
Abstract
Objectives
Decreased natural killer (NK) cells have been reported in systemic lupus erythematosus (SLE) patients. However, the role of NK cells in the pathogenesis of SLE is not well understood. In this study, we aimed to characterize NK cell subsets, phenotypes, and cytokine-secreting functions and investigate the clinical relevance of NK cells in SLE patients.
Methods
Peripheral blood samples from 81 SLE patients and 59 healthy donors (HDs) were collected. The frequency and phenotype of NK cells were measured by flow cytometry. Intracellular interferon-γ (IFN-γ) production by NK cells was evaluated by flow cytometry after stimulation with interleukin-12 (IL-12) and IL-18.
Results
The percentages of NK cells in the peripheral blood of SLE patients were significantly lower than those in HDs, and the percentages of CD56dim NK cells among total NK cells showed a trend toward decrease. The CD56dim NK cells in SLE patients showed increased production of IFN-γ and displayed relatively activated phenotypic characteristics, including significant increases in NKp44, NKp46, and CD69 and decreased expression of CD16 and CD158a/h/g. Furthermore, CD56dim NK cells in active SLE patients had higher percentages of NKp44+ cells and lower percentages of CD158a/h/g+ cells than those in inactive SLE patients. The percentages of CD158a/h/g+ cells among CD56dim NK cells were negatively correlated with the systemic lupus erythematosus disease activity index (SLEDAI) and positively correlated with C3 and C4 levels.
Conclusion
CD56dim NK cells in SLE patients show a reduced proportion tendency among total NK cells and are activated, which partially reflects the disease activity. CD158a/h/g expression on CD56dim NK cells may be considered an index of disease activity.
Key Points • In patients with SLE, the proportion of CD56dim NK cells showed a decreased trend and CD56dim NK cells were phenotypically activated which partially reflects the disease activity. • CD158a/h/g expression on CD56dim NK cells were decreased which may be used as an indicator for evaluating disease activity in SLE patients. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Natural killer (NK) cells constitute approximately 5–15% of the lymphocytes in human peripheral blood. NK cells were first named for their capacity to mediate spontaneous “natural” cytotoxicity toward certain tumor- and virus-infected cells. NK cells are also rapid producers of interferon-γ (IFN-γ), as well as TNF-α, GM-CSF, and other cytokines and chemokines [1]. NK cells traditionally belong to the innate immunity system, but it is now accepted that these cells shape and influence adaptive immunity and participate in autoimmune diseases [2, 3]. Two major subsets of NK cells are well established in human subjects: CD56dim and CD56bright cells. CD56dim NK cells are fully mature, predominant in the peripheral blood, and primarily mediate cytotoxic responses. In contrast, CD56bright NK cells are more immature, predominant in the tissues, and secondary lymphoid organs and have been considered primarily as cytokine producers that play a limited role in cytolytic responses [1]. Although CD56bright NK cells are more efficient at producing cytokines overall, CD56dim NK cells can also contribute significantly to early cytokine production because these cells are a significantly greater fraction of the total NK cell pool and can more rapidly secrete cytokines [4]. NK cells can produce a variety of inflammatory cytokines in response to activating receptor stimulation as well as inflammatory cytokine-induced activation signaling [5]. The combination of interleukin-12 (IL-12) and IL-18 is the best combination for inducing strong IFN-γ production [6].
NK cell activation and function are controlled by several germline-encoded membrane receptors which are generally divided into inhibitory and activating receptors. Activating receptors include FcγRIIIA (CD16), activating forms of killer cell immunoglobulin-like receptors (KIRs, KIR2DS and KIR3DS), 2B4, NKG2D, and the natural cytotoxicity receptors (NCRs) NKp30, NKp44, and NKp46. Inhibitory receptors comprise NKG2A and KIRs, such as KIR2DL1 (CD158a) [7]. CD56dim NK cells bear the low affinity Fc-γ-receptor CD16, which plays a prominent role in activating CD56dim NK cells [8]. Following stimulation, CD16 is downregulated by shedding and intracellular uptake [9]. Under certain activation conditions, NK cells are activated and express TRAIL, which interacts with TNF-related apoptosis-inducing ligand-receptor (TRAIL-R) to induce apoptosis of target cells [5].
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of autoantibodies. Increasing evidence indicates that NK cells are involved in the pathogenesis of SLE. Several studies showed that SLE patients had decreased NK cell numbers and proportions in the peripheral blood, impaired NK cell cytotoxic functions, impaired NK cell differentiation, altered phenotypes, and altered NK cell cytokine production [10,11,12,13,14,15,16,17]. In addition, studies conducted in a model of lupus also revealed the contribution of NK cells to lupus pathogenesis [18, 19]. Although it is not known whether these abnormalities are a cause or consequence of the pathological process underlying SLE, NK cells clearly contribute to the pathology. However, the relationship of NK cells with the clinical parameters of SLE has not been extensively studied. In addition, we recently discovered that blood IFNG expression levels correlate positively with anti-double-stranded DNA (ds-DNA) autoantibody levels and SLE activity [20]. Additionally, a study by Huang et al. also reported that NK cells participate in the pathogenesis and development of SLE by activation and production of IFN-γ in an amyloid-induced experimental lupus-like syndrome model [19]. Therefore, in this study, we investigated the phenotypes and IFN-γ-secreting function of NK cells and their relationship with the clinical parameters of the disease to provide comprehensive insights into the changes in NK cells in SLE patients.
