
Abstract
Objectives
To investigate the role of methylation levels of the IFN regulatory factor 8 (IRF8) gene promoter in the development of ankylosing spondylitis (AS).
Methods
In this study, we compared the methylation levels of the IRF8 gene promoter between 99 AS patients and 99 healthy controls using MethylTarget approach. Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) was performed to compare the mRNA levels of the IRF8 gene in the other 19 AS patients and 19 healthy controls.
Results
Differential methylation was found in 91 CpG sites of the IRF8 gene promoter, and 4 CpG regions were highly methylated in AS patients compared to healthy controls (p < 0.05). In the verification stage, we found that the mRNA levels of the IRF8 gene in AS patients were significantly lower than that in controls (AS 0.77 (0.39–1.74), P = 0.038). Positive correlations between methylation of the IRF8 gene and the duration of disease, BASFI, and ESR were observed in AS patients.
Conclusions
We found a significant hypermethylation of the IRF8 gene promoter and a downregulation of the mRNA levels of the IRF8 gene in AS patients. This suggests that aberrant methylation of the IRF8 gene promoter may probably contribute to the development and pathogenesis of AS through regulating the expression of mRNA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ankylosing spondylitis (AS) is the most common form of spondyloarthropathy (SpA). AS is characterized by long-term chronic inflammation in the spine and the sacroiliac joints and predominantly affects males [1]. The prevalence of AS varies from 0.1% to 1.6% worldwide, and the number is 0.22% in the Chinese population [2, 3]. It is well accepted that genetic factors are critical for the development and pathogenesis of AS [4, 5]. The strongest genetic risk factor for AS is human leucocyte antigen (HLA)-B27, and approximately 95% of AS patients are HLA-B27-positive [6]. However, HLA-B27 and other susceptible genes can only explain around 30% of the total genetic effect of AS [7]. Thus, other factors such as epigenetics may be related to AS susceptibility. Recent evidence suggests that environmental components, including drinking [8], diet [9, 10], and infections [11], may play a role in AS by combining with susceptible genes.
Epigenetic modification is an intermediary between environmental factors and gene expression, which includes DNA methylation, histone modification, and RNA interference [12]. DNA methylation adds a methyl group to the fifth carbon position of cytosines with Sadenosyl-methionine (SAM) as the methyl donor [13]. DNA methylation of gene promoter regions often results in transcriptional gene repression, which was mediated by DNA methyltransferase (DNMT) enzymes. Substantial evidence has indicated the presence of aberrant methylation in autoimmune diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and AS [14,15,16,17,18]. Previous study has demonstrated that methylation status of the interleukin(IL)-6 gene promoter was correlated with IL6 messenger RNA levels and RA [19]. Moreover, DNA methylation alterations were associated with SLE in various immune cell populations [20]. In the genome-wide DNA methylation profile analysis, 1915 differentially methylated CpG sites in AS have been detected [21], suggesting that aberrant methylation may act as a pivotal part in the development and pathogenesis of AS.
IFN regulatory factor 8 (IRF8), a member of the IRF family, plays a role in the differentiation and function of B cell [22], Th1/Th17 [23, 24], dendritic cell (DC) [25], and osteoclast [26]. T helper (Th) type 1 and Th17 cell are important effector T cell subsets. Th1 mainly secretes IL-2, IL-12, and interferon-γ (IFN-γ) and mediates cytotoxic T cells and immune responses [27]. Th17 predominantly produces IL-17 that can drive inflammation response [28, 29]. DCs belong to the professional antigen-presenting cells (APCs) that regulate the activation of different effector T cell subsets. DCs were related to a number of autoimmune diseases, including AS [30]. In addition, IRF8 can activate or suppress the expression of some critical immune response genes through interacting with other factors [10]. Thus, we hypothesized that IRF8 is related to the pathogenesis of AS.
Recent genetic association studies showed that genetic variation of IRF8 was associated with multiple autoimmune diseases, such as SLE [31], multiple sclerosis [32], autoimmune thyroid disease [33], and Behçet’s disease (BD) [34]. Moreover, abnormal methylation of the IRF8 gene has also involved in the development of autoimmune diseases, including BD [35] and Vogt-Koyanagi-Harada (VKH) disease [36]. As an autoimmune disease, AS may have a common pathogenesis with BD and VKH, so we hypothesized that methylation levels of the IRF8 gene promoter are associated with AS.
