
Abstract
The effects of substituting nitrogen atoms on the stability of novel singlet (s) and triplet (t) forms of germylenes (1–20) are compared and contrasted, at B3LYP/AUG-cc-pVTZ level of theory. Every one of the 40 new divalents scrutinized appears as a minimum on its energy surface, for showing no negative force constant. Also, every singlet (1s–20s) appears more stable than its corresponding triplet (1t–20t). The highest stability (ΔEs-t) is achieved by germylene (11) where all the three nitrogens are bonded to the central boron atom. The EHOMO slightly decreases when the number of electronegative, σ-acceptor nitrogen atoms increases, and also causes it to be less electron-rich. Germylene 16s with low stability (ΔEs–t = 17.19 kcal/mol), bond gap (ΔEHOMO-LUMO = 57.46 kcal/mol−1), and atomic charge on -G̈e- (+ 0.9012), has high electrophilicity (ω = 3.78 eV) and nucleophilicity (N = 3.87 eV). Germylenes 8s, 14s, and 19s with coordinate covalent bond between nitrogen (N(Y)) and germylene center have low ω and high ΔEHOMO-LUMO. The purpose of the present work was, therefore, to assess the influence of nitrogen substituents on the stability (ΔΕs–t), band gaps (ΔΕHOMO–LUMO), N, ω, and heat of hydrogenation (ΔEH). This investigation is aimed to introduce novel germylenes that can be applied as cumulated multi-dentate NHG̈e ligands.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The divalent germylenes, GeR2, have gained much attention over the last four decades because of their electron-deficient radicals applied in chemical vapor deposition, semiconductor manufacturing, the photonics, and aerospace industries and other roles [1,2,3].
NHGe derivatives with the coordination of electronegative and π-electron-donating heteroatoms have high reactivity [3,4,5,6,7,8,9,10]. The NHGe has a weaker pπ-pπ interaction between nitrogen and germylene center than the corresponding NHC because germanium is less electronegative and larger than carbon. This leads to a reduction in the π electron density on the germylene center which makes the NHGe to be a better π acceptor [11, 12]. In 1982, the first N-heterocyclic germylene with four-membered ring was reported by M. Veith [13]. The systematic theoretical studies employing correlated wave functions on R2Ge have shown a strong tendency for germylenes to have singlet ground states and a substantial electronic effect of different substituents on the ΔEs–t of divalent species [14,15,16,17]. The electronegative substituents at germylenes increase the ΔEs–t gap, whereas the electropositive ones reduce it [18].
Interestingly, many organogermanium compounds have biological activities that have attracted much attention. In addition, Heremann reported saturated and unsaturated five-membered NHGe compounds, [19] which could be used as the original body to prepare the Ge-film by chemical vapor deposition (VCD) [20]. Therefore, the studies on germylenes and germylene reactions have important theoretical as well as practical significance. The aim of our work is to answer the question that arises whether novel singlet and triplet germylenes are researchable and how nitrogen substitutions may influence their stability, multiplicity (singlet (s) vs. triplet (t)), band gap (ΔEHOMO–LUMO), nucleophilicity (N), and electrophilicity (ω) at B3LYP/AUG-cc-pVTZ//B3LYP/6–311++G** level of theory. In addition, a number of them show prospects of being employed as multi-dentate NHGe ligands.
Computational methods
Our computational study, due to its excellent performance-to-cost ratio as compared with the correlated wave function theory, is confined to B3LYP calculations [21], while some recent reports have questioned the reliability of the most popular density functional, B3LYP [22]. We used B3LYP with the 6–311++G** basis set that is prevalent in many other papers on germylenes [11, 23, 24]. Triplet states were calculated using the unrestricted broken spin-symmetry UB3LYP/6–311++G** method implemented in the GAMESS software package [25, 26]. The vibrational frequency computations are applied to characterize the nature of stationary points, as true minima only real frequency values (with a positive sign) or the transition states only one imaginary frequency value (with a negative sign) is accepted respectively [27, 28].
