
Abstract
The complexes formed between TX3–ZX2 (T = C, Si, Ge; Z = P, As, Sb; X = F, Cl) and NH3 were studied at the MP2/aug-cc-pVTZ(PP) level. For each TX3–ZX2, two types of complex were obtained. For CX3-ZX2, NH3 is inclined to approach the σ-hole on the Z atom, forming a pnicogen bond. For TX3–ZX2 (T = Si and Ge), however, the base favors engaging in a tetrel bond with the σ-hole on the T atom although the corresponding pnicogen-bonded complex is also stable. When NH3 approaches the CX3 terminal of CX3–ZX2, weak interactions are observed that may be classified as van der Waals interactions. The relative stability of both types of complexes is not affected by the substituent X. The tetrel bond is very strong and the largest interaction energy is up to −144 kJ mol−1. Dispersion is dominant in the weak van der Waals complexes, while tetrel- and pnicogen-bonded complexes are dominated by electrostatic interactions, with comparable contributions from polarization.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Non-covalent interactions play an important role in supramolecular chemistry [1], molecular recognition [2], and material science [3]. This motivates people to find and understand more new types of non-covalent interactions. Researchers now have a good knowledge of the formation, properties, nature, and applications of hydrogen bonding. Besides hydrogen bonding, there are other types of non-covalent interactions such as halogen bonding [4], chalcogen bonding [5, 6], pnicogen bonding [7, 8], and tetrel bonding [9, 10], which correspond to the interaction of a group VII–IV atom with a base, respectively. Recently, there has been a growing focus on the applications of halogen and chalcogen bonding interactions since their formation, properties, and nature are now deeply understood. In contrast, pnicogen and tetrel bonding interactions still require much attention to understand their formation, properties, and nature in different systems [11,12,13,14,15,16,17,18,19,20,21].
The pnicogen atom in pnicogen bonding and the tetrel atom in tetrel bonding have a similarity in their hybridization. Specially, they can be sp3- and sp2-hybridized, and the corresponding acidic centers are called σ-hole and π-hole, respectively. The σ-hole refers to a region with positive molecular electrostatic potential (MEP) at the end of a covalent bond [22], while the π-hole is vertical to the plane of a molecular framework or a group [23]. For a given base, the π-hole interaction is usually stronger than the σ-hole interaction in most cases. The MEP on a σ-hole is greater for a heavier pnicogen/tetrel atom [24, 25], and it is further magnified when the pnicogen/tetrel atom adjoins with strong electron-withdrawing groups [26, 27]. On the other hand, the sp3-hybridized pnicogen and tetrel atoms show some difference in the formation of a σ-hole interaction. The sp3-hybridized tetrel atom is tetravalent with four atoms/groups, which would hinder a base from approaching the tetrel atom. To facilitate the approach, the tetrahedral structure of the tetrel donor molecule deforms to resemble a trigonal bipyramid to a certain extent. This phenomenon is particularly prominent in silicon-containing compounds in tetrel bonding. A similar hindrance does often not occur in pnicogen-bonded complexes. The sp3-hybridized pnicogen atom, however, possesses a lone pair that would cause repulsion with a base, which affects the directionality of pnicogen bonding.
Researchers are interested in comparing the strength of different non-covalent interactions [28,29,30,31,32,33,34,35,36,37,38,39,40,41,42] since this can hint at competition and molecular recognition in chemical and biological systems. The σ-hole tetrel bond was compared with hydrogen bonds in complexes of HArF with TH3X (X = halogen, T = C and Si) [37], halogen bonds in complexes of DMSO with TF3X (T = C and Si; X = halogen) [38], and chalcogen bonds in complexes of N-methylacetamide with some cationic sulfur-containing compounds [39]. The σ-hole pnicogen bond was compared with hydrogen bonds in complexes of ZH4+ (Z = N, P, As) and their fluoro derivatives with HCN or LiCN [40], halogen bonds in complexes of HOX (X = halogen) with PH2Y (Y = H, F, Cl, Br, CH3, NH2, OH, and NO2) [41], and chalcogen bonds in complexes of XHS-PH2X (X = F, Cl, CCH, COH, CH3, OH, OCH3 and NH2) [42]. Their relative strength depends on the nature of the acid center and its substituents as well as the identity of the base. However, studies performed to compare σ-hole pnicogen bonds and σ-hole tetrel bonds are scarce.