Therefore, the aims of this study were to evaluate the methylation levels of the IRF8 gene promoter and their effect on the transcript levels of the IRF8 gene.
Materials and methods
Subjects
AS patients enrolled were diagnosed according to the 1984 Modified New York criteria. The exclusion criteria for the cases were as follows: (1) had other autoimmune diseases; (2) had malignant tumors, neurodegenerative diseases, or mental disease; and (3) had chronic diseases or systemic infections. Age- and sex-matched healthy controls were also recruited for this study. The exclusion criteria for the controls were as follows: (1) had a family history of rheumatism, (2) used drugs (e.g., hormone, immunosuppressive drugs) in the last month, (3) a history of surgery within the last 6 months. A total of 99 AS patients and 99 healthy controls were enrolled from the First Affiliated Hospital of Anhui Medical University for DNA methylation testing, and the other 19 AS patients and 19 healthy controls were enrolled to test mRNA expression level. Five milliliters of peripheral blood was obtained from each participant, and a questionnaire was collected only in AS patients. All participants provided written informed consent. The research was approved by the Local Ethics Research Committee of Anhui Medical University, and all procedures have complied with the 1964 Declaration of Helsinki. Systemic inflammation indexes such as erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) were measured. Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Functional Index (BASFI), and Ankylosing Spondylitis Disease Activity Score (ASDAS) were used to assess disease severity and functional disabilities.
DNA extraction and CpG sites selection
Genomic DNA was extracted from 5 ml of whole blood using a QIAGEN kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions and was stored at − 80 °C before the detection of DNA methylation. Quality control was applied for DNA samples. Methylation analysis was performed with a working concentration of 20 ng/μL. CpG islands located in the promoter of the IRF8 gene were selected from 2 k upstream of transcriptional start site(TSS) to 1 k downstream of first exon according to the following criteria: (1) the ratio of observed/expected dinucleotides CpG should be 0.60 or higher, (2) 200 bp minimum length, and (3) the content of GC should be no less than 50%. Finally, we selected 4 CpG regions of the IRF8 gene promoter including 91 CpG sites (Fig. 1). The details of the CpG regions are listed in Table 1.

CpG regions sequenced around promoter of IRF8. Red lines indicate four CpG regions analyzed in this study. Range of each region is indicated by its relative distance (in bp) to TSS
Bisulfite conversion and multiplex amplification
The methylation levels of the IRF8 gene promoter were analyzed by MethylTarget™ (Genesky Biotechnologies Inc., Shanghai, China), an NGS-based multiple methylation-specific PCR analysis method. Specifically, the selected genomic regions were analyzed and converted into bisulfite-converted DNA sequences by geneCpG software. We designed the PCR primers using primer3 software (http://primer3.ut.ee/) from the bisulfite-converted DNA.
Bisulfite conversion of genomic DNA (400 ng) was conducted using the EZ DNA Methylation™-GOLD Kit (Zymo Research), which converts unmethylated cytosine in genomic DNA to uracil. The samples with a rate of bisulfite conversion of DNA less than 98% were firstly filtered out. The multiple-PCR was performed to amplify the bisulfite-modified DNA sequence using indexed primers. PCR amplicons (170 bp–270 bp) were separated by agarose electrophoresis and purified using QIAquick Gel Extraction kit (QIAGEN).
Methylation detection of IRF8
The detection of IRF8 methylation was performed on Illumina Hiseq/Miseq 2000 using bidirectional sequencing verification with 2x150bp sequencing mode according to the manufacturer’s protocol. The methylation levels of the 91 CpG sites from + 78 to − 736 with respect to TSS were measured (additional file: Table S1). The methylation levels of each CpG were equal to the ratio of methylated cytosine to total cytosine.
