
Abstract
The mechanism of the cycloaddition reaction between singlet H2Ge=Ge: and ethene has been investigated by the B3LYP/6-311 ++G** method. From the potential energy profile and change of Gibbs free energy, it could be predict that the reaction has only one dominant reaction pathway at 298 K and 149.825 kPa. The reaction rule presented is that the two reactants first form a four-membered Ge-heterocyclic ring germylene through the [2 + 2] cycloaddition reaction; because of the 4p unoccupied orbital of Ge: atom in the four-membered Ge-heterocyclic ring germylene and the π orbital of ethene forming a π → p donor–acceptor bond, the four-membered Ge-heterocyclic ring germylene further combines with ethene to form an intermediate; and because the Ge: atom in intermediate happens sp3 hybridization after transition state, then the intermediate isomerizes to a spiro-Ge-heterocyclic ring compound via a transition state.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since unsaturated carbene [XYC=C: (X, Y=H, F, Cl, Me, Ph, ……)] was recognized as an active intermediate in the 1960s, it has not only attracted much attention from theoretic chemists but also been practically applied to the organic chemistry [1, 2]. For example, it has been proved that unsaturated carbene can provide a simple and direct way for synthesizing the small-ring, highly strained compounds as well as those that can hardly be synthesized through conventional ways [2]. So far, in depth exploration has been done on the rearrangement reaction of alkylidene carbene [3, 4], and the insertion reaction of alkylidene carbene has also been studied [5, 6]. Apeloig et al. [7] and Fox et al. [8] have made experimental and theoretic studies on the 3-dimensional selectivity of substituting groups from the products of the vinylidene-olefins addition reaction of alkylidene carbene. Meanwhile, we have done some relatively systematic theoretic investigations on the cycloaddition reaction of alkylidene carbene [9–13]. However, there has been no published reports about analog XYGe=Ge: (X, Y=H, F, Cl, Me, Ph, ……) of unsaturated carbene, and they are new study field of unsaturated germylenes chemistry. It is quite difficult to investigate the mechanisms of the cycloaddition reaction by experimental methods directly due to the high activity of unsaturated germylenes; therefore, the theoretic study is more practical. To explore the rules of the cycloaddition reactions between unsaturated germylene [XYGe=Ge: (X,Y=H, F, Cl, Me, Ph, ……)] and the symmetric π-bonded compounds, H2Ge=Ge: and ethene were selected as model molecules, the cycloaddition reaction mechanism (considering the H transfer simultaneously) was investigated and analyzed theoretically. The results show that the cycloaddition reaction consists of two possible pathways, as follows:


The research result indicates the laws of cycloaddition reaction between H2Ge=Ge: and its derivatives and the symmetric π-bonded compounds, which is significant for the synthesis of small-ring and spiro-Ge-heterocyclic compounds. The study extends research area and enriches research content of germylene chemistry; it opened up a new research field for germylene chemistry.
Calculation methods
B3LYP/6-311 ++G** [14] implemented in the Gaussian 09 package [15] is employed to locate all the stationary points along the reaction pathways. Full optimization and vibrational analysis are done for the stationary points on the reaction profile. In order to explicitly establish the relevant species, the intrinsic reaction coordinate (IRC) [16, 17] is also calculated for all the transition states appearing on the cycloaddition energy surface profile.
Results and discussion
Reaction (1): channels of forming the four-membered Ge-heterocyclic ring germylene (INT1), H transfer products (P1.1, P1.2 and P1.3)
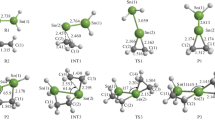
Theoretic researches show that the ground state of H2Ge=Ge: is a singlet state. The geometrical parameters of the intermediate (INT1), transition state (TS1.1, TS1.2, TS1.3), and product (P1.1, P1.2, P1.3) which appear in reaction (1) between H2Ge=Ge: and ethene are given in Fig. 1, the energies and Gibbs free energy are listed in Table 1, and the potential energy profile for the cycloaddition reaction is shown in Fig. 2. According to Fig. 2, it can be seen that the reaction (1) consists of four steps: the first one is that the two reactants (R1, R2) form a four-membered Ge-heterocyclic ring germylene (INT1), which is a barrier-free exothermic reaction of 104.8 kJ/mol; the second, third, and fourth steps are that the INT1 undergoes H transfer via transition states TS1.1, TS1.2, and TS1.3 with energy barriers of 63.6, 89.9, and 160.1 kJ/mol, respectively, resulting in the formation of products P1.1, P1.2, and P1.3. According to Table 1, the ΔG of R1 + R2 → INT1 is −38.9 kJ/mol, so R1 + R2 → INT is thermodynamics of spontaneous reaction at 298 K and 101.325 kPa because of the ΔG of INT1 → P1.1, INT1 → P1.2, and INT1 → P1.3 are 37.6, 1.65, and 65.3 kJ/mol, respectively; INT1 → P1.1, INT1 → P1.2, and INT1 → P1.3 are prohibited in thermodynamics at 298 K and 101.325 kPa, and reaction (1) will end in product INT1.