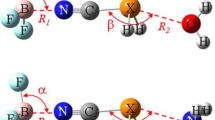
In this work, the complexes of a perfluoro or a perchloro molecule TX3-ZX2 (T = C, Si, Ge; Z = P, As, Sb; X = F, Cl) with NH3 were used to study competition between σ-hole pnicogen and σ-hole tetrel bonds. NH3 often serves as a base in studying non-covalent interactions. Here, we selected perfluoromethylphosphine CX3–PX2 to interact with NH3. For comparison, its heavy analogues were also studied. The corresponding diagrams and designations are shown in Fig. 1. Are there two interaction modes between the two molecules? Which interaction mode is stronger? Is one interaction mode always stronger than other mode, regardless of substituents? How does their relative strength rely on the nature of tetrel and pnicogen? What is the origin of both interactions? This work attempts to answer these questions by means of quantum chemical calculations.

Diagrams of two types of complex between TX3–ZX2 and NH3
Methods
All complexes and isolated molecules were optimized using second-order Møller–Plesset perturbation theory (MP2) [43] and the Dunning-type aug-cc-pVTZ basis set [44]. For Sb, the aug-cc-pVTZ-PP pseudopotential was used to incorporate relativistic effects. Frequency calculations were carried out at the same computational level to confirm that the obtained structures corresponded to energetic minima. The interaction energy (∆E) of the complex was calculated as a difference between the energy of the complex and the sum of energies of the monomers with their geometries frozen in the complex. The interaction energies were corrected for the basis set superposition error (BSSE) by the standard counterpoise method [45]. All calculations were performed with the Gaussian 09 set of codes [46].
The MEP maps of TX3-ZX2 were plotted at the 0.001 au isodensity surfaces using the wave function analysis-surface analysis suite (WFA-SAS) program [47]. The quantum theory of atoms in molecules (QTAIM) [48] was utilized to analyze bond critical points (BCPs) in terms of electron density, its Laplacian, and total energy density. The QTAIM calculations were performed with the use of the AIM2000 program [49]. Non-covalent interaction (NCI) maps were plotted using the VMD program [50]. Natural bond orbital (NBO) analysis was performed via NBO 3.1 program [51] implemented in Gaussian 09 to obtain charge transfer at the HF/aug-cc-pVTZ(PP) level. Energy decomposition analysis (EDA) was performed at the MP2/aug-cc-pVTZ(PP) level with the localized molecular orbital energy decomposition analysis (LMOEDA) method [52] using the GAMESS program [53]. This method decomposed the interaction energy into five terms including electrostatic (ES), exchange (EX), repulsion (REP), polarization (POL), and dispersion (DISP).
Results and discussion
MEPs of TX3-ZX2
Figure 2 shows the MEP maps of TX3-ZX2. Red regions with positive MEPs (σ-holes) are found at the both ends of the T–Z bond. The σ-holes on the T and Z atoms are thus able to engage in a σ-hole tetrel bond (TB) and a σ-hole pnicogen bond (ZB) with NH3, respectively. The most positive MEPs (Vmax) on the tetrel and pnicogen atoms in TX3–ZX2 are collected in Table 1. Generally, some regular variations are obtained. For a given TX3, the MEP of the σ-hole on the Z atom is larger for the heavier pnicogen atom. This is attributed primarily to the smaller electronegativity and the larger polarization of the heavier pnicogen atom. For a given ZX2, however, the MEP of the σ-hole on the T atom is larger in the order T = C < Ge < Si, which is inconsistent with the order in the periodic table. Even so, it accords with the change of the T electronegativity. Whether the σ-hole on the T atom or the σ-hole on the Z atom, their MEPs are greater when they adjoin with the stronger electron-withdrawing group F. On the other hand, some irregular changes are observed. The MEP of the σ-hole on the C atom is larger from CX3-PX2 to CX3-SbX2 to TCX3-AsX2, while the MEP of the σ-hole on the heavier tetrel atom increases in the sequence of TX3-SbX2 < TX3-AsX2 < TX3-PX2. The former disagrees with the pnicogen electronegativity. The MEP of the σ-hole on the C and Ge atoms is much smaller than that on the Z atom, while the MEP of the σ-hole on the Si atom is larger than that on the Z atom except in the case of SiCl3–SbCl2. Both types of σ-holes are able to participate in a tetrel bond and a pnicogen bond with NH3. However, the corresponding tetrel-bonded complexes are not obtained for CX3–ZX2. This was often reported in the complexes involving –CF3 group [17].