The reactivity parameters are estimated via following the expressions: N = EHOMO(Nu) − EHOMO(TCNE);(tetracyanoethylene (TCNE) is preferred as the reference); ω = (μ2/2η), where μ is the chemical potential (μ ≈ (EHOMO + ELUMO)/2) and η is chemical hardness (η = ELUMO − EHOMO) at the same level of theory [29].
To reach more accurate energetic data, single point calculations are accomplished B3LYP/AUG-cc-pVTZ (correlation consistent polarized valence triple zeta) based on the B3LYP/6–311++G**geometries [30].
Results and discussion
We have compared and contrasted novel singlet (s) and triplet (t) germylenes (1s–20s vs. 1t–20t) with regard to their geometrical parameters (Table 1); second-order perturbation stabilization energies (E(2)) (Table 2); occupancy numbers (Table 3); relative stability (ΔΕs-t = Εt−Es) and ΔEH (Table 4); the frontier molecular orbital energies (HOMO and LUMO) for singlet germylenes along with their band gaps (ΔΕHOMO–LUMO) (Table 5) at B3LYP/AUG-cc-pVTZ//B3LYP/6–311++G** level of theory.
The range of bond angle ∠ZG̈eV for our germylenes is from 67.29° to 83.78°. The singlet state of our germylenes has longer bond angle (∠ZG̈eV) than their corresponding triplets. The optimized bond lengths for (Z-G̈e or G̈e-V) 1s–20s vs. 1t–20t vary in a range of 1.93 to 2.82 Å. The Z-G̈e or G̈e-V bond lengths of our singlet germylenes, except 18s, depend on the π-bonds and π-donor interactions (LPN → LP*G̈e) nature of the nitrogen adjacent to germylene center. For instance, the G̈e–C bond lengths in germylene 6s with G̈e=N bond and 2s with high LPN(Z)→LP*G̈e interaction (E(2) = 13.65 kcal/mol−1) are 0.03 Å and 0.08 Å longer than G̈e–N bond lengths, respectively (Tables 1 and 2).
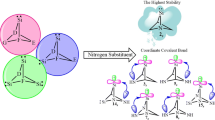
Germylenes 6s, 8s, 14s, and 19s have coordinate covalent bonds between nitrogen and boron or germylene atoms. They do not show any occupancy number for a lone pair on nitrogen at situation V or Y but display occupancy number for π(N(V)-B) or σ(G̈e-N(Y)) bonds (Table 3).
Our germylenes have singlet ground state, so every triplet germylene (1t–20t) appears at a higher level of energy than its corresponding singlet (1s–20s). For instance, 1s appears at almost 26.52 kcal/mol−1 lower in energy than its corresponding 1t. Our highest and lowest stable germylenes are 11 (ΔEs–t = 34.27 kcal/mol−1) and 15 (ΔEs–t = 14.87 kcal/mol−1), respectively. The overall stability order of our germylenes based on their ΔEs-t values is 11 > 7 > 5 > 4 > 18 > 9 > 20 > 3 > 2 > 1 > 14 > 17 > 12 > 8 > 6 > 19 > 10 > 13 > 16 > 15. This stability can be related to our imposed structures. Germylene 18s have high stability (ΔEs–t = 32.29 kcal/mol−1), vibrational frequencies (υmin = 220.60 cm−1), and dipole moment (D = 3.71). Interestingly, germylene 16s with low stability (ΔEs–t = 17.19 kcal/mol−1) has high dipole moment (D = 3.80) (Table 4).
The electrostatic potential (ESP) map is related to the electronic density and is considered a fundamental determinant of atomic and molecular properties [31]. Therefore, ESP has largely been used as a molecular descriptor of the chemical reactivity, which takes part in both electrophilic and nucleophilic reactions. For investigation, ESP surfaces are plotted over the optimized electronic structures of our germylenes using density functional B3LYP method with 6–311++G** basis set because the computationally or experimentally observed ESP surface directly provides information about the electrophilic (electronegative charge region) and nucleophilic (most positive charge region) regions (Table 3). The ESP map shows that the negative potential sites are on nitrogen atoms. The red and blue regions indicate the lowest and highest electrostatic potential energy values, respectively [31].
