
Abstract
Substituent effects on stability (assumed as the singlet and triplet energy gaps, ΔΕS-T) of novel 1,4-disubstituted-tetrazole-5-vinylidene germylenes (normal, 1R) and their corresponding 1,3-disubstituted-tetrazole-5-vinylidene germylenes (abnormal, 2R) are computed and compared, at B3LYP/6-311++G** and M06/6-311++G**, where R = H, CN, CF3, F, SH, C6H6, OMe, and OH. Interestingly, every triplet vinylidene germylene shows more stability than its corresponding singlet. Also, every triplet abnormal isomer (2R) emerges to be more stable than its corresponding normal (1R). All abnormal 2R isomers show broader band gaps (ΔEHOMO–LUMO) and higher nucleophilicity (N), but less electrophilicity (ω) than their corresponding normal 1R isomers. The NICS (nuclear-independent chemical shift) results indicate that every 1R (except singlet 2Ph) emerges more aromatic than its corresponding 2R. Our Hammet studies indicate that 1R is more sensitive to the electronic effects of substituents, R, than 2R. Electron-donating species increase N in both 1R and 2R, while electron-withdrawing groups increase stability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
N-heterocyclic carbenes (NHCs) are used as organocatalysts to bind every transition metal and form strong metal complexes in the fields of organic and inorganic chemistry [1,2,3,4,5]. Studies on NHCs date back to the research of Wanzlick in the 1960s [6]. In 1991, the first stable NHC was reported by Arduengo and coworkers [7, 8]. They synthesized and characterized colorless crystals of 1,3-di-l-adamantylimidazol-2-ylidene. The synthesis of NHC consisting of pendant alkenes was reported by Furstner and coworkers [9]. Recently, Bertrand reported a novel procedure to the synthesis of the stable six-membered NHC [10,11,12]. After these successful researches and some similar works, many N-heterocyclic carbenes, silylenes (NHSi), and germylenes (NHGes) were reported [13,14,15]. NHGes are very versatile ligands and the heavier homologs of the NHCs. Meller and Gräbe reported the first NHGe from some imidazoles, in 1985 [16]; after the discovery of Arduengo et al.’s NHC in 1991 [7], the first post-Arduengo NHGe being reported in 1992 [17]; then in 1994, Heinemann et al.’s [18] study on the stability of Arduengo-type NHGe and NHSi and compare them with the NHC. Since germanium is less electronegative and larger than carbon, the corresponding NHGe has a weaker N-Ge pπ-pπ interaction than NHC. This leads to a reduction in the π-electron density on germanium which makes the NHGe to be a better π acceptor [19]. After several years, theoretical researchers found that triplet N-heterocyclic silylenes and germylenes could also be stable [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. In fact, due to the importance of triplet ground–state silylenes and germylenes in chemical semiconductor manufacturing, vapor deposition, and the aerospace and photonics industries, the preparation and theoretical calculation of these isomers have become one of the most important topics in modern organosilicon and germanium chemistry [25].
In 2011, Momeni et al. researched on triplet NH-vinylidene silylenes with (NRCH2-CH2NR)Si=Si and (NRCHCHNR)Si=Si structures [28]. In our continued quest for novel divalent NH-vinylidene germylene species [29,30,31,32,33,34,35,36,37,38], the question immediately arises whether normal and abnormal tetrazol-5-vinylidene germylenes are researchable and how H, CN, CF3, F, SH, C6H6, OMe, and OH groups with different electronic effects may influence their stability, multiplicity (singlet (s) vs. triplet (t)), band gap (ΔEHOMO–LUMO), aromaticity (NICS), nucleophilicity (N), electrophilicity (ω), and geometrical parameters (Scheme 1). Clearly, it will be shown that appropriate substitutions with different electronic effects in normal and abnormal forms of tetrazol-5-vinylidene germylenes decrease the ΔES-T value of NH-vinylidene germylene enough to bring the resulting novel triplet germylene, which are more stable than the results of other workers.

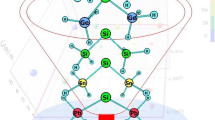
Scrutinized normal and abnormal tetrazol-5-vinylidene germylenes where R = H, CN, CF3, F, SH, C6H6, OMe, and OH
Computational methods
All geometry optimizations are carried out without any symmetry constraints by means of hybrid functional B3LYP [39,40,41,42,43] and the standardized 6-311++G** basis set, by using GAMMES package of programs [44].
