
Abstract
The influence of hybridization, substitution, and solvation on the triel bond has been investigated in the complexes of RZH2···NH3 (Z = B and Al). The magnitude of the π-hole on the triel atom is related to the nature of the Z atom and the hybridization of R. CH3BH2 has the largest π-hole among RBH2, while for RAlH2 the largest π-hole is found in CH≡CAlH2. The interaction energy is partly inconsistent with the magnitude of the π-hole on the triel atom and the orbital interaction from the N lone pair of NH3 into the empty p orbital of the triel atom. The strongest B···N triel bond is found in CH≡CBH2···NH3, while the weakest Al···N triel bond is in CH3AlH2···NH3. The strength of the triel bond is increased in solvents, and its enhancement is prominent with the increase of solvent polarity. Solvents also change the nature of the Al···N triel bond from an electrostatic interaction to a partially covalent one. The F substituent in the triel donor strengthens the triel bond, depending on the substitution position and number.

The π-hole triel bonded complexes between RZH2 (Z =B and Al) and NH3 have been investigated. We focused on the effects of hybridization, solvent, and substitution on the strength and nature of π-hole triel bond
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Different Lewis acid–base interactions are ubiquitous in various chemical, physical, and biological processes [1, 2]. Owing to the diversity of Lewis acid centers, these Lewis acid–base interactions are diverse, including hydrogen, halogen, and tetrel bonds, but most of them can be classified as σ-hole bonds [3,4,5]. The σ-hole is a region of depletion of the electron in the outer lobe of the half-filled p orbital that is involved in a covalent bond [3,4,5]. Another type of Lewis acid–base interaction is a π-hole bond, where the π-hole is a region of electron deficiency situated in a direction perpendicular to a center of a planar portion of a molecular framework [6,7,8]. An interesting issue is to compare the strength of σ-hole and π-hole bonds in different systems [9,10,11,12]. It was concluded that in most cases the π-hole bond is stronger than the σ-hole bond. This has an important implication in molecular recognition since the latter is partly dependent on the interaction strength. It was demonstrated that the π-hole bonds play important roles in crystal engineering [14] and biological systems [15, 16]. Therefore, more attention was paid to the π-hole bonds particularly those involving the C=O group [17,18,19,20]. Another usual π-hole bond is the triel bond, which is an attractive and highly directional Lewis acid–base interaction between a π-hole on the group III atom and a Lewis base [21]. Previously, complexes combined by this interaction were called partially bonded complexes since their binding distances are in the intermediate range between a van der Waals interaction and a covalent bond [22]. It should be noted that a triel atom also provides a σ-hole if it is bonded with four atoms or groups [23].
There are many theoretical and experimental studies on triel bonding [24,25,26,27,28,29,30,31,32,33,34] because it is involved in some important chemical processes particularly in the release of molecular hydrogen in hydrogen storage materials [35, 36]. Besides molecules with lone pairs, π systems [37, 38], metal hydrides [30, 39], and radicals [13] also engage in a triel bond. Some previous studies focused on the condensed-phase effects on the structural properties in triel-bonded complexes with experimental and theoretical methods [24, 27]. This is because triel bonding shows some abnormal larger shortening in the binding distance when it combines with another weak interaction [40]. On the other hand, triel bonding can compete with other types of interactions. For example, BX3 (X = F, Cl, Br, and I) can participate in lone pair···π, halogen···π, and triel bonding interactions with ethene, 1,2-difluoroethene, and perfluoroethene, but triel bonding dominates over the other two interactions [41].
The strength of a Lewis acid–base interaction is affected by factors other than the nature of both the Lewis acid and base, such as hybridization, solvation, and substitution. Usually, when the hybridization of R adjoined to the Lewis acid varies from sp3 to sp2 to sp, the stronger interaction is obtained. This conclusion has been confirmed in the C–H···Y (Y is a Lewis base) hydrogen bond [42,43,44], C–X···Y (X = halogen) halogen bond [45], and C–SiF3···NH3 tetrel bond [10]. Solvents have complicated effects on the strength of a Lewis acid–base interaction, which is dependent on the type and strength of this interaction as well as the polarity of solvent [46,47,48]. Theoretical studies showed that increasing the polarity of solvent leads to strengthened halogen bonding and weakened hydrogen bonding [46]. Then experimental evidence confirmed that hydrogen-bonded co-crystals are favorable in less polar solvents, while halogen-bonded co-crystals in more polar solvents [47]. This switching by solvents depends on the relative strengths of both interactions [47]. Very recently, a theoretical study unveiled the mechanism of solvent effect on the strength of halogen bond [48]. Su and co-authors thought that the influence of solvent on the strength of halogen bond is mainly realized by modulating its polarization contribution since polarization is more sensitive to the solvent effects than the other interaction terms [48]. F substitution in a Lewis acid would enhance the strength of hydrogen bond [49], halogen bond [50], and tetrel bond [10]. Thus, there appears an issue: How do the hybridization, solvent, and substituent influence the strength and nature of triel bonding?