Materials and methods
Patients and healthy donors
A total of 81 patients with SLE (93.8% female, median age: 35 (10–70) years) and 59 healthy donors (HDs) (79.7% female, median age: 33 (23–67) years) were included in this study. All SLE patients were recruited from the inpatient department of Xin Hua Hospital affiliated to Shanghai Jiao Tong University School of Medicine and met the 1982 American College of Rheumatology revised criteria for the classification of SLE [21]. No other exclusion criteria were applied. Clinical and laboratory data were documented at the time of inclusion. Disease activity was assessed using the SLEDAI 2000, and a score ≥ 5 was considered active SLE [22, 23]. Of the 81 patients with SLE, six were sampled twice during the course of induction therapy. HDs were selected from the Department of Medical Examination Center and did not have a history of autoimmune diseases, infectious diseases, malignancies, or immunosuppressive therapy. The study was approved by the ethics committee of Xin Hua Hospital and was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants. The clinical and laboratory characteristics of the patients and HDs are summarized in Table 1.
Isolation of peripheral blood mononuclear cells and flow cytometry
Peripheral blood mononuclear cells (PBMCs) were isolated by density-gradient centrifugation using the lymphoprep premium kit (StemCell Technologies). The cells were stained with combinations of the appropriate antibodies (Abs) against different cell surface markers for 30 min on ice. Stained cells were analyzed on a four-laser flow cytometer (Gallios, Beckman Coulter), and the data were processed using FlowJo version 10.1 software (TreeStar). The Abs used in this study included anti-CD3 FITC (Clone: OKT3), anti-CD56 PE-Cyanine5.5 (CMSSB), anti-CD16 APC (eBioCB16, CB16), anti-NKp30 PE-Cyanine7 (AF29-4D12), anti-NKp44 APC (44.189), anti-NKp46 eFluor 450 (9E2), anti-CD158a/h/g eFluor 450 (HP-MA4), anti-TRAIL PE (RIK-2), anti-CD69 eFluor 450 (FN50), and anti-IFN-γ PE (4S.B3) (eBioscience).
Intracellular IFN-γ detection
Freshly isolated PBMCs were cultured in RPMI 1640 containing 10% FBS in the presence of IL-12 (20 ng/ml) and IL-18 (25 ng/ml) (R&D Systems) with GolgiPlug (Becton Dickinson) for 4 h. The cells were harvested and stained with anti-CD3 FITC, anti-CD16 APC, and anti-CD56 PE-Cyanine5.5 for 30 min on ice. After fixation and permeabilization with the cytofix/cytoperm kit (Becton Dickinson), the cells were stained with anti-IFN-γ PE for 30 min on ice, washed with permeabilization buffer, and analyzed by flow cytometry.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 6 software. All group results are expressed as the mean ± SEM. Differences between groups were evaluated by an unpaired t test for continuous parametric variables and by a nonparametric Mann-Whitney U test for skewed-distribution variables. The Wilcoxon signed rank test for matched pairs was used to evaluate SLE patients who had completed two visits. A spearman’s test was used to test for correlation. Two-tailed p values below 0.05 were considered statistically significant.
Results
SLE patients showed a trend toward reduced proportion of circulating CD56dim NK cells
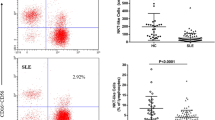
The flow cytometry gating strategy for the analysis of NK cells and their subsets are shown in Fig. S1A. The percentages of NK cells in lymphocytes were markedly reduced in SLE patients (mean ± SEM; 6.679 ± 0.6661) compared with those in HDs (mean ± SEM; 14.36 ± 1.206; p < 0.0001) (Fig. 1a, left). In addition, though they were not statistically significant, SLE patients showed a tendency of having lower proportion of CD56dim NK cells (mean ± SEM; HD: 95.17 ± 0.6845, SLE: 89.94 ± 1.650; p = 0.0721) and higher proportion of CD56bright NK cells (mean ± SEM; HD: 4.876 ± 0.6831, SLE: 10.14 ± 1.660; p = 0.0702) among total NK cells compared with HDs (Fig. 1a, middle and right). Therefore, a decrease in the CD56dim subset probably was responsible for the decrease in total NK cell numbers in SLE patients. The percentages of NK cells were lower in both inactive (mean ± SEM; 5.782 ± 1.063; p < 0.0001) and active SLE patients (mean ± SEM; 7.315 ± 0.8481; p < 0.0001) compared with those in HDs (mean ± SEM; 14.36 ± 1.206) but were similar between inactive and active SLE patients. No difference was observed in CD56dim NK cell proportions and CD56bright NK cell proportions among HDs and inactive and active SLE patients (Fig. 1b). There was a trend toward a decrease in CD56bright NK cell proportions and a trend toward an increase in CD56dim NK cell proportions after treatment or follow-up in six SLE patients, but no significant differences were found (Fig. S1B).