Our computational study is confined to B3LYP calculations due to its excellent performance-to-cost ratio as compared with the correlated wave function theory [45,46,47,48]. While recently, some reports appeared to develop the controversies on the reliability of most of the popular density functional, B3LYP, [15] in parallel to many other papers on germylenes, in this work, B3LYP with the 6-311++G** basis set is employed as the method of choice [22, 25, 49,50,51,52,53]. To obtain more accurate energetic data, the M06 calculation is performed as a different density functional [41]. Singlet states are considered with spin-restricted wave function and triplet states are studied by using the unrestricted formalisms [54, 55]. The spin-restricted and spin-unrestricted (with broken symmetry) calculations give the identical values of the singlet ground–state energy (Table S1). Thus, the restricted wave functions were employed to investigate the possible pathways of the singlet germylenes [56]. After spin annihilation, an ideal value (2.001) is indicated for the S2 expectation values of these divalents, so that their geometries are reliable for our study. The frequency calculations are applied to characterize the structures as minima (the number of imaginary frequencies (NIMAG = 0) or transition states (NIMAG = 1)) [57]. The global electrophilicity (ω) has been computed as ω = (μ2/2η), where μ is the chemical potential (μ ≈ (EHOMO + ELUMO)/2) and η is the chemical hardness (η = ELUMO − EHOMO) [58]. The nucleophilicity index N [59] has been considered as N = EHOMO (Nu) − EHOMO (TCNE), where tetracyanoethylene (TCNE) is utilized as the reference.
Results and discussion
Band gap (ΔE HOMO–LUMO), electrophilicity (ω), and nucleophilicity (N) of normal (1 R) vs. abnormal (2 R) vinylidene germylenes
The excellent σ-donor capacities of NH-silylenes and germylenes make them very useful ligands for research on the variety of organometallic catalysts [60, 61]. Here, we have considered ΔEHOMO–LUMO, N, and ω indices for our singlet and triplet germylenes, at B3LYP/6–311++G**. Abnormal singlet and triplet germylenes (2R) have higher ΔEHOMO–LUMO and nucleophilicity but lower electrophilicity than the corresponding normal (1R) isomers (Table 1). For instance, the singlet abnormal 2OH (N = 4.69 eV) appears more nucleophile than its corresponding singlet normal 1OH (N = 4.45 eV).
Such differences may be explained in terms of the difference between the occupancy of their vacant π*Ge(2′)-Ge(1′) orbitals (Scheme 1).
The electron donation of the N(4) atom with negative charge is much more efficient in the abnormals than the corresponding normal isomers. The computed NBO results indicate that the donation of LPN(4 (π*Ge(2′)-Ge(1′) in the abnormal germylenes is approximately twice of that on the normal (Table 1, Scheme 1).
E. Kühn et al. also have reported that variation of substitution pattern in normal and abnormal isomers effects on σ-donor capacities of the species. It is found that the abnormal tetrazolylidene with 1,3-substitutions is considered a better electron donor than its corresponding normal isomer [30, 62].
This is also a good reason why in going from normal to abnormal, N increases as a function of π-donor or π-acceptor abilities of the employed substituents. Between normal and abnormal, 2Ph and 1CN are the most and the least nucleophilic groups (Table 2). Hence, the trend of N values in singlets 1 and 2 is 1CN < 1F < 1CF3 < 1SH < 1OH < 1Ph < 1OMe < 1H; and 2CN < 2F < 2CF3 < 2OH < 2SH < 2OMe < 2Ph < 2H. In triplets, the trend is 1CN < 1CF3 < 1F < 1SH < 1OH < 1OMe < 1H < 1Ph and 2CN < 2F < 2CF3 < 2OH < 2SH < 2OMe < 2H < 2Ph. Plotting nucleophilicity as a function of σm gives negative ρ of − 5.07 and − 4.46 with a correlation factor of R2 = 0.90 and 0.88, for (1R) and (2R), respectively (Fig. 1). This indicates that N is highly sensitive to the electronic effects of substituents and that electron-donating groups enhance N. In contrast, plotting ΔΕS-T as a function of σp gives positive ρ of 12.94 and 14.97 with R2 of 0.9 and 0.6, for (1R) and (2R), respectively (Fig. 2). Hence, stability of our germylenes appears to increase as a function of electron-withdrawing effects of R. These data indicate that both (1R) and (2R) show high stability in nucleophilicity (Table 2). Here, 1R demonstrates a higher sensitivity toward electronic effects of R than 2R.

