderosis (1979-1994): prolonged survival with low-dose corticosteroid therapy. Pediatr Pulmonol 27(3):180–184
Kjellman B, Elinder G, Garwicz S, Svan H (1984) Idiopathic pulmonary haemosiderosis in Swedish children. Acta Paediatr Scand 73(5):584–588
Kuromaru R, Kohno H, Hara T (2002) Changes in adiposity and excess body weight correlate with growth responses but not with decreases in low-density lipoprotein cholesterol levels during GH treatment in GH-deficient children. Clin Endocrinol (Oxf) 56(6):799–803
Le Clainche L, Le Bourgeois M, Fauroux B, Forenza N, Dommergues JP, Desbois JC, Bellon G, Derelle J, Dutau G, Marguet C, Pin I, Tillie-Leblond I, Scheinmann P, De Blic J (2000) Long-term outcome of idiopathic pulmonary hemosiderosis in children. Med (Baltimore) 79(5):318–326
Luo XQ, Ke ZY, Huang LB, Guan XQ, Zhang XL, Zhu J, Zhang YC (2008) Maintenance therapy with dose-adjusted 6-mercaptopurine in idiopathic pulmonary hemosiderosis. Pediatr Pulmonol 43(11):1067–1071
Mizushima Y, Hamano T, Yokoyama K (1982) Tissue distribution and anti-inflammatory activity of corticosteroids incorporated in lipid emulsion. Ann Rheum Dis 41(3):263–267
Nuesslein TG, Teig N, Rieger CH (2006) Pulmonary haemosiderosis in infants and children. Paediatr Respir Rev 7(1):45–48
Ohga S, Miyazaki S (1997) Diagnosis and management of IPH—idiopathic pulmonary hemosiderosis—current diagnosis and management. Int J Pediat Hem Onc 4(2):161–170
Ohga S, Nomura A, Suga N, Hikino S, Kira R, Matsuzaki A, Masuda K, Ueda K (1994) Liposteroid against refractory pulmonary haemorrhage in idiopathic pulmonary haemosiderosis. Eur J Pediatr 153(9):687–690
Ohga S, Takahashi K, Miyazaki S, Kato H, Ueda K (1995) Idiopathic pulmonary haemosiderosis in Japan: 39 possible cases from a survey questionnaire. Eur J Pediatr 154(12):994–995
Poggi V, Lo Vecchio A, Menna F, Menna G (2011) Idiopathic pulmonary hemosiderosis: a rare cause of iron-deficiency anemia in childhood. J Pediatr Hematol Oncol 33(4):e160–e162
Saeed MM, Woo MS, MacLaughlin EF, Margetis MF, Keens TG (1999) Prognosis in pediatric idiopathic pulmonary hemosiderosis. Chest 116(3):721–725
Sakurai Y, Kamisue S, Shima M, Yoshioka A (1995) Successful treatment with liposteroid in an infant case of idiopathic pulmonary hemosiderosis. Jpn J Pediatr Hematol 9(1):42–46
Soergel KH, Sommers SC (1962) Idiopathic pulmonary hemosiderosis and related syndromes. Am J Med 32(4):499–511
Suwa S, Tachibana K (1993) Standard growth charts for height and weight of Japanese children from birth to 17 years based on a crosssectional survey of national data. Clin Pediatr Endocrinol 2:87–97
Tang LF, Huang XY, Chen ZM, Du LZ (2010) Matrix metalloproteinase-9 and its inhibitor in idiopathic pulmonary hemosiderosis. Indian J Pediatr 77(5):581–582
Yao TC, Hung IJ, Jaing TH, Yang CP (2002) Pitfalls in the diagnosis of idiopathic pulmonary haemosiderosis. Arch Dis Child 86(6):436–438
Yao TC, Hung IJ, Wong KS, Huang JL, Niu CK (2003) Idiopathic pulmonary haemosiderosis: an Oriental experience. J Paediatr Child Health 39(1):27–30
Yokoyama A, Kondo K, Nakajima M, Matsushima T, Takahashi T, Nishimura M, Bando M, Sugiyama Y, Totani Y, Ishizaki T, Ichiyasu H, Suga M, Hamada H, Kohno N (2006) Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 11(2):164–168
Yokoyama K, Okamoto H, Watanabe M, Suyama T, Mizushima Y (1985) Development of a corticosteroid incorporated in lipid microspheres (liposteroid). Drugs Exp Clin Res 11(9):611–620
Zaki M, Al Saleh Q, Al Mutari G (1995) Effectiveness of chloroquine therapy in idiopathic pulmonary hemosiderosis. Pediatr Pulmonol 20(2):125–126
Acknowledgments
We thank Dr. Akihiko Nomura and Dr. Naohiro Suga (Department of Pediatrics, Graduate School or Medical Sciences, Kyushu University, Fukuoka, Japan) for the clinical supports and helpful discussion. This work was partly supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and from the Ministry of Health, Labour and Welfare of Japan. This work was supported in part by a grant from the Ministry of Health, Labour and Welfare of Japan.
Conflict of interest
The authors have no financial relationship with the organization that sponsored the research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary figure
Overall survival rate of IPH patients treated with or without liposteroid therapy. A (○): The actuarial overall survival rate of 9 patients who underwent liposteroid therapy after the disease onset is estimated by Kaplan–Meier method (p < 0.05, log-rank test). B (▲): A historical control group of 36 Japanese patients who did not receive liposteroid. Thirteen patients deceased (9 from acute massive bleeding, and 4 from chronic bleeding). The survival period, 11.3±1.3 years (mean±SE). (PDF 101 kb)
Rights and permissions
About this article
Cite this article
Doi, T., Ohga, S., Ishimura, M. et al. Long-term liposteroid therapy for idiopathic pulmonary hemosiderosis. Eur J Pediatr 172, 1475–1481 (2013). https://doi.org/10.1007/s00431-013-2065-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-013-2065-9