Molecular electrostatic potential (MEP) maps of TX3–ZX2 (T = C, Si, Ge; Z = P, As, Sb; X = F, Cl). Color ranges (kJ mol−1): red > 52.51, yellow between 52.51 and 0, green between 0 and − 13.13, blue < −13.13.
Interaction energy and geometries
Table 2 presents the interaction energies of both types of complexes. The interaction energy of pnicogen bond varies in a wide range of 15–68 kJ mol−1. The pnicogen bond is stronger for the heavier pnicogen atom. The stronger electron-withdrawing group F corresponds to a stronger pnicogen bond. The strength of pnicogen bond is also related to the TX3 group, increasing from SiX3 to CX3 to GeX3. Comparison for the interaction energy of the pnicogen bond and the positive MEP on the Z atom shows that they have a consistent change, confirming the role of electrostatic interaction in the formation of a pnicogen bond. The interaction energy of tetrel bond is comparable for the SiF3 and GeF3 donors, whereas it has a larger difference for the SiCl3 and GeCl3 donors. GeCl3 forms a stronger tetrel bond than SiCl3 when ZX2 is PCl2, but the reverse result is obtained for AsCl2 and SbCl2. The dependence of tetrel bonding energy on the Z atom is irregular for X = F; however, it increases for the heavier pnicogen atom if X = Cl. The interaction energy is very small in HB-1 to HB-6, where more than one interaction is present according to the following AIM and NCI analyses. That is, each interaction in HB-1 to HB-6 is actually very weak and its interaction energy may be in a range of van der Waals interactions, supported also by the long distance. CCl3–ZX2 has more stability than the CF3–ZX2 analogue.
For CX3–ZX2, the pnicogen-bonded complex is more stable than its van der Waals counterpart. Namely, the former is dominant for CX3–ZX2. For TX3–ZX2 (T = Si and Ge), the tetrel-bonded complex shows greater larger stability than the pnicogen-bonded analogue. This result is incompletely consistent with the positive MEPs on the T and Z atoms. For example, the MEP of the σ-hole on the Ge atom is smaller than that on the Sb atom in GeCl3–SbCl2; however, the corresponding tetrel bond is stronger than the pnicogen bond. We partly ascribe it to the larger deformation of TX3 in tetrel bonds, witnessed by the angle change in Table S1. The pnicogen bond is always stronger than van der Waals interaction for CX3–ZX2, while the tetrel bond is always stronger than the pnicogen bond for TX3–ZX2 (T = Si and Ge), regardless of substituents. Their relative strength does not rely on the nature of tetrel and pnicogen atoms.
The interaction energy was calculated to be less than 19 kJ mol−1 in F3P⋯NH3 and Cl3P⋯NH3 [54]. This value is comparable with that in TX3–ZX2⋯NH3. That is, the electron-withdrawing ability is similar for F and TX3. The interaction energy was 44.27 and 70.10 kJ mol−1 in F4Si⋯NH3 and F4Ge⋯NH3, respectively [20]. It is much smaller than that in NH3⋯TX3–ZX2 (T = Si and Ge), indicating that the ZX2 group has a larger electron-withdrawing ability than F.
For the van der Waals complexes, the C⋯N distance is listed in Table 2 since there is more than one interaction. The Si/Ge⋯N distance is shorter than 2.1 Å, much smaller than the sum of the van der Waals radii of both atoms. In spite of the smaller atomic radius of Z, the Z⋯N distance is much longer than the T⋯N distance due to the weaker pnicogen bond.
In the van der Waals complexes, the angle Z–T–X has a slight change (Table S1). However, this angle shrinks greatly in the tetrel-bonded complexes (by more than 13°). For the pnicogen-bonded complexes, the angle T–Z–X also has an observed shrink with one exception in ZB-1. The shrink of both the angle Z-T-X in the tetrel-bonded complex and the angle T-Z-X in the pnicogen-bonded complex increases in the order Z = P < As < Sb for the given T and X. The larger angle shrink corresponds to the larger deformation of TX3–ZX2 monomer in the complexes, implying the larger contribution of deformation energy in stabilizing the complex.