Correlation between nucleophilicity (N) and Hammett substituent constants (σm) for our normal (1R) vs. abnormal (2R) tetrazole-5-vinylidene germylenes

Correlations between ΔΕS-T and Hammett substituent constants (σp) for our normal (1R) vs. abnormal (2R) tetrazole-5-vinylidene germylenes
Abnormal vinylidene germylenes due to their nucleophilicity and 1,3-substitution patterns are stronger σ-electron donors than their corresponding normal congeners [62]. In this case, the N(4) α-nitrogen atom nearby the Ge(2′) is able to transport a higher electron density on germanium than corresponding normal isomers (Table 1, Scheme 1). Consequently, Ge(2′)-Ge(1′) bond length decreases and N(1)-Ge(2′)–N(4) bond angle increases in abnormal isomers more than the corresponding normals (Table 3).
As to the electronic effects of various types of substituents, Ge(2′)–Ge(1′) bond lengths analyses do not show notable a difference between the normal (1R) and abnormal (2R) germanium-germylene bonding mode. Furthermore, stereoelectronic effects of the substituents modify the N(1)–Ge(2′)–N(4) bond angles. For example, benzo substituent features the significantly largest N(1)–Ge(2′)–N(4) bond angles for abnormal and normal germylenes (81.45 (1) Å vs. 83.90 (2) Å in singlets and 80.48 (1) Å vs. 83.25 (2) Å in triplets). Likewise, fluorine substituent makes the smallest N(1)–Ge(2′)–N(4) bond angle in both abnormal and normal germylenes (75.03 (1) Å vs. 80.93 (2) Å in singlets and 73.45 (1) Å vs. 79.94 (2) Å in triplets) (Table 3).
Substituent effects on normal (1 R) and abnormal (2 R) vinylidene germylenes
Worthington and Cramer in 1997 claimed that vinylidene (H2C=C) could have two symmetry-distinct triplet states with similar energy that in one of them, an electrone from the C=C σ (sp) orbital promotes to the vacant p orbital and in the other one electron from π orbital promote to the p orbital [29, 60]. In 2011, Momeni et al. studied triplet NHC vinylidene silylenes and confirmed their latter choice for the silylidene structures. They indicated that silylidene preferre to form π1p1 triplet state over n1p1, due to the high energy gap between the 3s and 3p orbitals of Si (Fig. 3) [29]. So, due to the high similarity between silylene and gemylene, we guess that vinylidene germylenes behave in the same manner trans to silylene and our results confirm this.

Scrutinized N-heterocyclic vinylidene silylenes in Momeni et al. studies [22]
Our NBO analysis shows that the formal charge of Ge(1′) in the triplets, because of electron transfer from the π orbital to the vacant p orbital, is more than their corresponding singlets (Table 4).
Also, on the basis of the NBO analysis, the triplet state of vinylidene germylenes has a semioccupied 4Py orbital with occupancy number of 0.5–0.8 e, where 4Py occupancy number in singlet is about zero (Table 1).
So, α-germanium substituent due to its electropositive character is a triplet state stabilizer and permits the promotion of an electron from a π orbital to the vacant p orbital (Scheme 2). The molecular electrostatic potential (MEP) and HOMO-LUMO molecular orbitals for singlet and triplet germylenes are shown (Fig. 4).

The π1p1 triplet germanium germylene

Shapes of electrostatic potential maps (EMP) and HOMO-LUMO molecular orbitals for singlet and triplet a normal 1R and b abnormal 2R vinylidene germylenes
MEP may be used to predict reactive sites for electrophilic and nucleophilic attack and can be determined experimentally by X-ray diffraction techniques or theoretical calculations. It has its origin in the charge distribution within the molecule. The backbone is formed by the atoms whose nuclei are the centers of positive charge. The electrons with a negative charge are distributed around these nuclei occupying their respective orbitals [63,64,65,66,67].
In the MEP, the color coding from blue to red regions correspond to highly electron deficient to electron-rich regions while green regions suggest almost electrically neutral region. MEP (Fig. 4) of title molecule visibly suggests that the major negative potential region lies around oxygen and nitrogen atoms, while germanium (Ge(2′)) atoms of tetrazol rings bear maximum burnt of positive potential.