Germylene 5s with two nitrogens adjacent to its germylene center has more positive atomic charges on -G̈e- (+ 1.1876) and B (+ 0.8576) than 2s which has one nitrogen (-G̈e- = + 1.0487 and B = + 0.7724). Also, germylene 11s with three nitrogens adjacent to its boron has high positive atomic charge on its B (+ 0.9043) than 1s which has no nitrogen (B = + 0.5960). The atomic charges of the singlet germylene centers are significantly high positive compared to their corresponding triplets (Figure 1). Germylene 5s with the lowest vibrational frequencies (υmin = 24.12 cm−1) has high positive atomic charge on the B (+ 0.8576) and -G̈e- (1.1876) (Tables 4 and 5).

Schematic EHOMO, ELUMO (eV), and ΔEHOMO-LUMO (kcal/mol−1) for singlet silylenes at the B3LYP/6311++G** level of theory. IsoValue = 0.02 and the density = 0.0004
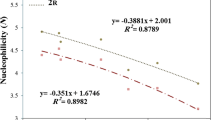
The crucial factor for stability of germylenes is nucleophilicity index, N, which was introduced by Domingo et al. [29] The nucleophilicity of our germylenes is decreased when their energy of the highest occupied molecular orbital (EHOMO) is decreased [32]. For instance, germylene 10s has the highest nucleophilicity (3.98 eV) and EHOMO (− 5.48 eV) (Table 5).
As EHOMO slightly decreased, the number of σ-acceptor nitrogen atoms increased, also caused less electron-rich. For example, germylene 19s with four nitrogens has lower EHOMO (− 6.09 kcal/mol−1) than 2s (− 5.73 kcal/mol−1) which has one nitrogen (Table 6). Germylene 11 with high stability (34.27 kcal/mol−1) and ω (3.56 eV) has the lowest N (2.98 eV) (Tables 5, 6, and Figure 1).
Germylenes 1s, 3s, 10s, and 16s regardless of LPG̈e → σ* interactions have high N because they do not have any nitrogen adjacent to the germylene center. Germylene 2s despite nitrogen adjacent to the germylene center has high N (3.72 eV) for high LPN(Z) → LP*G̈e interaction (E(2) = 13.65 kcal/mol−1). Two factors that have an effect on our ω and ΔEHOMO-LUMO are σ-bond (σ(G̈e-N(Y))) and LPN(W or Y) → LP*G̈e interactions. Hence, germylenes 8s, 14s, and 19s with σ-bond between nitrogen (N(Y)) and germylene center have low ω and high ΔEHOMO-LUMO. This σ-bond arose from a tendency of nonbonding electrons of nitrogen to empty p orbital of the germylene center. Also, germylene 18s with LPN(Y) → LP*G̈e (E(2) = 47.25 kcal/mol−1) and 20s with LPN(W) → LP*G̈e (E(2) = 42.73 kcal/mol−1) interaction have low ω and high ΔEHOMO-LUMO.
Germylene 20s despite LPN(W) → LP*G̈e (E(2) = 42.73 kcal/mol−1) interaction has similar ω with 3s which do not have any LPN(Z) and LPN(V) → LP*G̈e interactions (Tables 2 and 6).
Recently, we have reached novel borastannylenes that have similar structures with our germylenes, but they have different properties at geometrical parameters, stability (ΔΕs-t), the heat of hydrogenation (ΔEH), nucleophilicity (N), and electrophilicity (ω). Such various properties have attributed to the effect of our imposed topology and LPN(V) interactions [33].