The complexation leads to charge transfer (Table 2). In HB-1 to HB-6, charge transfer is negative, indicating that it moves from CX3–ZX2 to NH3. However, it is very small and close to zero in HB-1 to HB-6, thus it may not provide reliable information for the presence of any complex. Charge transfer is very large in the tetrel bond (>0.17e). Interestingly, the TCl3–ZCl2 (T = Si and Ge) complex has larger charge transfer than the TF3–ZF2 counterpart despite the weaker tetrel bond in the former. The charge transfer in the pnicogen bond is much smaller than that in the tetrel bond, and it shows a linear relationship with the interaction energy (Fig. S1).
Topological analyses
AIM analysis, to a great extent, provides reliable information for the presence of non-covalent interactions by revealing the existence of a BCP between two molecules. Figure 3 shows the AIM diagrams of three representative complexes. For HB-4, there are two Cl⋯H BCPs and one Cl⋯N BCP. However, this does not necessarily imply the existence of any directional interaction since AIM bond paths are not infallible indicators of bonds, as some have pointed out [55, 56]. For TB-4, a N⋯Si bond path is used to characterize the N⋯Si tetrel bond. For ZB-4, the pnicogen bond is featured with a N⋯P path with a curve near the P atom. Bond paths in other tetrel- and pnicogen-bonded complexes are similar, but they have some differences in van der Waals complexes.

Atoms in molecules (AIM) diagrams of three types of complexes
Table 3 presents the electron density, Laplacian and energy density at the intermolecular BCP. The electron density at the H⋯X BCP is very small (<0.006 au), and the three terms are positive. This indicates that the van der Waals interaction is very weak, corresponding to a closed-shell interaction. For the Si/Ge⋯N BCP, the electron density is very large (>0.06 au) with positive Laplacian and negative energy density. This confirms that the tetrel bond is very strong with a nature of a partially covalent interaction [57]. The electron density at the Z⋯N BCP varies from 0.0144 to 0.1180 au, the corresponding Laplacian is positive, and the energy density is negative in most complexes. This implies that most pnicogen bonds are also strong and have properties of a partially covalent interaction. For most P⋯N pnicogen bonds excluding ZB-1 and HB-13, the energy density is positive, corresponding to a closed-shell interaction. For tetrel and pnicogen bonds, different correlations between the electron density and the binding distance are found (Fig. S2).
Relative to the AIM diagrams, NCI maps are more intuitive when studying non-covalent interactions. Figures 4 and 5 are the NCI maps of both types of complex. In HB-1 to HB-6, there are at least three green regions between the two molecules, corresponding to three interactions. However, each interaction in these complexes is much weaker since the total interaction energy is less than 7 kJ mol−1 in HB-1 to HB-6. The green regions in HB-4,5,6 are larger than those in HB-1,2,3, consistent with the larger interaction energy in the former. For TB-1,2,3, the region between the Si and N atoms is very similar, characterized with different colors, where blue corresponds to a strong interaction. For TB-4,5,6, an irregular region with different colors surrounds the Si atom. The Ge⋯N interaction is characterized by different regions from the Si⋯N interaction. The NCI region in the Z⋯N pnicogen bond has a similar shape in all complexes. For the Sb⋯N pnicogen bond, the NCI region is partly blue, consistent with the stronger pnicogen bond.

Non-covalent interaction (NCI) diagrams of van der Waals and tetrel-bonded complexes. Interactions: Red Strong repulsion, blue strong attractive, other areas weak attractive

NCI diagrams of pnicogen-bonded complexes. Interactions: Red Strong repulsion, blue strong attractive, other areas weak attractive
One can see from Fig. 4 that the Si–Z and Ge–Z bonds seem to be broken. Therefore, we focus on the comparison for their bond length with the Si⋯N and Ge⋯N distances. The Si–Z and Ge–Z bond lengths are 2.31–2.60 and 2.36–2.64 Å, respectively. Clearly, they are longer than the Si⋯N and Ge⋯N distances. However, the Si–Z and Ge–Z bond lengths are 2.28–2.60 Å and 2.33–2.65 Å, respectively, in the monomers. As a result, the illusion of the Si–Z and Ge–Z bond fracture in Fig. 4 is attributed mainly to their long bond length.