Frequencies of NK cells and NK cell subsets in the peripheral blood samples of SLE patients. Freshly isolated peripheral blood mononuclear cells (PBMCs) were stained with antibodies against CD3, CD56 and CD16 and analyzed by flow cytometry. (a) Percentages of total NK cells (left) or the proportions of CD56dim (middle) and CD56bright NK cells (right) in healthy donors (HDs) (n = 29) and SLE patients (n = 41). (b) SLE patients were categorized into two groups, inactive and active patients, according to disease activity. ****p < 0.0001, by Mann–Whitney U-test.
CD56dim NK cells but not CD56bright NK cells were phenotypically activated in active SLE patients
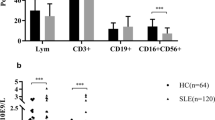
We further analyzed the expression of activating and inhibitory NK cell receptors including NKp30, NKp44, NKp46, and CD158a/h/g on both CD56dim and CD56bright NK cells. The percentages of NKp44+ cells among CD56dim NK cells were increased in active SLE patients (mean ± SEM; 1.173 ± 0.2700) compared with those in inactive SLE patients (mean ± SEM; 0.7319 ± 0.3844; p = 0.0424) and HDs (mean ± SEM; 0.2917 ± 0.04276; p = 0.0005) (Fig. 2a). Additionally, CD56dim NK cells from inactive SLE patients (mean ± SEM; 59.38 ± 4.414) had a significantly higher proportion of NKp46+ cells than those from HDs (mean ± SEM; 43.28 ± 4.140; p = 0.0190) (Fig. 2a). The expression of NKp30 on CD56dim NK cells was not different between SLE patients and HDs (data not shown). A lower frequency of CD158a/h/g+ cells among CD56dim NK cells was found in active SLE patients (mean ± SEM; 10.99 ± 1.489) compared with those in inactive SLE patients (mean ± SEM; 20.27 ± 3.852; p = 0.0413) and HDs (mean ± SEM; 20.45 ± 2.915; p = 0.0082) (Fig. 2b). We also analyzed the expression of TRAIL, CD16, and CD69, which are all signs of NK cell activation. CD56dim NK cells from active SLE patients had higher percentages of TRAIL+ cells (mean ± SEM; 10.70 ± 2.161) compared with those from HDs (mean ± SEM; 5.742 ± 1.333; p = 0.0059) (Fig. 2c left). The activation of NK cells via CD16 leads to the downregulation of surface CD16. The proportions of CD16+ cells among CD56dim NK cells were significantly lower in inactive (mean ± SEM; 79.86 ± 4.186; p < 0.0001) and active SLE patients (mean ± SEM; 81.50 ± 2.966; p < 0.0001) compared with those in HDs (mean ± SEM; 93.92 ± 1.090) (Fig. 2c middle). Additionally, CD69 is an early cell activation marker, and we found that CD56dim NK cells from inactive (mean ± SEM; 6.551 ± 1.835; p < 0.0001) and active SLE patients (mean ± SEM; 6.108 ± 1.006; p < 0.0001) more frequently expressed CD69 compared with those from HDs (mean ± SEM; 1.324 ± 0.1989) (Fig. 2c right). Intriguingly, the expression of NKp30, NKp44, NKp46, CD158a/h/g, TRAIL, and CD16 on CD56bright NK cells did not differ between SLE patients and HDs, except CD69, which was increased in SLE patients (data not shown). Together, CD56dim NK cells but not CD56bright NK cells were phenotypically activated in SLE patients, especially in active patients.

Relatively activated phenotype of CD56dim NK cells in active SLE patients. Freshly isolated PBMCs were stained with antibodies against CD3, CD56 and the natural killer receptors and analyzed by flow cytometry. (a–b) Frequencies of CD56dim NK cells expressing NKp44, NKp46, CD158a/h/g, TRAIL, CD16, and CD69 in SLE patients (n = 40 ~ 45) grouped by disease activity and HDs (n = 26 ~ 30). *p < 0.05, ** p < 0.01, ***p < 0.001, ****p < 0.0001, by Mann–Whitney U test
Increased IFN-γ production by CD56dim but not CD56bright NK cells in SLE patients
IFN-γ secretion was one of the main functions of activated NK cells. We examined the IFN-γ production of spontaneous and IL-12/18-stimulated activated NK cells. The median fluorescence intensity (MFI) of IFN-γ in both spontaneous and stimulated CD56dim NK cells was higher in inactive (mean ± SEM; 664.9 ± 38.08, 939.1 ± 107.2; p = 0.0023, p = 0.0029, respectively) and active SLE patients (mean ± SEM; 657.8 ± 38.60, 822.3 ± 54.62; p = 0.0058, p = 0.0047, respectively) compared with those in HDs (mean ± SEM; 534.3 ± 16.95, 646.3 ± 26.17, respectively) (Fig. 3a). As shown in Fig. 3b, SLE patients with hypocomplementemia displayed significantly higher IFN-γ expression in both spontaneous (mean ± SEM; 675.5 ± 33.38; p = 0.0004) and stimulated CD56dim NK cells (mean ± SEM; 925.0 ± 65.84; p < 0.0001) compared with those from HDs (mean ± SEM; 534.3 ± 16.95, 646.3 ± 26.17, respectively) and slightly increased IFN-γ expression in stimulated CD56dim NK cells compared with those from SLE patients without hypocomplementemia (mean ± SEM; 699.2 ± 66.77, p = 0.0507, not statistically significant). Anti-dsDNA did not affect CD56dim NK cell secretion of IFN-γ protein (data not shown). For CD56bright NK cells, there were no significant differences in both spontaneous and stimulated IFN-γ production between cells from SLE patients and HDs (data not shown). Overall, these data demonstrated that the numerically decreased CD56dim NK cells but not CD56bright NK cells in SLE patients were activated and secreted IFN-γ. Thus, the subsequent analysis focused on CD56dim NK cells.