In fact, we have introduced 4,6-diaza-7-boratricyclo[1.1.1.01,7.07,3.07,5]hexa-2-stannylene (10s) with high stability and N that can be applied as accumulated multi-dentate ligands. But, for this purpose, we found that singlet 5-aza-7-boratricyclo[1.1.1.01,7.07,3.07,5]hexa-2-germylene (4s) and 1,3-diraaza-7-boratricyclo[1.1.1.01,7.07,3.07,5]hexa-2-germylene (5s) are suitable.
The heats of hydrogenation for our germylene were calculated at B3LYP/AUG-cc-pVTZ level. The calculated heat of hydrogenation [33] is a thermodynamic method to estimate the relative stability of germylenes. For instance, germylene 18s with high stability (ΔEs–t = 32.29 kcal/mol−1) has low heat of hydrogenation (ΔEH = − 3.87 kcal/mol−1). Also, germylene 16s with low stability (ΔEs–t = 17.19 kcal/mol−1) has the highest heat of hydrogenation (ΔEH = − 20.88 kcal/mol−1). Our triplet germylenes have higher heats of hydrogenation than their corresponding singlets. For instance, the heat of hydrogenation 2s and 2t are − 18.19 and + 12.98 kcal/mol−1, respectively (Table 4).
We have employed the NBO analysis to stress the roles of intermolecular orbital interactions through second-order perturbation theory. The NBO analysis provides significant evidence for the nature of our hydrogenated germylenes. The nonbonding electrons at the nitrogen appear to have a tendency to make a coordinate covalent bond with the empty p orbital of boron atom. This is demonstrated by hydrogenated germylenes 2′s, 6′s, 8′s, 12′s, 13′s, 14′s, and 18′s, for showing π(N-B) occupancy number. Interestingly, hydrogenated germylenes with the nitrogen attached to boron have π(N-B) or LPN → LP*B interactions. For example, hydrogenated germylenes 2′s with π(N-B) and 4′s with high LPN(X) → LP*B interactions (E(2) = 11.64 kcal/mol−1) have one nitrogen attached to boron. Hydrogenated germylene 20′s has the lowest ΔEH (− 1.79 kcal/mol−1) for LPN(W) → LP*B interaction. This interaction has caused to decrease in the stability of 20′s (Tables 7 and 8).
Conclusions
In this research, we have studied thermodynamical and geometrical parameters for investigation of the effects of nitrogen substitution on the stability, multiplicity, and reactivity of novel singlet and triplet germylenes (1s–20s and 1t–20t, respectively), all of which appear as minima on their potential energy surfaces at B3LYP/AUG-cc-pVTZ//B3LYP/6-311G** level of theory. The germylene 11 with the enormous steric strain for their cubic structure has the highest stability (ΔEs-t = 34.27 kcal/mol−1). The EHOMO slightly decreases when the number of electronegative, σ-acceptor nitrogen atoms increases, also causes it to be less electron-rich. We have employed the NBO analysis to stress the roles of intermolecular orbital interactions through second-order perturbation theory. The NBO analysis provides significant evidences for the nature of our germylenes. Based on the following arguments, two factors that have the effect on our ω and ΔEHOMO-LUMO are σ-bond (σ(G̈e-N(Y))) and LPN(W or Y) → LP*G̈e interactions. Germylenes 8s, 14s, and 19s with σ-bond between nitrogen (N(Y)) and germylene center have low ω and high ΔEHOMO-LUMO. Germylenes 18s and 20s with LPN(W or Y) → LP*G̈e interactions have low ω and high ΔEHOMO-LUMO. The nucleophilicity index, N, is a crucial factor for showing the aptitude of our germylenes for coordination to transition metal complexes. So, we introduce germylenes 4s and 5s with high stability (ΔEs-t = 32.95 and 33.60 kcal/mol−1, respectively) and N (3.49 and 3.45 eV) that can be applied as multi-dentate ligands.