Energy decomposition
To gain a deeper understanding of the origin of interactions in these complexes, we obtained the three attractive terms of electrostatic, polarization and dispersion using the GAMESS program (Table 4). For the weak van der Waals complexes, the greatest stability comes from dispersion, particularly in the Cl-substituted complexes where dispersion amounts to two to three times as much as electrostatic; polarization has the smallest contribution. It should be noted that such decomposed results may be meaningless and unreliable for this weak complexes due to the possible error. For the strong tetrel-bonded complexes, electrostatic is dominant, corresponding to ~60% of the total attractive energy, while polarization also has an important contribution, accounting for a third of the total attractive energy. Dispersion can be ignored in the strong tetrel-bonded complexes and it is even positive in the F-substituted complexes. The three attractive terms show similar contributions in the pnicogen-bonded complexes with those in the tetrel-bonded complexes. Of course, the relative contribution is different for both types of complexes. For instance, polarization has a larger contribution in the tetrel bond than that in the pnicogen bond. The former is consistent with the greater deformation in the tetrel bond. The positive dispersion in some systems is attributed mainly to differences in the intra- and intermolecular correlation energy on going from noninteracting to interacting molecules [52]. Politzer and co-authors [58] claimed that noncovalent bonding is in nature coulombic interactions, which encompass polarization and therefore dispersion, based on the Hellmann-Feynman theorem [59, 60]. Politzer and Murray [61] pointed out that polarization is an intrinsic part of an electrostatic interaction at most cases since the electric fields of the positive σ-hole and the negative site can induce some rearrangement of the electronic densities of both sites and there is no actual physical distinction between polarization and charge transfer.
Conclusions
Quantum chemical calculations have been performed for the complexes of TX3–ZX2 (T = C, Si, Ge; Z = P, As, Sb; X = F, Cl) and NH3. MEP analysis shows two σ-holes at both ends of the T–Z bond. The σ-hole on the Z atom engages in a pnicogen bond, while the σ-hole on the heavier T atom participates in a tetrel bond and the X atom of CX3 forms van der Waals interactions. For CX3–ZX2, the pnicogen-bonded complex is more stable than the van der Waals complex. For TX3-ZX2 (T = Si and Ge), the tetrel-bonded complex is dominant over the pnicogen-bonded complexes. For each TX3–ZX2, the X group has no effect on the relative stability of both types of complexes. TX3–ZX2 (T = Si and Ge) is a good tetrel donor since the interaction energy is larger than 90 kJ mol−1 in magnitude, up to ~144 kJ mol−1. The larger interaction energy of the tetrel bond is accompanied with a bigger charge transfer (>0.17e). It has a nature of a partially covalent interaction with a positive Laplacian and a negative energy density. The strong tetrel bond is dominated by electrostatic interaction with a comparable contribution from polarization. The pnicogen bond varies from a moderate interaction to a strong one, depending on the pnicogen atom. It also shows a nature of a partially covalent interaction in most pnicogen-bonded complexes. The similar energy contributions are also found in the pnicogen-bonded complexes.
References
Schneider HJ (2009) Angew Chem Int Ed 48:3924–3977
Hunter CA, Sanders JKM (1990) J Am Chem Soc 112:5525–5534
Vickaryous WJ, Herges R, Jonhson DW (2004) Angew Chem Int Ed 43:5831–5833
Legon AC (2010) Phys Chem Chem Phys 12:7736–7747
Iwaoka M, Takemoto S, Tomoda S (2002) J Am Chem Soc 124:10613–10620
Murray JS, Lane P, Clark T, Politzer P (2007) J Mol Model 13:1033–1038
Scheiner S (2013) Acc Chem Res 46:280–288
Murray JS, Lane P, Politzer P (2007) Int J Quantum Chem 107:2286–2292