In this study, we selected RZH2···NH3 (Z = B and Al) as a model to investigate the effect of hybridization on the strength of triel bonding. These complexes were also explored in solvents (heptanes and dimethyl sulfoxide (DMSO)) to study the effect of solvents on the strength and nature of triel bonding. From CH2=CHZH2···NH3, the hydrogen atoms of the CH2=CH group were replaced by different numbers of F atoms to probe the substitution effect on the strength of triel bonding. These complexes were investigated in view of geometrics, interaction energy, charge transfer, orbital interactions, and electron density.
Computational details
The structures of the monomers and complexes were first optimized at the MP2/aug-cc-pVDZ level. Frequency calculations at the same level confirmed that all structures are energetic minima since no imaginary frequencies were observed. These structures were then re-optimized at the MP2/aug-cc-pVTZ level to obtain more accurate results. The interaction energies (ΔE) were calculated as differences between the energy of the complex and the sum of energies of monomers with their geometries in the complex, and they were corrected for the basis set superposition error (BSSE) by the counterpoise method [51]. All calculations were carried out within the framework of the Gaussian 09 set of codes [52].
The atoms in molecules (AIM) analysis was used to find the intermolecular bond critical points (BCPs) and to calculate the corresponding topological parameters, including electron density, its Laplacian, and total energy density. The AIM analyses were performed with the use of the AIM2000 program [53]. The molecular electrostatic potentials (MEPs) of the isolated monomers have been analyzed with the Wave Function Analysis-Surface Analysis Suite (WFA-SAS) program [54] on the 0.001 a.u. electron density isosurface. The natural bond orbital (NBO) method [55] implemented in Gaussian 09 was applied to analyze orbital interactions and charge transfer at the HF/aug-cc-pVTZ level. The localized molecular orbital-energy decomposition analysis (LMO-EDA) method [56] was used to decompose the interaction energy of the complex using the GAMESS program [57] at the MP2/aug-cc-pVTZ level. An analysis of natural orbital for chemical valence (NOCV) was performed at the GGA-BPE-D3/QZ2P level with the ADF program [58].
Results and discussion
MEPs
An analysis of MEP can provide some useful information for interaction sites between two molecules and the corresponding interaction strength. Figure 1 illustrates the MEP maps on the 0.001 au molecular surfaces of monomers. Obviously, two red areas with positive MEPs (π-holes) are found on the triel atom, and their maximal values are also marked in Fig. 1. The heavier triel atom has the bigger π-hole than the lighter analogue due to its larger polarization and less electronegativity. Thus, these π-holes act as a Lewis acid to bind with a base such as NH3. When the hybridization of the triel atom is varied, the π-hole is biggest in CH≡CAlH2 (CH3BH2) but smallest in CH2=CHZH2. The biggest π-hole in CH≡CAlH2 can be attributed to the greatest electronegativity of the acetenyl group, while both the biggest π-hole in CH3BH2 and the smallest one in CH2=CHZH2 cannot be explained with electronegativity since the vinyl group has greater electronegativity than the methyl group.