Increased IFN-γ production by CD56dim NK cells in SLE patients. PBMCs were cultured with or without IL-12 (20 ng/ml) and IL-18 (25 ng/ml) in the presence of GolgiPlug for 4 h, stained with antibodies against CD3, CD56, CD16 and IFN-γ, and analyzed by flow cytometry. Median fluorescence intensity (MFI) of IFN-γ in CD56dim NK cells without (left) and with (right) IL-12/IL-18 stimulation from healthy donors (n = 26 ~ 36) and SLE patients (n = 47 ~ 48) grouped by disease activity (a) and complement levels (b), respectively. ** p < 0.01, ***p < 0.001, ****p < 0.0001, by Mann–Whitney U test.
Decreased expression of CD158a/h/g on CD56dim NK cells was associated with disease activity and low complement levels
We observed a negative correlation between the SLEDAI and the percentages of CD158a/h/g+ cells among CD56dim NK cells (r = −0.4779, p = 0.0016) (Fig. 4a). The percentages of CD158a/h/g+ cells among CD56dim NK cells were lower in SLE patients with hypocomplementemia (mean ± SEM; 12.64 ± 1.604; p = 0.0269) compared with those in HDs (mean ± SEM; 20.45 ± 2.915), but no such difference was observed compared with those in SLE patients without hypocomplementemia (mean ± SEM; 20.83 ± 5.511) (Fig. 4b). Serum C3 levels (r = 0.4533, p = 0.0038), as well as C4 levels (r = 0.4110, p = 0.0115), were positively correlated with the percentages of CD158a/h/g+ cells among CD56dim NK cells (Fig. 4c). Anti-dsDNA antibody levels did not impact the CD158a/h/g expression of CD56dimNK cells (data not shown). There was no relationship between the lupus activation marker, SLEDAI, anti-dsDNA antibody, or complements and other receptors or activation markers of CD56dim NK cells, including NKp44, NKp46, TRAIL, CD16, and CD69 (data not shown).

The expression of CD158a/h/g on CD56dim NK cells was negatively correlated with disease activity and positively correlated with complement levels. (a) Correlations between systemic lupus erythematosus disease activity index (SLEDAI) scores and percentages of CD158a/h/g+ cells among CD56dim NK cells (n = 41). (b) Percentages of CD158a/h/g+ cells among CD56dim NK cells from healthy donors (n = 29) and SLE patients without (n = 11) or with (n = 30) decreased complement levels (Mann–Whitney U-test). (c) Correlations between complement 3 (left, n = 39) or complements 4 (right, n = 37) levels and the percentages of CD158a/h/g+ cells among CD56dim NK cells. Symbols represent individual subjects. *p < 0.05, ** p < 0.01, correlations were analyzed using Spearman’s correlation analysis
CD56dim NK cell phenotype and IFN-γ secretion in relation to immunosuppressive treatment
The changes in CD56dim NK cells may be affected by medication, and so we compared the phenotype and IFN-γ secretion of CD56dim NK cells after subgrouping according to different treatments, including glucocorticoids (GCs), hydroxychloroquine (HCQ), and immunosuppressants (IS). Lower frequencies of CD16+ CD56dim NK cells were found in the high-dose group (GCs > 30 mg/d) (mean ± SEM; 73.38 ± 4.620) compared with those in the naive group (mean ± SEM; 86.98 ± 6.828; p = 0.0167) and the low to moderate dose group (GCs ≤ 30 mg/d) (mean ± SEM; 83.22 ± 2.970; p = 0.0344), as well as in patients taking HCQ (mean ± SEM; 76.68 ± 3.569) compared with those in patients that were not taking HCQ (mean ± SEM; 85.81 ± 2.888; p = 0.0314) (Fig. 5a). In addition, patients taking IS (mean ± SEM; 21.41 ± 4.048) had a higher frequency of CD158a/h/g+ cells among CD56dim NK cells compared with those in patients that were not taking IS (mean ± SEM; 11.43 ± 1.777; p = 0.0040) (Fig. 5b). Patients taking HCQ had slightly decreased IFN-γ MFI in stimulated CD56dim NK cells compared with those from patients that were not taking HCQ (Fig. S2, p = 0.0668, not statistically significant). NKp44, NKp46, TRAIL, and CD69 expression on CD56dim NK cells did not change among the treatments (data not shown).