Singlet-triplet energy gap (ΔE S–T) for normal (1 R) and abnormal (2 R) vinylidene germylenes
The singlet-triplet splittings are predicted as the energy differences between the neutral ground state and the lowest triplet state. Stability of every germylene is assumed to depend on its ΔΕS-T values which is calculated at B3LYP/6-311++G** and M06/6-311++G** levels of theory (Table 2). In fact, in germylenes, the np valence electrons are spatially separated from ns valence electrons, as a result of Pauli repulsion with the (n–1)p electrons in the inner shells. On the other hand, the electron-electron repulsion between the paired electrons in germylenes is less than their analogues which is due to the larger size of the lone pair orbital that makes the singlet state for these elements to be favored. So germanium prefers to have nonbonding electrons in atomic orbitals with a higher percentage of s character and we can say that the higher s character in the germylenes suggests the stabilization of the singlet state, leading to an increase in singlet-triplet energy gap (ΔES-T) value as observed [24]. Consequently, the probability of triplet stabilization has been investigated through a decrease in ΔES-T. So, the variation of ΔES-T can provide useful means in determining the stability of the triplet states compared with the singlets.
Interestingly, a total of sixteen triplet germylenes are encountered as global minimum with rather high stabilities. In accordance with Worthington and Cramer [60] statements and in contrast to Momeni’s [29], findings on the stabilization of vinylidene silylenes, our results indicate that in the case of vinylidene germylenes, substituent effect on vinylidene multiplet splitting and the stabilizing PGe(1′) → σ*N(4)-Ge(2′) interaction is detectable in our triplets (Table 1). So, we confirm that σ-electron-withdrawing interactions bring effects on the germylene (Ge (1′)) p orbital stabilization, preferentially.
Every triplet abnormal is more stable than its corresponding normal. The ΔΕS-T (kcal/mol) for our scrutinized normal vinylidene germylenes immerge consistent with the order of 1CF3 (− 9.58) > 1CN (− 10.23) > 1Ph (− 11.91) > 1SH (− 12.07) > 1F (− 13.90) > 1H (− 14.54) > 1OH (− 14.77) > 1OMe (− 15.03). This order is maintained for abnormal vinylidene germylenes: 2CF3 (− 13.02) > 2CN (− 13.35) > 2Ph (− 13.40) > 2SH (− 14.89) > 2F (− 15.84) > 2OH (− 15.85) > 2H (− 16.17) > 2OMe (− 16.25). Clearly, normal isomers have a rather manifest consistency trends with the abnormals. However, every triplet abnormal isomer shows more stability than its corresponding normal. For instance, stability of triplet 2CF3 (∆ES-T = − 13.02 kcal/mol) is more than that of its corresponding 1CF3 (∆ES-T = − 9.58 kcal/mol). In abnormal isomers, the N(4) nitrogen atom (adjacent to the Ge(2′)) does not carry any steric effects due to substituent. So, it’s ability to stabilize with PGe(1′) → σ*N(4)-Ge(2′) interaction is higher than that in the case of normal isomers with 1,4-substitution pattern. In the confirmation, NBO analyses show that the donation of PGe(1′) → σ*N(4)-Ge(2′) in the abnormal germylenes is more than that of PGe(1′) → σ*N(1)-Ge(2′). Another factor that has an effect on triplet abnormal stabilization is the donation of LPN4 → π*Ge(2′)-Ge(1′) interaction (Scheme 2), which in the abnormal germylenes is approximately twice of that on the normals (Table 1).
The ΔΕS-T also changes as a function of different electronic effects of various types of substituents (1R and 2R). In our results, a decrease in ΔΕS-T via an increase in the π-electron-donating effect of substituents is detected. Clearly, 1R and 2R substituents affect the triplet state through π-donating LPN(1, 4) → π*Ge(2′)-Ge(1′) interactions (Table 1, Scheme 3, a → m and a′ → m′). Among normal germylenes, 1OMe is the most stable (ΔΕS-T = − 15.03 kcal/mol) structure, while the least stable structure is 1CF3 with ΔΕS-T = − 9.58 kcal/mol. Also in abnormal germylenes, the highest ΔES–T is detectable for 2OMe with ΔΕS-T = − 16.25 kcal/mol, while the lowest is detectable for 2CF3 with − 13.02 kcal/mol (Table 2).