Quantitative real-time PCR (qRT-PCR)
Peripheral blood mononuclear cells (PBMCs) from peripheral blood were isolated using Ficoll-Hypaque density gradient centrifugation method. Total cellular RNA was extracted using miRNeasy Mini Kit (Qiagen, Germany). The quality of RNA was examined by NanoDropTM 2000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA), and then RNA was reverse-transcribed to complementary DNA (cDNA) using a PrimeScript™ RT reagent kit (Takara Bio Inc., Japan). The mRNA expression levels of IRF8 were measured using the quantitative Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) and SYBR Green (Takara Bio Inc., Japan) master mix. Data were normalized to the internal control β-actin and each reaction was run in duplicate. The forward and reverse primers of IRF8 and β-actin were as follows: IRF8, forward: 5′-GAAGACGAGGGTTACGCTGTG-3′, reverse: 5′-TCCTCAGGAACAATTCGGTAA-3′; β-actin, forward: 5′-TACTCATACTCCTTGTTGTCCC-3′, reverse: 5′-AGTTGAAGGTAGTTTCGTGGAT-3′. Relative quantification was achieved using 2−∆∆Ct method as described previously.
Statistical analysis
Data analysis was carried out using SPSS software version 23.0 (SPSS, Chicago, IL, USA). The data did not meet the normality assumption; therefore, the results were described as median (max-min). Mann–Whitney U test was used to compare methylation and mRNA levels of the IRF8 gene between AS patients and healthy controls. Spearman correlations were adopted for bivariate correlation analyses. To draw the graphs, we used the GraphPad Prism version 7 for windows (GraphPad Software, La Jolla, CA USA, www.graphpad.com). P value ≤ 0.05 was considered statistically significant.
Results
Study population characteristics
Demographic and clinical features of the 99 AS patients and 99 healthy controls at the first stage are listed in Table 2. There were no statistically significant differences in sex and age between AS patients and healthy controls (age 31.14 ± 9.93 vs. 31.77 ± 8.69 years, P = 0.635; male/female 77/22 vs. 83/16, P = 0.353, respectively.). Eighty-three HLA-B27-positive individuals were observed in the AS group, and the ESR and CRP levels were elevated (ESR 20.71 ± 19.38 mm/h; CRP 18.56 ± 27.83 mg/L, respectively.). The mean disease duration of AS cases was 7.26 ± 6.36 year. In AS patients, BASDAI, BASFI, and ASDAS scores were 2.37 ± 1.52, 1.50 ± 1.70, and 3.63 ± 4.46, respectively.
Methylation levels of IRF8 promoter
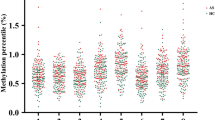
Differential methylation analyses were performed for the 91 CpG sites, and the results showed a significant change of DNA methylation in the IRF8 gene promoter in AS patients (additional file: Table S2). Four CpG regions of the IRF8 gene promoter were highly methylated in AS patients compared to healthy controls (AS vs. HC: CpG-1 1.53% (1.03–2.17%) vs. 1.40% (0.30–8.57%), P < 0.001; CpG-2 3.43% (2.66–4.54%) vs. 3.20% (2.12–10.37%), P < 0.001; CpG-3:1.23% (0.80–1.87%) vs. 1.16% (0.81–11.75%), P < 0.001; CpG-4 1.40% (0.92–2.15%) vs. 1.34% (0.79–11.85%), P = 0.02, respectively) (Fig. 2).

The methylation levels of IRF8 promoter region in AS: The methylation levels of CpG_1 (a), CpG_2 (b), CpG_3 (c) and CpG_4 (d) in AS were significantly higher than that observed in healthy controls
Subgroup analysis of IRF8 methylation
Previous reports indicated that drugs could reverse the methylation levels of DNA. Furthermore, the most important heritability of AS comes from the HLA-B27 antigen [37, 38]. Therefore, subgroup analysis by drug (received or not) and HLA-B27 (positive or negative) were performed. The results showed that AS patients who received treatments had lower methylation levels compared to those who did not (IRF8: 2.42% (1.67–12.60%) vs. 2.23% (1.58–3.08%), P = 0.012) (Fig. 3a). The methylation levels of IRF8 in HLA-B27-positive groups and HLA-B27-negative patients were higher than that in healthy controls, but there were no significant differences between the two groups.(IRF8: HLA-B27(−) vs. HLA-B27(+) 2.44% (1.90–4.26%) vs. 2.28% (1.58–12.60%), P = 0.091; HC vs. HLA-B27(−) 1.44% (1.16–7.67%) vs. 2.44% (1.90–4.26%), P < 0.001; HC vs. HLA-B27(+) 1.44% (1.16–7.67%) vs. 2.28% (1.58–12.60%), P < 0.001, respectively) (Fig. 3b–d).