Optimized B3LYP/6-311 ++G** geometrical parameters of INT1, TS1.1, TS1.2, TS1.3. P1.1, P1.2, P1.3, and the atomic numbering for these species in cycloaddition reaction (1). Bond lengths and angles are in Å and (°)

The potential energy profile for the cycloaddition reactions between H2Ge=Ge: and H2C=CH2 with B3LYP/6-311 ++G**
Reaction (2): channel of forming a spiro-Ge-heterocyclic ring compound (P2)
In reaction (2), the four-membered Ge-heterocyclic ring germylene (INT1) further reacts with ethene (R2) to form a spiro-Ge-heterocyclic ring compound (P2). The geometrical parameters of intermediate (INT2), transition state (TS2), and product (P2), which appear in reaction (2) are given in Fig. 3. The energy and Gibbs free energy are listed in Table 1, and the potential profile for the cycloaddition reaction is shown in Fig. 2. According to Fig. 2, it can be seen that the process of reaction (2) as follows: on the basis of INT1 formed from the reaction (1) between R1 and R2, the INT1 further reacts with ethene to form an intermediate (INT2), which is a barrier-free exothermic reaction of 54.4 kJ/mol; next, the intermediate (INT2) isomerizes to a spiro-Ge-heterocyclic ring compound (P2) via a transition state (TS2) with an energy barrier of 8.2 kJ/mol. According to Table 1, the ΔG of INT1 + R2 → INT2, INT1 → P2, and INT1 + R2 → P2 are 8.5, −0.8, and 7.6 kJ/mol, respectively, so the reactions INT1 + R2 → INT2 and INT1 + R2 → P2 at 298 K and 101, 325 Pa are prohibited in thermodynamics and the spontaneous tendency of INT1 → P2 is very small. Considering INT1 + R2 → INT2 and INT1 + R2 → P2 are volume reduced reaction to make the reaction goes on increasing the pressure of the system can make INT1 + R2 → P2 complete. According to the formula:

Optimized B3LYP/6-311 ++G** geometrical parameters of INT2, TS2, P2 and the atomic numbering for cycloaddition reaction (2). Bond lengths and angles are in Å and (°)
We can know that when the pressure of the system is 149.825 kPa at 298 K, the ΔG of INT1 + R2 → INT2, INT1 + R2 → P2, and R1 + R2 → INT1 are −40.0, −40.9, and −87.4 kJ/mol, respectively; so \( {\text{R}1} + {\text{R}2} \to {\text{INT}1}\mathop{\longrightarrow}\limits^{+R2} {\text{INT2}}\mathop{\longrightarrow}\limits^{TS2}{\text{P2}} \) will be the dominant reaction pathway of cycloaddition reaction between singlet H2Ge=Ge: and ethane.
Theoretic analysis and explanation of the dominant reaction channel
According to the above analysis, the dominant reaction channel of the cycloaddition reaction between singlet H2Ge=Ge: and ethane at 298 K and 149.825 kPa is as follows:
In the reaction, the frontier molecular orbitals of R2 and INT1 are shown in Fig. 4. According Fig. 4, the frontier molecular orbitals of R2 and INT1 can be expressed in schematic diagram 5. The mechanism of the reaction could be explained with the molecular orbital diagram (Fig. 5) and Figs. 1 and 3. According to Fig. 1, as H2Ge=Ge: initially interacts with ethene, the [2 + 2] cycloaddition of two bonding π-orbitals firstly results in a four-membered Ge-heterocyclic ring germylene (INT1). Because the INT1 is an active intermediate, it could further reacts with ethene (R2) to form a spiro-Ge-heterocyclic ring compound (P2). The mechanism of the reaction could be explained with Figs. 1, 3, and 5, according to orbital symmetry matching condition, when INT1 interacts with ethene (R2), the 4p unoccupied orbital of the Ge(2) atom in INT1 will insert the orbital of ethene, the 4p unoccupied orbital of the Ge(2) atom in INT1 will insert the π orbital of ethane, then the shift of π-electrons to the p unoccupied orbital gives a π → p donor–acceptor bond, leading to the formation of intermediate (INT2). As the reaction goes on, because of C(3)–Ge(2) bond (INT2: 2.435 Å, TS2: 2.117 Å, P2: 1.972 Å) gradually shorten, ∠C(2)Ge(2)C(1) (INT2: 90.2°, TS2:103.3°, P2: 127.80°) increase gradually, ∠C(4)C(3)Ge2Ge1(INT2: 167.0°, TS2: 150.4°, P2: 126.9°) decrease gradually, and the C(4)C(3) bond (INT2: 1.393 Å, TS2: 1.466Å, P2: 1.534Å) elongate gradually, the Ge(2) in INT2 happens sp3 hybridization after the transition state (TS2), forming a spiro-Ge-heterocyclic ring compound (P2).

The frontier molecular orbitals of R2, INT1

A schematic diagram for the frontier orbitals of INT1 and H2C=CH2 (R2)
Conclusion
On the basis of the potential energy profile and Gibbs free energy (G) obtained with the B3LYP/6-311 ++G** method for the cycloaddition reaction between singlet H2Ge=Ge: and ethene, it can be predicted that the reaction (2) is one dominant reaction pathway of cycloaddition reaction between singlet H2Ge=Ge: and ethene at 298 K and 149.825 kPa. The process of reaction (2) consists of three steps: the first step is that the two reactants (R1, R2) form a four-membered Ge-heterocyclic ring germylene (INT1), which is a barrier-free exothermic reaction of 104.8 kJ/mol; the second step is that intermediate (INT1) further reacts with ethene (R2) to form an intermediate (INT2), which is also a barrier-free exothermic reaction of 54.4 kJ/mol; and the third step is that intermediate (INT2) isomerizes to a spiro-Ge-heterocyclic ring compound (P2) via a transition state (TS2) with an energy barrier of 8.2 kJ/mol.
References
Stang P (1982) Acc Chem Res 15:348
Stang P (1978) Chem Rev 78:384
Krishnan R, Frisch MJ, Pople JA (1981) Chem Phys Lett 79:408
Frisch MJ, Krishnan R, Pople JA (1981) Chem Phys Lett 81:421
Wardrop DJ, Zhang W (2002) Tetrahedron Lett 43:5389
Feldman KS, Perkins AL (2001) Tetrahedron Lett 42:6031
Apeloig Y, Karni M, Stang PJ, Fox DP (1983) J Am Chem Soc 105:4781
Fox DP, Stang PJ, Apeloig Y, Karni M (1986) J Am Chem Soc 108:750
Lu XH, Wang YX (2003) J Phys Chem 107:7885
Lu XH, Wang YX (2004) J Mol Struct Theochem 686:207
Lu XH, Wu WR, Yu HB, Yang XL, Xu YH (2005) J Mol Struct Theochem 755:39
Lu XH, Yu HB, Wu WR, Xu YH (2007) Int J Quantum Chem 107:451
Lu XH, Xiang PP, Wu WR, Che X (2008) J Mol Struct Theochem 853:82
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, revision B.01. Gaussian Inc, Wallingford
Fukui K (1970) J Phys Chem 74:4161
Ishida K, Morokuma K, Komornicki A (1981) J Chem Phys 66:2153
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bao, W., Li, Y. & Lu, X. Density functional theory study of mechanism of forming a spiro-Ge-heterocyclic ring compound from H2Ge=Ge: and ethene. Struct Chem 24, 1615–1619 (2013). https://doi.org/10.1007/s11224-013-0199-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-013-0199-z