Electron delocalizations through seven sets of possible canonical forms (I–VII) for our a normal 1R and b abnormal 2R germylenes
Nucleus-independent chemical shift (NICS) for normal (1 R) and abnormal (2 R) vinylidene germylenes
The nucleus-independent chemical shift (NICS) is a computational method that evaluates the absolute magnetic shielding at the center of a ring. The NICS values are reported with a reversed sign to make them compatible with the chemical shift conventions of NMR spectroscopy. Positive NICS values indicate antiaromaticity while rather high negative values point out aromaticity [68]. NICSs are calculated at 0.5, 1, 1.5, and 2 Å above the ring center for singlet and triplet normal and abnormal vinylidene germylenes, at B3LYP/6-311++G** level of theory. Our NICS calculations show that every normal 1R congres is more aromatic than its corresponding abnormal, 2R. However, singlet state of 2Ph shows the smallest NICS value (Table 2).
Negative charge of Ge(1′) in singlet state of 2Ph is less than singlet state of 1Ph, which is in contrast to the other structures (Table 4). We found that 2Ph in singlet state has LPGe(1′ → (σ*C-C (0.75 kcal/mol) and LPGe(1′ → (σ*C-H (1.91 kcal/mol) electron donation interactions which make Ge(1′)–Ge(2′) to be more skew than that of triplet and normal isomers (Fig. 5). In this case, the antiaromaticity value decreases and on the other hand, there is no antiaromaticity in singlet state of 2Ph.

Shapes of total density EMP and contours for singlet and triplet 1Ph and 2Ph
The resonance hybrid structures (Scheme 3) for vinylidene germylenes coupled with the electrostatic potential maps (Fig. 4) reveal the electronic effects on (a) normal 1R and (b) abnormal 2R isomers (Fig. 6).

The π-interaction between germanium and the adjacent nitrogens
Conclusions
We have reached at two sets of normal and abnormal exocyclic vinylidene triplet germylenes (1R and 2R) that appear rather more stable than several other calculated triplet germylenes. Investigating the effects of the R groups (R = H, CF3, CN, C6H6, SH, F, OMe, and OH) leads us to the ΔES-T values of − 9.58, − 10.23, − 11.91, − 12.07, − 13.90, − 14.77, and − 15.03 kcal/mol for normal vinylidene germylenes and − 13.02, − 13.35, − 13.40, − 14.89, − 15.84, − 15.85 and − 16.25 kcal/mol for abnormal isomers, respectively, at the B3LYP/6–311++G** level. Clearly, every triplet abnormal isomer shows more stability than its corresponding normal. Variation of the substitution pattern in normal (1R) and abnormal (2R) isomers effects on σ-donor capacities of the species. It is found that the abnormal isomers with 1,3-substitutions are stronger electron donors than their normal congeners. Also, in our results, a decrease in ΔΕS-T via an increase in π-electron-donating effect of substituents is detected. The geometrical parameters (bond lengths, angles) of the reported germylenes are performed at B3LYP/6-311++G** level of theory. NICS calculations show that every normal isomer appears more aromatic than its corresponding abnormal, except for singlet state of 2Ph which appears less antiaromatic than the others. All the results indicate the role of aromaticity and delocalization of the π-system as a stabilizing factor. Furthermore, all abnormal 2R isomers have wider band gaps and more nucleophilicity (N) but less electrophilicity (ω) than their normal 1R isomers. Our Hammet studies indicate that 1R is more sensitive to the electronic effects of substituents, R, than 2R. Electron-donating species increase N in both 1R and 2R, while electron-withdrawing groups increase stability.
Consequently, we have found abnormal forms of tetrazol-5-vinylidene germylenes with electron-donating substitutions could decrease the ΔES-T value enough to bring the resulting novel stable triplet germylenes which indicate more stability and nucleophilicity (N) than their corresponding normals.