Subgroup analysis of IRF8 methylation based on therapy and HLA-B27 antigen. a Untreated AS patients vs. treated AS patients. b HLA(−) AS patients vs. HLA(+) AS patients. c HLA(−) AS patients vs. HC. d HLA(+) AS patients vs. HC
Correlation of methylation with clinical manifestations
We analyzed the correlations of the methylation levels of the IRF8 gene promoter with ESR, CRP, duration, BASDAI, BASFI, and ASDAS. Positive correlations between methylation of the IRF8 gene promoter and disease duration and BASFI in CpG-3 (duration: r = 0.222, P = 0.032; BASFI: r = 0.260, P = 0.010) and CpG-4 (duration: r = 0.322, P = 0.001; BASFI: r = 0.261, P = 0.009) were observed (Table 3). Moreover, a significant correlation was identified between ESR and methylation of CpG-2. These results suggested that the methylation levels of the IRF8 gene were associated with the degree of inflammation and functional disabilities in AS.
qRT-PCR validation
To further validate the functional relevance of the IRF8 gene for AS, we compared the mRNA levels of the IRF8 gene in PBMCs between 19 AS patients and 19 healthy controls. Gender and age between the two groups showed no significant differences (age: 32.21 ± 7.58 vs. 32.05 ± 7.75 years, P = 0.950; male/female: 13/6 vs. 14/5, P = 0.721, respectively.). The mRNA levels of the IRF8 gene in AS patients were significantly lower than that in controls. The average mRNA level of IRF8 in AS is 0.77 (0.39–1.74), P = 0.038 (Fig. 4).

The results of IRF8 mRNA expression levels. P < 0.05 was considered significant
Discussion
Accumulating evidence shows that aberrant DNA methylation plays an important role in the development and pathogenesis of AS. Aberrant methylation of SOCS-1, BCL11B, and DNMT1 could be observed in AS patients [39,40,41]. In addition, a genome-wide DNA methylation profile analysis has detected 1915 differentially methylated CpG sites in AS [21]. Previous reports demonstrated that methylation of the IRF8 gene promoter was related to autoimmune disease, so we hypothesized that methylation of the IRF8 gene promoter is associated with the pathogenesis of AS.
Consistent with our hypothesis, the differential methylation was found in 91 CpG sites of the IRF8 gene promoter and 4 CpG regions were highly methylated in AS patients compared with healthy controls. This was the first report that aberrant DNA methylation of the IRF8 gene was found in AS, and thus it can further support that aberrant DNA methylation plays an important role in the development and pathogenesis of AS. In the verification stage, we found that the mRNA level of the IRF8 gene in AS was significantly lower than that in controls. On one hand, IRF8 can stimulate the expression of many important immune response genes including IL-12 [42]. Increased expression of these genes could contribute to the breakdown of immune tolerance and lead to chronic inflammation. On the other hand, IRF8 mutation resulted in a complex immunodeficiency syndrome with DC deficiency, monocytopenia, and immune dysregulation [43]. This has been confirmed by animal studies showing that IRF8−/− mice impaired T cell function and resulted in Th1 polarization defects of early immune response [44]. Therefore, hypermethylation of the IRF8 gene promoter, alongside with gene silencing [41], may contribute to the pathogenesis of AS.
Previous studies have shown an association between DNA methylation and clinical manifestation [39, 45], so we analyzed the correlations of methylation of the IRF8 gene promoter with ESR, CRP, duration, BASDAI, BASFI, and ASDAS. The positive correlations of methylation of IRF8 with duration, ESR, and BASFI further verified that methylation of the IRF8 gene promoter was related to inflammation and disease severity of AS, indicating a role of IRF8 in AS pathogenesis.
Subgroup analysis showed that treated groups had a lower methylation compared with untreated groups in AS. This suggests that therapeutics may affect the methylation status of gene promoters. Consistent with our findings, Kim et al. identified that genomic DNA hypomethylation was associated with inflammatory arthritis, and it could be reversed by methotrexate [38]. In cancer, celecoxib was found to be associated with a reduced DNA methylation and an increased mRNA expression [46]. In this study, most of the AS patients had been treated with celecoxib; this further demonstrated that DNA methylation levels can be reduced by celecoxib. The methylation levels of IRF8 in HLA-B27-positive groups and HLA-B27-negative patients were higher than that in healthy controls, but there were no significant differences between the two groups. Although it is well known that the most important heritability of AS comes from the HLA-B27 antigen, the results suggested that HLA-B27 status may not be related to the expression of methylation.