CD16 and CD158a/h/g expression on CD56dim NK cells in SLE patients in relation to immunosuppressive treatment. (a) We divided the SLE patients receiving glucocorticoids into two groups according to their prednisone dose: a low to moderate dose (≤ 30 mg/day) group and a high dose (> 30 mg/day) group. Percentages of CD16+ cells among CD56dim NK cells from naive patients (n = 5), the low to moderate dose (n = 25) and high-dose groups (n = 13). (b) Percentages of CD16+ cells among CD56dim NK cells from SLE patients who were not taking (n = 20) and who were taking hydroxychloroquine (HCQ) (n = 24). (c) Percentages of CD158a/h/g+ cells among CD56dim NK cells from SLE patients who were not taking (n = 27) and who were taking immunosuppressants (IS) (n = 14). *p < 0.05, ** p < 0.01, by Mann-Whitney U test
Discussion
Consistent with almost all previous studies, we confirmed that NK cell percentages were lower in SLE patients than in HDs, which might be a common phenomenon in SLE. Furthermore, we showed a downward trend in the proportion of CD56dim NK cells among total NK cells in SLE patients, but it was not statistically significant, which may be related to small sample size. Interestingly, the proportion of CD56dim NK cells in SLE patients was significantly decreased when direct comparison using the unpaired t test was performed (p = 0.0129). There are several possible mechanisms that might explain the reduction in peripheral blood total NK cells and CD56dim NK cells, including activation-induced cell death, impaired differentiation, cell apoptosis induced by reactive oxygen species (ROS), and the use of medication [9]. The follow-up during induction therapy suggested a trend toward an intraindividual increase in the proportion of CD56dim NK cells over time. Our data are partially consistent with a previous observation that showed higher proportions of CD56bright NK cells in SLE patients compared with those in HDs [24], but our results were slightly different from two previous reports that showed that the distribution of NK cells into the CD56bright and CD56dim NK subsets was not significantly different between SLE patients and HDs [13, 25]. Together, our results support the possibly inverse correlation between NK cells, specifically CD56dim NK cells, and the disease course. High expression of the early cell activation marker CD69 on CD56dim NK cells from SLE patients revealed that those cells were strongly activated, which was consistent with most previous studies. It has been shown that activation of CD56dim NK cells results in the downmodulation of CD16 by shedding or internalization [9, 26]. We showed that expression of CD16 on CD56dim NK cells was strongly reduced in SLE patients, which was also found in Huang et al. [12], indicating the activation state of CD56dim NK cells. NKp44 is expressed by IL-2-activated NK cells but not by resting NK cells. We found increased proportions of NKp44+ CD56dim NK cells in active SLE patients compared with those in inactive SLE patients and HDs, which indicated that CD56dim NK cells are activated in active SLE patients. TRAIL is another marker of NK cell activation, and the increased expression of TRAIL on CD56dim NK cells indicated the activation of these cells in SLE patients. The increase in activating receptors and the decrease in inhibitory receptors might affect the immune response in patients with SLE. However, the phenotypic analyses of NK cells in SLE patients in our study have yielded some different results with others. For instance, we reported increased NKp46 expression, which was found in Lin et al. [16], but not in Hervier et al. [11]. Above differences might be reasonably caused by the different techniques used, patients’ selection, eventual pre-treatments, and size of the population analyzed. But overall, results of previous research and our work indicate that CD56dim NK cells in SLE patients showed relatively activated phenotypic characteristics. Taken together, our data suggest that CD56dim NK cells are mainly responsible for the decrease in total NK cells, and CD56dim NK cells from SLE patients, especially those from active SLE patients, are significantly activated. What is noteworthy is that almost all previous studies confirmed impaired cytotoxicity of circulating CD56dim NK cells in SLE patients. A possible explanation for the discrepancy between activated phenotype and reduced cytotoxic ability of CD56dim NK cells in SLE patients was that NK cells were activated to promote autoimmune response following induction of autoimmunity before they were suppressed and degenerated [27, 28]. In addition, dysfunctional KIR expression and NK education might also partially explain the impaired cytotoxicity of NK cells [29].