References
Lee HM, Zeng JY, Hu CH, Lee MT (2004) A new tridentate pincer phosphine/N-heterocyclic carbene ligand: palladium complexes, their structures, and catalytic activities. Inorg. Chem. 43(21):6822–6829
Skander M, Retailleau P, Bourrié B, Schio L, Mailliet P, Marinetti A (2010) N-heterocyclic carbene-amine Pt (II) complexes, a new chemical space for the development of platinum-based anticancer drugs. J. Med. Chem. 53(5):2146–2154
Raynaud J, Liu N, Fèvre M, Gnanou Y, Taton D (2011) No matter the order of monomer addition for the synthesis of well-defined block copolymers by sequential group transfer polymerization using N-heterocyclic carbenes as catalysts. Polym. Chem. 2(8):1706–1712
Budagumpi S, Endud S (2013) Group XII metal–N-heterocyclic carbene complexes: synthesis, structural diversity, intramolecular interactions, and applications. Organometallics 32(6):1537–1562
Telitel S, Schweizer S, Morlet-Savary F, Graff B, Tschamber T, Blanchard N, Fouassier JP, Lelli M, Lacôte E, Lalevée J (2013) Soft photopolymerizations initiated by dye-sensitized formation of NHC-boryl radicals under visible light. Macromolecules 46(1):43–48
Wanzlick HW, Schikora E (1960) Ein neuer Zugang zur Carben-Chemie. Angew. Chem. 72(14):494–494
Arduengo III AJ, Harlow RL, Kline M (1991) A stable crystalline carbene. J. Am. Chem. Soc. 113(1):361–363
Arduengo III AJ, Dias HVR, Harlow RL, Kline M (1992) Electronic stabilization of nucleophilic carbenes. J. Am. Chem. Soc. 114(14):5530–5534
Furstner A, Krause H, Ackermann L, Lehmann CW (2001) N-heterocyclic carbenes can coexist with alkenes and C–H acidic sites. Chem. Commun. 21:2240–2241
Otto M, Conejero S, Canac Y, Romanenko VD, Rudzevitch V, Bertrand G (2004) Mono-and diaminocarbenes from chloroiminium and-amidinium salts: synthesis of metal-free bis (dimethylamino) carbene. J. Am. Chem. Soc. 126(4):1016–1017
Igau A, Baceiredo A, Trinquier G, Bertrand G (1989) [Bis (diisopropylamino) phosphino] trimethylsilylcarbene: a stable nucleophilic carbene. Angew Chem Int Ed Engl 28(5):621–622
Igau A, Grutzmacher H, Baceiredo A, Bertrand G (1988) Analogous. alpha.,. alpha.’-bis-carbenoid, triply bonded species: synthesis of a stable. lambda. 3-phosphino carbene-. lambda. 5-phosphaacetylene. J. Am. Chem. Soc. 110(19):6463–6466
Herrmann WA, Denk M, Behn J, Scherer W, Klingan FR, Bock H, Solouki B, Wagner M (1992) Stable cyclic germanediyls (“cyclogermylenes”): synthesis, structure, metal complexes, and thermolyses. Angew. Chem. Int. Ed. 31(11):1485–1488
Denk M, Lennon R, Hayashi R, West R, Belyakov AV, Verne HP, Haaland A, Wagner M, Metzler N (1994) Synthesis and structure of a stable silylene. J. Am. Chem. Soc. 116(6):2691–2692
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41(2):157–167
Meller A, Gräbe CP (1985) Synthese und Isolierung neuer Germanium (II)-Verbindungen und freier Germylene. Chem Ber 118(5):2020–2029
Herrmann WA, Denk M, Behm J, Scherer W, Klingan FR, Bock H, Solouki B, Wagner M (1992) Stabile, cyclische Germandiyle (“cyclogermylenes”): Herstellung, Molekülstruktur, Metallkomplexe und Thermolysen. Angew. Chem. 104(11):1489–1492
Heinemann C, Herrmann WA, Thiel W (1994) Theoretical study of stable silylenes and germylenes. J. Organomet. Chem. 475(1–2):73–84
Kühl O (2004) N-heterocyclic germylenes and related compounds. Coordin Chem Rev 248(5–6):411–427
Bertrand G, Melaimi M, Soleilhavoup M (2010) Stable cyclic carbenes and related species beyond diaminocarbenes. Angew Chem Int Edn 49(47):8810–8849
Veszprémi T, Nyulászi L, Kárpáti T (1996) Toward stable silylenes. J. Phys. Chem. 100(15):6262–6265
Kassaee MZ, Buazar F, Soleimani-Amiri S (2008) Triplet germylenes with separable minima at ab initio and DFT levels. J. Mol. Struct. THEOCHEM 866(1–3):52–57