This study has some potential limitations. First, this work did not analyze methylation levels of the IRF8 gene promoter among different cell subtypes in PBMCs. It has been reported that methylation profiles were highly specific for individual cell types [47]. Second, we only measured IRF8 methylation levels in AS patients but not in RA and osteoarthritis (OA). However, our next study will address this issue by adding two more arms involving RA and OA as examples of autoimmune disease and non-inflammatory disease, respectively.
In conclusion, we found a hypermethylation of the IRF8 gene promoter and a downregulation of the mRNA levels of IRF8 in AS patients compared with healthy controls. This suggests that aberrant methylation of the IRF8 gene promoter may probably contribute to the development and pathogenesis of AS through regulating the expression of mRNA.
References
Braun J, Sieper J (2007) Ankylosing spondylitis. Lancet 369(9570):1379–1390. https://doi.org/10.1016/S0140-6736(07)60635-7
Bakland G, Nossent HC, Gran JT (2005) Incidence and prevalence of ankylosing spondylitis in northern Norway. Arthritis Care Res 53(6):850–855. https://doi.org/10.1002/art.21577
Xiang Y-J, Dai S-M (2008) Prevalence of rheumatic diseases and disability in China. Rheumatol Int 29(5):481–490. https://doi.org/10.1007/s00296-008-0809-z
Mowla K, Rajaei E, Jalali MT, Zayeri ZD (2018) Threatening biomarkers in lupus pregnancy: biochemistry and genetic challenges. Front Biol 13(1):28–35. https://doi.org/10.1007/s11515-017-1477-8
Mowla K, Saki MA, Jalali MT, Zayeri ZD (2017) How to manage rheumatoid arthritis according to classic biomarkers and polymorphisms? Front Biol 12(3):183–191. https://doi.org/10.1007/s11515-017-1452-4
Brown MA, Kennedy LG, MacGregor AJ, Darke C, Duncan E, Shatford JL, Taylor A, Calin A, Wordsworth P (1997) Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum 40(10):1823–1828. https://doi.org/10.1002/1529-0131(199710)40:10<1823::AID-ART15>3.0.CO;2-1
Hanson A, Brown MA (2017) Genetics and the causes of ankylosing spondylitis. Rheum Dis Clin N Am 43(3):401–414. https://doi.org/10.1016/j.rdc.2017.04.006
Ge R, Pan F, Liao F, Xia G, Mei Y, Shen B, Zhang T, Gao J, Zhang L, Duan Z, Xu S, Xu J (2011) Analysis on the interaction between IL-1F7 gene and environmental factors on patients with ankylosing spondylitis: a case-only study. Mol Biol Rep 38(4):2281–2284. https://doi.org/10.1007/s11033-010-0359-9
Ding N, Hu Y, Zeng Z, Liu S, Liu L, Yang T, Wu S, Fan D, Xu S, Xu J, Wang J, Pan F (2015) Case-only designs for exploring the interaction between FCRL4 gene and suspected environmental factors in patients with ankylosing spondylitis. Inflammation 38(2):632–636. https://doi.org/10.1007/s10753-014-9970-6
Jia Y, Han S, Li J, Wang H, Liu J, Li N, Yang X, Shi J, Han J, Li Y, Bai X, Su L, Hu D (2017) IRF8 is the target of SIRT1 for the inflammation response in macrophages. Innate Immun 23(2):188–195. https://doi.org/10.1177/1753425916683751
Feng XG, Xu XJ, Ye S, Lin YY, Chen P, Zhang XJ, Lin GY, Lin XQ (2011) Recent chlamydia pneumoniae infection is highly associated with active ankylosing spondylitis in a Chinese cohort. Scand J Rheumatol 40(4):289–291. https://doi.org/10.3109/03009742.2011.560891
Zhao M, Wang Z, Yung S, Lu Q (2015) Epigenetic dynamics in immunity and autoimmunity. Int J Biochem Cell Biol 67:65–74. https://doi.org/10.1016/j.biocel.2015.05.022