References
Vessally E (2008). Heteroat. Chem. 19(3):245–251
Heaven MW, Metha GF, Buntine MA, Phys J (2001). Chem. A. 105(7):1185–1196
Biswas AK, Ganguly B (2017). Chem. Eur. J. 23(11):2700–2705
Hlina J, Baumgartner J, Marschner C, Albers L, Müller T, Jouikov VV (2014). Chem. Eur. J. 20(30):9357–9366
Mansikkamäki A, Power PP, Tuononen HM (2013). Organometallics 32(22):6690–66700
Wilfling P, Schittelkopf K, Flock M, Herber RH, Power PP, Fischer RC (2015). Organometallics 34(11):2222–2232
Akbari A, Golzadeh B, Arshadi S, Kassaee MZ (2015). RSC Adv 5:43319–43327
Su MD, Chu SY, Chin J (2000). Chem. Soc 47(1):135–139
Neumann WP (1991). Chem. Rev 91(3):311–334
Becerra R, Boganov SE, Egorov MP, Nefedov OM, Walsh R (1997). Mendeleev Commun. 7(3):87–88
Ashenagar S, Kassaee MZ, Cummings PT (2019). J. Mol. Model. 25:371–383
Kühl O (2004). Coord. Chem. Rev. 248(5–6):411–427
Veith M, Grosser M (1982). Zeitschrift für Naturforschung B 37(11):1375–1381
Barthelat JC, Roch BS, Trinquier G, Satge J, Am J (1980). Chem. Soc. 102(12):4080–4085
Mizuhata Y, Sasamori T, Tokitoh N (2009). Chem. Rev. 109:3479–3511
Kirilchuk AA, Rozhenko AB, Leszczynski J (2017). Comput. Theor. Chem. 1103:83–91
Hadlington TJ, Driess M, Jones C (2018). Chem. Soc. Rev. 47:4176–4197
Schreiner PR, Reisenauer HP, Allen WD, Sattelmeyer KW (2004). Org. Lett. 6:1163–1166
Herrmann WA, Denk M, Behm J, Scherer W, Klingan FR, Bock H, Solouki M, Wagner M (1992). Angew. Chem. 104(11):1489–1492
Lu XH, Xu YH, Yu HB, Lin H (2005). Chin. J. Chem. 23(10):1339–1342
Adamo C, di Matteo A, Barone V (2000) Adv. Quantum Chem. 36:45–75
Zhao Y, Truhlar DG (2008). Acc. Chem. Res. 41(2):157–167
Bao W, Li Y, Lu X (2013). Struct. Chem. 24:1615–1619
Aysin RR, Bukalov SS, Leites LA, Zabula AV (2017). Dalton Trans. 46:8774–8781
Hoffmann R, Schleyer PVR, Schaefer HF (2008). Angew. Chem., Int. Ed. 47(38):7164–7167
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993). J. Comput. Chem. 14:1347–1363
Soleimani Purlak N, Kassaee MZ (2020). J. Phys. Org. Chem. 33(6)
Kassaee MZ, Ashenagar S (2018). J. Mol. Model. 24:2–11
Domingo LR, Chamorro E, Perez P (2008). J. Org. Chem. 73:4615–4624
Kassaee MZ, Shakib FA, Momeni MR, Ghambarian M, Musavi SM (2010). J. Org. Chem. 75:2539–2545
Haerizadea BN, Kassaee MZ, Zandib H, Koohi M, Ahmadi AA (2014). J. Phys. Org. Chem. 27:902–908
Martin D, Baceiredo A, Gornitzka H, Schoeller WW, Bertrand G (2005). Angew. Chem. Int. Ed. 44:1700–1703
Abedini N, Kassaee MZ (2020). Comput. Theor. Chem.:1190
Funding
The authors received support from Tarbiat Modares University (TMU).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 6921 kb)
Rights and permissions
About this article

Cite this article
Abedini, N., Kassaee, M.Z. A theoretical investigation into novel germylenes: effects of nitrogen substitution on stability and multiplicity. J Mol Model 26, 325 (2020). https://doi.org/10.1007/s00894-020-04570-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04570-7