Gaspar PP, Xiao M, Pae DH, Berger DJ, Haile T, Chen T, Lei D, Winchester WR, Jiang P (2002) The quest for triplet ground state silylenes. J. Organomet. Chem. 646(1–2):68–79
Kassaee MZ, Ghambarian M, Musavi SM (2005) In search of triplet ground state GeCNX germylenes (X= H, F, Cl, and Br): an ab initio and DFT study. J. Organomet. Chem. 690(21–22):4692–4703
Kassaee MZ, Ashenagar S (2018) Theoretical descriptions of novel triplet germylenes M1-Ge-M2-M3 (M1= H, Li, Na, K; M2= Be, Mg, Ca; M3= H, F, Cl, Br). J. Mol. Model. 24(2):49
Driess M, Yao S, Brym M, van Wüllen C (2006) A heterofulvene-like germylene with a betain reactivity. Angew. Chem. Int. Ed. 45(26):4349–4352
Driess M, Yao S, Brym M, van Wüllen C, Lentz D (2006) A new type of N-heterocyclic silylene with ambivalent reactivity. J. Am. Chem. Soc. 128(30):9628–9629
Pintér B, Veszprémi T (2008) Synthesizability of the heavy analogues of disubstituted cyclopropenylidene: a theoretical study. Organometallics 27(21):5571–5576
Momeni MR, Shakib FA (2011) Theoretical description of triplet silylenes evolved from H2Si= Si. Organometallics 30(18):5027–5032
Rezaee N, Ahmadi A, Kassaee MZ (2016) Nucleophilicity of normal and abnormal N-heterocyclic carbenes at DFT: steric effects on tetrazole-5-ylidenes. RSC Adv. 6(16):13224–13233
Kassaee MZ, Musavi SM, Ghambarian M, Buazar F (2005) Multiplicity vs. stability in C2HP carbenes and their halogenated analogues: an ab initio and DFT study. J. Mol. Struct. THEOCHEM 726(1–3):171–181
Kassaee MZ, Ghambarian M, Musavi SM (2007) Halogen switching of azacarbenes C2NH ground states at ab initio and DFT levels. Heteroat. Chem. 19(4):377–388
Kassaee MZ, Musavi SM, Buazar F (2005) An ab initio and DFT comparative study of electronic effects on spin multiplicities and structures of X–C2N carbenes. J. Mol. Struct. THEOCHEM 728(1–3):15–24
Kassaee MZ, Ghambarian M, Musavi SM, Shakib FA, Momeni MR (2009) A theoretical investigation into dimethylcarbene and its diamino and diphosphino analogs: effects of cyclization and unsaturation on the stability and multiplicity. J. Phys. Org. Chem. 22(10):919–924
Kassaee MZ, Momeni MR, Shakib FA, Ghambarian M, Musavi SM (2010) Novel α-spirocyclic (alkyl)(amino) carbenes at the theoretical crossroad of flexibility and rigidity. Struct. Chem. 21(3):593–598
Kassaee MZ, Shakib FA, Momeni MR, Ghambarian M, Musavi SM (2010) Carbenes with reduced heteroatom stabilization: a computational approach. J Org Chem 75(8):2539–2545
Kassaee MZ, Ghambarian M, Shakib FA, Momeni MR (2011) From acyclic dialkylcarbene to the unsaturated cyclic heteroatom substituted ones: a survey of stability. J. Phys. Org. Chem. 24(5):351–359
Kassaee MZ, Momeni MR, Shakib FA, Najafi Z, Zandi H (2011) Effects of α-cyclopropyl on heterocyclic carbenes stability at DFT. J. Phys. Org. Chem. 24(11):1022–1029
Kassaee MZ, Najafi Z, Shakib FA, Momeni MR (2011) Stable silylenes with acyclic, cyclic, and unsaturated cyclic structures: effects of heteroatoms and cyclopropyl α-substituents at DFT. J. Organomet. Chem. 696(10):2059–2064
Kendali RA, Dunning Jr TH, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. Chem. Phys. 96(9):6796–6806
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Account 120(1–3):215–241
Becke AD (1998) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38(6):3098
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98(7):5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37(2):785
Chong DP (ed) (1997) Recent advances in density functional methods, parts I and II. World Scientific, Singapore
Barone V, Bencini A (eds) (1999) Recent advances in density functional methods, part III. World Scientific, Singapore
Adamo C, di Matteo A, Barone V (1999) From classical density functionals to adiabatic connection methods. The state of the art. Adv. Quantum Chem. 36:45–75