Jones PA, Takai D (2001) The role of DNA methylation in mammalian epigenetics. Science (New York, NY) 293(5532):1068–1070. https://doi.org/10.1126/science.1063852
Chavez-Valencia RA, Chiaroni-Clarke RC, Martino DJ, Munro JE, Allen RC, Akikusa JD, Ponsonby AL, Craig JM, Saffery R, Ellis JA (2018) The DNA methylation landscape of CD4(+) T cells in oligoarticular juvenile idiopathic arthritis. J Autoimmun 86:29–38. https://doi.org/10.1016/j.jaut.2017.09.010
Ding W, Pu W, Wang L, Jiang S, Zhou X, Tu W, Yu L, Zhang J, Guo S, Liu Q, Ma Y, Chen S, Wu W, Reveille J, Zou H, Jin L, Wang J (2018) Genome-wide DNA methylation analysis in systemic sclerosis reveals hypomethylation of IFN-associated genes in CD4(+) and CD8(+) T cells. J Invest Dermatol 138(5):1069–1077. https://doi.org/10.1016/j.jid.2017.12.003
Hong KM, Kim HK, Park SY, Poojan S, Kim MK, Sung J, Tsao BP, Grossman JM, Rullo OJ, Woo JM, McCurdy DK, Rider LG, Miller FW, Song YW (2017) CD3Z hypermethylation is associated with severe clinical manifestations in systemic lupus erythematosus and reduces CD3zeta-chain expression in T cells. Rheumatology (Oxford, England) 56(3):467–476. https://doi.org/10.1093/rheumatology/kew405
Zhao M, Zhou Y, Zhu B, Wan M, Jiang T, Tan Q, Liu Y, Jiang J, Luo S, Tan Y, Wu H, Renauer P, Del Mar Ayala Gutierrez M, Castillo Palma MJ, Ortega Castro R, Fernandez-Roldan C, Raya E, Faria R, Carvalho C, Alarcon-Riquelme ME, Xiang Z, Chen J, Li F, Ling G, Zhao H, Liao X, Lin Y, Sawalha AH, Lu Q (2016) IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis 75(11):1998–2006. https://doi.org/10.1136/annrheumdis-2015-208410
Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, Shchetynsky K, Scheynius A, Kere J, Alfredsson L, Klareskog L, Ekstrom TJ, Feinberg AP (2013) Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 31(2):142–147. https://doi.org/10.1038/nbt.2487
Nile CJ, Read RC, Akil M, Duff GW, Wilson AG (2008) Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum 58(9):2686–2693. https://doi.org/10.1002/art.23758
Long H, Yin H, Wang L, Gershwin ME, Lu Q (2016) The critical role of epigenetics in systemic lupus erythematosus and autoimmunity. J Autoimmun 74:118–138. https://doi.org/10.1016/j.jaut.2016.06.020
Hao J, Liu Y, Xu J, Wang W, Wen Y, He A, Fan Q, Guo X, Zhang F (2017) Genome-wide DNA methylation profile analysis identifies differentially methylated loci associated with ankylosis spondylitis. Arthritis Res Ther 19(1):177. https://doi.org/10.1186/s13075-017-1382-1
Lu R (2008) Interferon regulatory factor 4 and 8 in B-cell development. Trends Immunol 29(10):487–492. https://doi.org/10.1016/j.it.2008.07.006
Lee W, Kim HS, Baek SY, Lee GR (2016) Transcription factor IRF8 controls Th1-like regulatory T-cell function. Cell Mol Immunol 13(6):785–794. https://doi.org/10.1038/cmi.2015.72
Ouyang X, Zhang R, Yang J, Li Q, Qin L, Zhu C, Liu J, Ning H, Shin MS, Gupta M, Qi CF, He JC, Lira SA, Morse HC 3rd, Ozato K, Mayer L, Xiong H (2011) Transcription factor IRF8 directs a silencing programme for TH17 cell differentiation. Nat Commun 2:314. https://doi.org/10.1038/ncomms1311
Meyer MA, Baer JM, Knolhoff BL, Nywening TM, Panni RZ, Su X, Weilbaecher KN, Hawkins WG, Ma C, Fields RC, Linehan DC, Challen GA, Faccio R, Aft RL, DeNardo DG (2018) Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat Commun 9(1):1250. https://doi.org/10.1038/s41467-018-03600-6