Another interesting finding was that the expression of CD158a/g/h on CD56dim NK cells was decreased in active SLE patients. Correlation analysis showed that the percentages of CD158a/h/g+ cells among CD56dim NK cells correlated inversely with disease activity and positively with serum C3 and C4 levels. KIR2DL1 (CD158a) polymorphisms have been identified as a risk factor for SLE [30]. The monoclonal antibody we used recognizes several CD158 proteins, also known as KIR2D, specifically KIR2DL1 (CD158a), KIR2DS1 (CD158h), KIR2DS3, and KIR2DS5 (CD158g). Thus, we were unable to accurately know which one or which specific receptors are involved. Bai et al. found that the expression levels of the KIR2DL1 were decreased, while the expression levels of the KIR2DS1 were increased in NK cells from SLE patients [31]. Hou et al. found a higher frequency of KIR2DS1 gene in patients with SLE [32]. Another study also showed decreased proportions of KIR2DL1/KIR2DS1+ NK cells in SLE patients [11]. Therefore, according to our results, we hypothesize that it is most likely that CD158a expression in CD56dim NK cells is reduced in SLE patients, which is consistent with the relatively activated phenotype of the CD56dim NK cells. Some studies conducted on the association of KIR with SLE suggested that individuals with more stimulatory receptors or fewer inhibitory receptors could be more susceptible to developing lupus [33]. The absence of inhibitory CD158a could potentially lower the threshold for activating receptor-mediated CD56dim NK cell activation, thereby contributing to the pathogenesis of SLE. Taken together, we showed that CD158a/h/g may be used as a clinical activity index for SLE and may be relevant to disease progression.
It has been reported that activated type II interferon signaling pathway might play a role in the pathogenesis of SLE by our and other group [34]. Although CD56bright NK cells are more efficient at producing cytokines overall, CD56dim NK cells can also contribute significantly to early cytokine production when activated, which was confirmed by our results (data not shown). Our data revealed that the expression levels of IFN-γ in spontaneous and stimulated CD56dim NK cells were higher both from active and inactive SLE patients compared with those of cells from HDs, which was not found in CD56 bright NK cells, suggesting that CD56dim NK cells in SLE patients were already activated in vivo and were able to respond to further stimulation in vitro. Our data are partially consistent with previous observations by Hervier et al. that showed a higher proportion of IFN-γ+ NK cells in patients with active SLE than in inactive SLE and controls after IL-12 and IL-18 stimulation, but the proportion of IFN-γ+ NK cells in inactive SLE patients was equivalent to that in controls [11]. However, our results are different from those of Ye et al., who showed no significant differences in the proportions of both spontaneous and inducible IFN-γ secretion by NK cells from SLE patients and controls [15]. The two studies above did not analyze the secretion of IFN-γ by the two NK subsets. Such differences may be due to the variable disease activity or treatments of patients.
Finally, in addition to the disease and/or its activity, phenotypic changes may be due to medication. A previous study has shown that classic therapies modulate the expression of several NK cell receptors in SLE patients who respond well to drugs [14]. Our analysis based on glucocorticoid treatment revealed that patients taking high doses of glucocorticoids showed lower frequencies of CD16+ CD56dim NK cells compared with those of patients not receiving or receiving low to moderate doses of steroids, which might mean more activated CD56dim NK cells in patients taking high doses of steroids. However, it is important to note that most those patients taking high doses of steroids were on intravenous steroids or even pulse therapy, which meant that they generally had high disease activity. Our data also suggested that the effect of hydroxychloroquine on the expression of CD16 and IFN-γ cannot be excluded. Intriguingly, the frequencies of CD158a/h/g+ CD56dim NK cells in patients taking IS were significantly higher than those of patients not taking IS, which indicated that IS may restore the expression of CD158a/h/g, thereby inhibiting the activation of CD56dim NK cells. Therefore, we should further explore the possibility that CD158a/h/g may serve as a biomarker to evaluate the therapeutic responses of SLE patients to IS.
Taken together, in SLE patients CD56dim NK cells showed a trend toward reduced proportion, were in activated status, and showed increased IFN-γ production. CD158 expression on CD56dim NK cells may be used as an indicator for evaluating disease activity in SLE patients. Exploring the detailed mechanisms underlying the alterations in CD56dim NK cells and understanding whether CD158a/h/g expression on CD56dim NK cells drives more pathogenic or protective functions in SLE will shed light on disease pathogenesis and may help to form a basis for new therapeutic strategies.