Ess DH, Houk KN (2005) Activation energies of pericyclic reactions: performance of DFT, MP2, and CBS-QB3 methods for the prediction of activation barriers and reaction energetics of 1,3-dipolar cycloadditions, and revised activation enthalpies for a standard set of hydrocarbon pericyclic reactions. J. Phys. Chem. A 109:9542–9553
Aysin RR, Bukalov SS, Leites LA, Zabula AV (2017) Optical spectra, electronic structure and aromaticity of benzannulated Nheterocyclic carbene and its analogues of the type C6H4(NR)2E: (E = Si, Ge, Sn, Pb). Dalton Trans. 46:8774–8781
Zhang MX, Zhang MJ, Li WZ, Li QZ, Cheng JB (2015) Structure of H2GeFMgF and its insertion reactions with RH (R = F, OH, NH2). J. Theor. Comput. Chem. 14:01–13
BaoW LY, Lu X (2013) Density functional theory study of mechanism of forming a spiro-Ge-heterocyclic ring compound from H2Ge=Ge: and ethane. Struct. Chem. 24(5):1615–1619
LiWZ YBF, Li QZ, Cheng JB (2013) The insertion reactions of the germylenoid H2GeLiF with CH3X (X = F, cl, Br). J. Organomet. Chem. 724:163–166
Yan B, LiW XC, Li Q, Cheng J (2013) A new reaction mode of germanium-silicon bond formation: insertion reactions of H2GeLiF with SiH3X (X = F, cl, Br). J. Mol. Model. 19(10):4537–4543
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL (1993) GAMESS program package. J. Comput. Chem. 14:1347–1363
Sobolewski AL, Domcke W (2002) Ab initio investigation of the structure and spectroscopy of hydronium− water clusters. J. Phys. Chem. A 106(16):4158–4167
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford Univ, New York, p 333
Sulzbach HM, Bolton E, Lenoir D, Schleyer PV, Schaefer HF (1996) Tetra-tert-butylethylene: an elusive molecule with a highly twisted double bond. Can it be made by carbene dimerization? J. Am. Chem. Soc. 118(41):9908–9914
Parr RG, Szentpaly LV, Liu S (1999) Electrophilicity index. J. Am. Chem. Soc. 121(9):1922–1924
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. Org. Chem. 73(12):4615–4624
Worthington SE, Cramer CJ (1997) Density functional calculations of the influence of substitution on singlet–triplet gaps in carbenes and vinylidenes. J. Phys. Org. Chem. 10(10):755–767
Akbari A, Golzadeh B, Arshadi S, Kassaee MZ (2015) A quest for stable 2, 5-bis (halobora) cyclopentenylidene and its Si, Ge, Sn and Pb analogs at theoretical levels. RSC Adv. 5(54):43319–43327
Schaper LA, Wei X, Altmann PJ, Öfele K, PÖthig A, Drees M, Mink J, Herdtweck E, Bechlars B, Herrmann WA, Kühn FE (2013) Synthesis and comparison of transition metal complexes of abnormal and normal tetrazolylidenes: a neglected ligand species. Inorg. Chem. 52(12):7031–7044
Murray JS, Sen K (eds.) (1996) Molecular electrostatic potentials: concepts and applications. (Vol. 3) Elsevier
Alkorta I, Perez JJ (1996) Molecular polarization potential maps of the nucleic acid bases. Int. J. Quantum Chem. 57(1):123–135
Scrocco E, Tomasi J, Lowdin P (1978) Advances in quantum chemistry, vol 2. Academic Press, New York
Luque FJ, Orozco M, Bhadane PK, Gadre SR (1993) SCRF calculation of the effect of water on the topology of the molecular electrostatic potential. J. Phys. Chem. 97(37):9380–9384
Šponer J, Hobza P (1996) DNA base amino groups and their role in molecular interactions: ab initio and preliminary density functional theory calculations. Int. J. Quantum Chem. 57(5):959–970
Schleyer PV, Maerker C, Dransfeld A, Jiao H, van Eikema Hommes NJ (1996) Nucleus-independent chemical shifts: a simple and efficient aromaticity probe. J. Am. Chem. Soc. 118(26):6317–6318
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 45 kb).
Rights and permissions
About this article

Cite this article
Ashenagar, S., Kassaee, M.Z. & Cummings, P.T. Novel triplet germylenes in focus: normal vs. abnormal triplet exocyclic tetrazol-5-vinylidene germylenes at DFT. J Mol Model 25, 371 (2019). https://doi.org/10.1007/s00894-019-4213-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4213-2