Saito E, Suzuki D, Kurotaki D, Mochizuki A, Manome Y, Suzawa T, Toyoshima Y, Ichikawa T, Funatsu T, Inoue T, Takami M, Tamura T, Inagaki K, Kamijo R (2017) Down-regulation of Irf8 by Lyz2-cre/loxP accelerates osteoclast differentiation in vitro. Cytotechnology 69(3):443–450. https://doi.org/10.1007/s10616-016-0013-z
Echigo T, Hasegawa M, Shimada Y, Inaoki M, Takehara K, Sato S (2006) Both Th1 and Th2 chemokines are elevated in sera of patients with autoimmune blistering diseases. Arch Dermatol Res 298(1):38–45. https://doi.org/10.1007/s00403-006-0661-5
Pernis AB (2009) Th17 cells in rheumatoid arthritis and systemic lupus erythematosus. J Intern Med 265(6):644–652. https://doi.org/10.1111/j.1365-2796.2009.02099.x
Rajaei E, Haybar H, Mowla K, Zayeri ZD (2018) Metformin one in a million efficient medicines for rheumatoid arthritis complications: inflammation, osteoblastogenesis, cardiovascular disease, malignancies. Curr Rheumatol Rev 14. https://doi.org/10.2174/1573397114666180717145745
Steinman RM (2003) The control of immunity and tolerance by dendritic cell. Pathologie-biologie 51(2):59–60
Cunninghame Graham DS, Morris DL, Bhangale TR, Criswell LA, Syvanen AC, Ronnblom L, Behrens TW, Graham RR, Vyse TJ (2011) Association of NCF2, IKZF1, IRF8, IFIH1, and TYK2 with systemic lupus erythematosus. PLoS Genet 7(10):e1002341. https://doi.org/10.1371/journal.pgen.1002341
Chrabot BS, Kariuki SN, Zervou MI, Feng X, Arrington J, Jolly M, Boumpas DT, Reder AT, Goulielmos GN, Niewold TB (2013) Genetic variation near IRF8 is associated with serologic and cytokine profiles in systemic lupus erythematosus and multiple sclerosis. Genes Immun 14(8):471–478. https://doi.org/10.1038/gene.2013.42
Lin JD, Wang YH, Liu CH, Lin YC, Lin JA, Lin YF, Tang KT, Cheng CW (2015) Association of IRF8 gene polymorphisms with autoimmune thyroid disease. Eur J Clin Investig 45(7):711–719. https://doi.org/10.1111/eci.12463
Jiang Y, Wang H, Yu H, Li L, Xu D, Hou S, Kijlstra A, Yang P (2016) Two genetic variations in the IRF8 region are associated with Behcet’s disease in Han Chinese. Sci Rep 6:19651. https://doi.org/10.1038/srep19651
Qiu Y, Zhu Y, Yu H, Yi S, Su W, Cao Q, Yuan G, Kijlstra A, Yang P (2017) Ocular Behcet’s disease is associated with aberrant methylation of interferon regulatory factor 8 (IRF8) in monocyte-derived dendritic cells. Oncotarget 8(31):51277–51287. https://doi.org/10.18632/oncotarget.17235
Qiu Y, Yu H, Zhu Y, Ye Z, Deng J, Su W, Cao Q, Yuan G, Kijlstra A, Yang P (2017) Hypermethylation of interferon regulatory factor 8 (IRF8) confers risk to Vogt-Koyanagi-Harada disease. Sci Rep 7(1):1007. https://doi.org/10.1038/s41598-017-01249-7
Costantino F, Talpin A, Said-Nahal R, Leboime A, Zinovieva E, Zelenika D, Gut I, Charon C, Izac B, Weissman M, Chiocchia G, Reveille J, Breban M, Garchon HJ (2017) A family-based genome-wide association study reveals an association of spondyloarthritis with MAPK14. Ann Rheum Dis 76(1):310–314. https://doi.org/10.1136/annrheumdis-2016-209449
de Andres MC, Perez-Pampin E, Calaza M, Santaclara FJ, Ortea I, Gomez-Reino JJ, Gonzalez A (2015) Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res Ther 17:233. https://doi.org/10.1186/s13075-015-0748-5
Lai NS, Chou JL, Chen GC, Liu SQ, Lu MC, Chan MW (2014) Association between cytokines and methylation of SOCS-1 in serum of patients with ankylosing spondylitis. Mol Biol Rep 41(6):3773–3780. https://doi.org/10.1007/s11033-014-3242-2