References
Campbell KS, Hasegawa J (2013) Natural killer cell biology: an update and future directions. J Allergy Clin Immunol 132(3):536–544. https://doi.org/10.1016/j.jaci.2013.07.006
Shi FD, Wang HB, Li H, Hong S, Taniguchi M, Link H, Van Kaer L, Ljunggren HG (2000) Natural killer cells determine the outcome of B cell-mediated autoimmunity. Nat Immunol 1(3):245–251. https://doi.org/10.1038/79792
Fogel LA, Yokoyama WM, French AR (2013) Natural killer cells in human autoimmune disorders. Arthritis Res Ther 15(4):216. https://doi.org/10.1186/ar4232
De Maria A, Bozzano F, Cantoni C, Moretta L (2011) Revisiting human natural killer cell subset function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma on activation. Proc Natl Acad Sci U S A 108(2):728–732. https://doi.org/10.1073/pnas.1012356108
Abel AM, Yang C, Thakar MS, Malarkannan S (2018) Natural killer cells: development, maturation, and clinical utilization. Front Immunol 9:1869. https://doi.org/10.3389/fimmu.2018.01869
Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J (2009) CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 126(4):458–465. https://doi.org/10.1111/j.1365-2567.2008.03027.x
Martinet L, Smyth MJ (2015) Balancing natural killer cell activation through paired receptors. Nat Rev Immunol 15(4):243–254. https://doi.org/10.1038/nri3799
Bryceson YT, March ME, Ljunggren HG, Long EO (2006) Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 107(1):159–166. https://doi.org/10.1182/blood-2005-04-1351
Grzywacz B, Kataria N, Verneris MR (2007) CD56dimCD16+ NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia 21(2):356–359. https://doi.org/10.1038/sj.leu.2404499
Park YW, Kee SJ, Cho YN, Lee EH, Lee HY, Kim EM, Shin MH, Park JJ, Kim TJ, Lee SS, Yoo DH, Kang HS (2009) Impaired differentiation and cytotoxicity of natural killer cells in systemic lupus erythematosus. Arthritis Rheum 60(6):1753–1763. https://doi.org/10.1002/art.24556
Hervier B, Beziat V, Haroche J, Mathian A, Lebon P, Ghillani-Dalbin P, Musset L, Debre P, Amoura Z, Vieillard V (2011) Phenotype and function of natural killer cells in systemic lupus erythematosus: excess interferon-gamma production in patients with active disease. Arthritis Rheum 63(6):1698–1706. https://doi.org/10.1002/art.30313
Huang Z, Fu B, Zheng SG, Li X, Sun R, Tian Z, Wei H (2011) Involvement of CD226+ NK cells in immunopathogenesis of systemic lupus erythematosus. J Immunol 186(6):3421–3431. https://doi.org/10.4049/jimmunol.1000569
Henriques A, Teixeira L, Ines L, Carvalheiro T, Goncalves A, Martinho A, Pais ML, da Silva JA, Paiva A (2013) NK cells dysfunction in systemic lupus erythematosus: relation to disease activity. Clin Rheumatol 32(6):805–813. https://doi.org/10.1007/s10067-013-2176-8
Ma H, Zhao L, Jiang Z, Jiang Y, Feng L, Ye Z (2014) Dynamic changes in the numbers of different subsets of peripheral blood NK cells in patients with systemic lupus erythematosus following classic therapy. Clin Rheumatol 33(11):1603–1610. https://doi.org/10.1007/s10067-014-2712-1
Ye Z, Ma N, Zhao L, Jiang ZY, Jiang YF (2016) Differential expression of natural killer activating and inhibitory receptors in patients with newly diagnosed systemic lupus erythematosus. Int J Rheum Dis 19(6):613–621. https://doi.org/10.1111/1756-185X.12289
Lin SJ, Kuo ML, Hsiao HS, Lee PT, Chen JY, Huang JL (2017) Activating and inhibitory receptors on natural killer cells in patients with systemic lupus erythematosis-regulation with interleukin-15. PLoS One 12(10):e0186223. https://doi.org/10.1371/journal.pone.0186223
Lin YL, Lin SC (2017) Analysis of the CD161-expressing cell quantities and CD161 expression levels in peripheral blood natural killer and T cells of systemic lupus erythematosus patients. Clin Exp Med 17(1):101–109. https://doi.org/10.1007/s10238-015-0402-1
Spada R, Rojas JM, Perez-Yague S, Mulens V, Cannata-Ortiz P, Bragado R, Barber DF (2015) NKG2D ligand overexpression in lupus nephritis correlates with increased NK cell activity and differentiation in kidneys but not in the periphery. J Leukoc Biol 97(3):583–598. https://doi.org/10.1189/jlb.4A0714-326R
Huang X, Li J, Dorta-Estremera S, Di Domizio J, Anthony SM, Watowich SS, Popkin D, Liu Z, Brohawn P, Yao Y, Schluns KS, Lanier LL, Cao W (2015) Neutrophils regulate Humoral autoimmunity by restricting interferon-gamma production via the generation of reactive oxygen species. Cell Rep 12(7):1120–1132. https://doi.org/10.1016/j.celrep.2015.07.021
Liu M, Liu J, Hao S, Wu P, Zhang X, Xiao Y, Jiang G, Huang X (2018) Higher activation of the interferon-gamma signaling pathway in systemic lupus erythematosus patients with a high type I IFN score: relation to disease activity. Clin Rheumatol 37(10):2675–2684. https://doi.org/10.1007/s10067-018-4138-7
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ (1982) The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25(11):1271–1277