Bauzá A, Mooibroek TJ, Frontera A (2016) Chem Rec 16:473–487
Murray JS, Lane P, Politzer P (2009) J Mol Model 15:723–729
Alkorta I, Elguero J, Del Bene JE (2013) J Phys Chem A 117:10497–10503
Li QZ, Li R, Liu XF, Li WZ, Cheng JB (2012) ChemPhysChem 13:1205–1212
Li QZ, Li R, Liu XF, Li WZ, Cheng JB (2012) J Phys Chem A 116:2547–2553
Del Bene JE, Alkorta I, Elguero J (2015) Phys Chem Chem Phys 17:30729–30735
Li QZ, Guo X, Yang X, Li WZ, Cheng JB, Li HB (2014) Phys Chem Chem Phys 16:11617–11625
Guo X, Liu YW, Li QZ, Li WZ, Cheng JB (2015) Chem Phys Lett 620:7–12
Liu MX, Li QZ, Scheiner S (2017) Phys Chem Chem Phys 19:5550–5559
Bauzá A, Frontera A, Mooibroek TJ (2016) Phys Chem Chem Phys 18:1693–1698
Legon AC (2017) Phys Chem Chem Phys 19:14884–14896
Scheiner S (2017) J Phys Chem A 121:5561–5568
Martín-Fernández C, Montero-Campillo MM, Alkorta I, Elguero J (2017) J Phys Chem A 121:7424–7431
Clark T, Hennemann M, Murray JS, Politzer P (2007) J Mol Model 13:291–296
Murray JS, Lane P, Clark T, Riley KE, Politzer P (2012) J Mol Model 18:541–548
Grabowski SJ (2014) Phys Chem Chem Phys 16:1824–1834
Bauzá A, Mooibroek TJ, Frontera A (2016) ChemPhysChem 17:1608–1614
Scheiner S (2011) J Phys Chem A 115:11202–11209
Mani D, Arunan E (2013) Phys Chem Chem Phys 15:14377–14383
Gao L, Zeng YL, Zhang XY, Meng LP (2016) J Comput Chem 37:1321–1327
Bauzá A, Frontera A (2015) ChemPhysChem 16:3108–3113
Zhou PP, Yang X, Ye WC, Zhang LW, Yang F, Zhou DG, Liu SB (2016) New J Chem 40:9139–9147
Wei YX, Li QZ (2018) Mol Phys 116:222–230
Xu HL, Cheng JB, Yang X, Liu ZB, Li WZ, Li QZ (2017) ChemPhysChem 18:2442–2450
Dong WB, Yang X, Cheng JB, Li WZ, Li QZ (2018) J Fluor Chem 207:38–44
Solimannejad M, Ramezani V, Trujillo C, Alkorta I, Sánchez-Sanz G, Elguero J (2012) J Phys Chem A 116:5199–5206
Lang T, Li XY, Meng LP, Zheng SJ, Zeng YL (2015) Struct Chem 26:213–221
Nziko Vde PN, Scheiner S (2016) Phys Chem Chem Phys 18:3581–3590
Liu MX, Li QZ, Li WZ, Cheng JB, McDowell SAC (2016) RSC Adv 6:19136–19143
Wei QC, Li QZ, Cheng JB, Li WZ, Li HB (2016) RSC Adv 6:79245–79253
Scheiner S (2015) J Phys Chem A 119:9189–9199
Grabowski SJ (2013) Chem Eur J 19:14600–14611
Li QZ, Zhu HJ, Zhuo HY, Yang X, Li WZ, Cheng JB (2014) Spectrochim Acta A 132:271–277
Esrafili MD (2016) Mol Phys 114:1847–1855
Møller C, Plesset MS (1934) Phys Rev 46:618–622
Woon DE, Dunning Jr TH (1993) J Chem Phys 98:1358–1371
Boys SF, Bernardi F (1970) Mol Phys 19:553–566
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Scalmani G, Cossi M, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VGDS, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Gonzalez C, Wong MW, Pople JA (2009) Gaussian 09, revision A02. Gaussian Inc., Wallingford
Bulat FA, Toro-Labbé A, Brinck T, Murray JS, Politzer P (2010) J Mol Model 16:1679–1691
Bader RFW (1990) Atoms in Molecules: A Quantum Theory. Oxford University Press, Oxford
Bader RFW (2000) AIM2000 Program, Version 2.0. McMaster University, Hamilton Canada
Humphrey W, Dalke A, Schulten K (1996) J Mol Graph 14:33–38
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Su PF, Li H (2009) J Chem Phys 13:014102
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993) J Comput Chem 14:1347–1363
Scheiner S (2011) Chem Phys 387:79–84
Keyvani ZA, Shahbazian S, Zahedi M (2016) Chem Eur J 22:5003–5009
Alkorta I, Sanchez-Sanz G, Elguero J (2014) J Phys Chem A 118:1527–1537
Arnold WD, Oldfield E (2000) J Am Chem Soc 122:12835–12841
Politzer P, Murray JS, Clark T (2015) J Mol Model 21:52
Hellmann H (1933) Z Phys 85:180–190
Feynman RP (1939) Phys Rev 56:340–343
Politzer P, Murray JS (2013) ChemPhysChem 14:278–294
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 169 kb)
Rights and permissions
About this article

Cite this article
Li, Y., Xu, Z. Competition between tetrel bond and pnicogen bond in complexes of TX3-ZX2 and NH3. J Mol Model 24, 247 (2018). https://doi.org/10.1007/s00894-018-3732-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-018-3732-6