Karami J, Mahmoudi M, Amirzargar A, Gharshasbi M, Jamshidi A, Aslani S, Nicknam MH (2017) Promoter hypermethylation of BCL11B gene correlates with downregulation of gene transcription in ankylosing spondylitis patients. Genes Immun 18(3):170–175. https://doi.org/10.1038/gene.2017.17
Aslani S, Mahmoudi M, Garshasbi M, Jamshidi AR, Karami J, Nicknam MH (2016) Evaluation of DNMT1 gene expression profile and methylation of its promoter region in patients with ankylosing spondylitis. Clin Rheumatol 35(11):2723–2731. https://doi.org/10.1007/s10067-016-3403-x
Masumi A, Tamaoki S, Wang IM, Ozato K, Komuro K (2002) IRF-8/ICSBP and IRF-1 cooperatively stimulate mouse IL-12 promoter activity in macrophages. FEBS Lett 531(2):348–353
Bigley V, Maisuria S, Cytlak U, Jardine L, Care MA, Green K, Gunawan M, Milne P, Dickinson R, Wiscombe S, Parry D, Doffinger R, Laurence A, Fonseca C, Stoevesandt O, Gennery A, Cant A, Tooze R, Simpson AJ, Hambleton S, Savic S, Doody G, Collin M (2018) Biallelic interferon regulatory factor 8 mutation: a complex immunodeficiency syndrome with dendritic cell deficiency, monocytopenia, and immune dysregulation. J Allergy Clin Immunol 141(6):2234–2248. https://doi.org/10.1016/j.jaci.2017.08.044
Tamura T, Ozato K (2002) ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J Interf Cytokine Res 22(1):145–152. https://doi.org/10.1089/107999002753452755
Guo S, Zhu Q, Jiang T, Wang R, Shen Y, Zhu X, Wang Y, Bai F, Ding Q, Zhou X, Chen G, He DY (2017) Genome-wide DNA methylation patterns in CD4+ T cells from Chinese Han patients with rheumatoid arthritis. Mod Rheumatol 27(3):441–447. https://doi.org/10.1080/14397595.2016.1218595
Liu JF, Li YS, Drew PA, Zhang C (2016) The effect of celecoxib on DNA methylation of CDH13, TFPI2, and FSTL1 in squamous cell carcinoma of the esophagus in vivo. Anti-Cancer Drugs 27(9):848–853. https://doi.org/10.1097/cad.0000000000000396
Mok A, Rhead B, Holingue C, Shao X, Quach HL, Quach D, Sinclair E, Graf J, Imboden J, Link T, Harrison R, Chernitskiy V, Barcellos LF, Criswell LA (2018) Hypomethylation of CYP2E1 and DUSP22 promoters associated with disease activity and erosive disease among rheumatoid arthritis patients. Arthritis Rheum 70(4):528–536. https://doi.org/10.1002/art.40408
Acknowledgments
We thank all patients and healthy subjects who made this study possible.
Funding
This study was supported by grants from the National Natural Science Foundation of China (Grant no. 81273169, 81573218 and 81773514), Funding for scientific research activities of academic and technical leaders in Anhui Province (Grant no. e2017D140), and Scientific Research Foundation of Translational Medicine of Anhui Province (Grant no. 2017zhyx03).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The study was approved by the Local Ethics Research Committee of Anhui Medical University, and all participants provided written informed consent. The procedures were in accordance with the 1964 Helsinki Declaration and ethical standards of the institutional.
Disclosures
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 24 kb)
Rights and permissions
About this article

Cite this article
Chen, M., Wu, M., Hu, X. et al. Ankylosing spondylitis is associated with aberrant DNA methylation of IFN regulatory factor 8 gene promoter region. Clin Rheumatol 38, 2161–2169 (2019). https://doi.org/10.1007/s10067-019-04505-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-019-04505-5