Gladman DD, Ibanez D, Urowitz MB (2002) Systemic lupus erythematosus disease activity index 2000. J Rheumatol 29(2):288–291
Franklyn K, Lau CS, Navarra SV, Louthrenoo W, Lateef A, Hamijoyo L, Wahono CS, Chen SL, Jin O, Morton S, Hoi A, Huq M, Nikpour M, Morand EF (2016) Definition and initial validation of a lupus low disease activity state (LLDAS). Ann Rheum Dis 75(9):1615–1621. https://doi.org/10.1136/annrheumdis-2015-207726
Schepis D, Gunnarsson I, Eloranta ML, Lampa J, Jacobson SH, Karre K, Berg L (2009) Increased proportion of CD56bright natural killer cells in active and inactive systemic lupus erythematosus. Immunology 126(1):140–146. https://doi.org/10.1111/j.1365-2567.2008.02887.x
Hudspeth K, Wang S, Wang J, Rahman S, Smith MA, Casey KA, Manna Z, Sanjuan M, Kolbeck R, Hasni S, Ettinger R, Siegel RM (2019) Natural killer cell expression of Ki67 is associated with elevated serum IL-15, disease activity and nephritis in systemic lupus erythematosus. Clin Exp Immunol 196(2):226–236. https://doi.org/10.1111/cei.13263
Lajoie L, Congy-Jolivet N, Bolzec A, Gouilleux-Gruart V, Sicard E, Sung HC, Peiretti F, Moreau T, Vie H, Clemenceau B, Thibault G (2014) ADAM17-mediated shedding of FcgammaRIIIA on human NK cells: identification of the cleavage site and relationship with activation. J Immunol 192(2):741–751. https://doi.org/10.4049/jimmunol.1301024
Zingoni A, Sornasse T, Cocks BG, Tanaka Y, Santoni A, Lanier LL (2004) Cross-talk between activated human NK cells and CD4+ T cells via OX40-OX40 ligand interactions. J Immunol 173(6):3716–3724. https://doi.org/10.4049/jimmunol.173.6.3716
Liu R, Van Kaer L, La Cava A, Price M, Campagnolo DI, Collins M, Young DA, Vollmer TL, Shi FD (2006) Autoreactive T cells mediate NK cell degeneration in autoimmune disease. J Immunol 176(9):5247–5254. https://doi.org/10.4049/jimmunol.176.9.5247
Segerberg F, Lundtoft C, Reid S, Hjorton K, Leonard D, Nordmark G, Carlsten M, Hagberg N (2019) Autoantibodies to killer cell immunoglobulin-like receptors in patients with systemic lupus Erythematosus induce natural killer cell hyporesponsiveness. Front Immunol 10. https://doi.org/10.3389/fimmu.2019.02164
Liang HL, Ma SJ, Tan HZ (2017) Association between killer cell immunoglobulin-like receptor (KIR) polymorphisms and systemic lupus erythematosus (SLE) in populations: a PRISMA-compliant meta-analysis. Medicine (Baltimore) 96(10):e6166. https://doi.org/10.1097/MD.0000000000006166
Bai Y, Zhang Y, Yang Q, Hou Y, Hu N, Wang D, Sun H (2014) The aberrant expression of stimulatory and inhibitory killer immunoglobulin-like receptors in NK- and NKT-cells contributes to lupus. Clin Lab 60(5):717–727
Hou YF, Zhang YC, Jiao YL, Wang LC, Li JF, Pan ZL, Yang QR, Sun HS, Zhao YR (2010) Disparate distribution of activating and inhibitory killer cell immunoglobulin-like receptor genes in patients with systemic lupus erythematosus. Lupus 19(1):20–26. https://doi.org/10.1177/0961203309345779
Hou Y, Zhang C, Xu D, Sun H (2015) Association of killer cell immunoglobulin-like receptor and human leucocyte antigen-Cw gene combinations with systemic lupus erythematosus. Clin Exp Immunol 180(2):250–254. https://doi.org/10.1111/cei.12582
Chasset F, Arnaud L (2018) Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun Rev 17(1):44–52. https://doi.org/10.1016/j.autrev.2017.11.009
Acknowledgments
This research was supported by the grants from the National Natural Science Foundation of China (31470854) and the Pujiang Talents Plan (16PJ1405600).
Funding
National Natural Science Foundation of China (Grants No. 31470854) and Pujiang Talents Plan (Grants No. 16PJ1405600).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Figure S1
Frequency of CD56dim NK cells in SLE patients before and after treatment or follow-up. (a) Flow cytometry gating strategy for the analysis of NK cells. (b) SLEDAI scores, frequency of total NK cells among lymphocytes and percentages of CD56bright NK cells and CD56dim NK cells among total NK cells from six SLE patients were determined at two points during induction therapy or follow-up (Wilcoxon signed rank test). The first and second measurements are depicted. Data from the same patient before and after treatment or follow-up are denoted by an individual line. (PNG 197 kb).
Figure S2
IFN-γ production by CD56dim NK cells from SLE patients not taking (n = 22) and taking HCQ (n = 25) with IL12/18 stimulation (unpaired t test) (PNG 11 kb).
Rights and permissions
About this article

Cite this article
Liu, M., Liu, J., Zhang, X. et al. Activation status of CD56dim natural killer cells is associated with disease activity of patients with systemic lupus erythematosus. Clin Rheumatol 40, 1103–1112 (2021). https://doi.org/10.1007/s10067-020-05306-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-020-05306-x