MEP maps of the monomers on the 0.001 a.u. electron density isosurface. Color ranges, in au, are: red, greater than 0.0193; yellow, between 0.0193 and 0.0108; green, between 0.0108 and −0.0030; and blue, less than −0.0030
How are these unexpected variations for the MEPs on these triel atoms explained? We fall back on orbital interactions in these molecules. In CH2=CHZH2, a conjugative effect occurs between the π electrons on the C=C bond and the empty p orbital of the triel atom, confirmed by an orbital interaction from the π occupied orbital to the empty p orbital. The corresponding perturbation energy is 36.48 and 13.90 kcal mol–1 in CH2=CHBH2 and CH2=CHAlH2, respectively. A similar orbital interaction is also present in CH≡CZH2 and the respective perturbation energy is 28.70 and 8.25 kcal mol–1 in CH≡CBH2 and CH≡CAlH2. Another type of conjugative effect between the C–H σ orbital and the empty p orbital of the triel atom is present in CH3BH2 and CH3AlH2 with the perturbation energy of 17.83 and 4.49 kcal mol–1. Such conjugative effect results in an increase of electron density on the triel atom and thus a smaller π-hole. It is found that the conjugative effect is more prominent in RBH2 than that in RAlH2. For a given triel center, the conjugative effect is dependent on its hybridization, bigger from sp3 to sp to sp2. The largest conjugative effect in CH2=CHZH2 is responsible for the smallest π-hole on the Z atom. Therefore, the magnitude of π-hole on the triel atom is a combinative result of the electronegativity of its adjoining group and the conjugative effect. In addition, it should be noted that positive electrostatic potential reflects the lower electronic density of the σ-hole as well as contributions from other portions of the molecule [59].
The MEP on the π-hole of BH3 and AlH3 is respectively 47.1 and 87.3 kcal mol–1 [13], which is larger than that in RZH2. This means that the alkyl group plays an electron-donating role in RZH2 and thus decreases its positive MEPs. If R is combined with a halogen atom, the positive MEP on the halogen atom increases from sp3 to sp2 to sp, which is different from that on the triel atom.
Hybridization
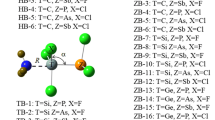
Figure 2 shows the optimized structures of the dyads with the binding distances. The binding distances are in the range of 1.648~1.660 Å and 2.057~2.076 Å in the B and Al complexes, respectively, which are much shorter than the sum of the van der Waals (vdW) radii of the corresponding atoms (3.4 Å for B and N; 3.7 Å for Al and N) [60]. The binding distance in the B complex is shorter from CH2=CHBH2···NH3 to CH3BH2···NH3 to CH≡CBH2···NH3, while the binding distance in the Al complex is shorter from CH3BH2···NH3 to CH2=CHBH2···NH3 to CH≡CBH2···NH3. Both of them are partly consistent with the positive MEP on the π-hole of RZH2.

The optimized structures of the dyads and binding distances are given in angstrom
One also finds that the planar structure of the triel center is distorted in the complex. This deformation can be estimated with deformation energy in Table 1. The deformation energy is defined as the energy difference between the isolated molecules and the molecules at the geometry of the complex. This term in the B complex is three times larger than that in the Al counterpart. Thus, the deformation has a larger contribution to the stability of the B complex.
From Table 1, it is seen that the interaction energy of the B complex depends on the B hybridization, being more negative from sp2 to sp3 to sp, while the interaction energy of the Al complex is also largest in CH≡CAlH2···NH3 but is almost equal in CH3AlH2···NH3 and CH2=CHAlH2···NH3. The interaction energy in the B complex is more negative than that in the Al analogue, inconsistent with the positive MEP on the π-hole of RZH2. This abnormal result is primarily attributed to the larger deformation energy in the B complex. The interaction energies of BH3···NH3 and AlH3···NH3 were calculated to be −40.09 and −32.87 kcal mol–1, respectively, which are more negative than that in CH≡CZH2···NH3 but less negative than that in CH3ZH2···NH3 and CH2=CHZH2···NH3. Obviously, only the former result is consistent with the positive MEP of the π-hole of the triel atom.
The formation of triel bonding leads to a big charge transfer within 0.14–0.38e, which is partly due to the large distortion of RZH2. The charge transfer in the B complex is bigger than that in the Al analogue, consistent with the interaction energy; specifically, the charge transfer in the B complex is more than twice that in the Al complex. However, the interaction energy in the former is one time larger than that in the latter. On the other hand, the charge transfer becomes bigger from CH3ZH2···NH3 to CH2=CHZH2···NH3 to CH≡CZH2···NH3. This order is in line with the interaction energy in the Al complex but some inconsistencies are found between them in the B complex.
Intermolecular orbital interactions were not discerned in NBO calculations; thus, their contribution was analyzed by natural orbital for chemical valence (NOCV) with the ADF program. Red and blue areas represent charge density decrease and increase, respectively. According to the color of deformation densities in Fig. 3, charge shifts from the N atom into the triel atom in all interactions. From the shape of deformation densities, we think that an orbital interaction between the lone pair orbital on the N atom and the empty p orbital of the triel atom is present. This orbital interaction contribution increases from sp3 to sp2 to sp, and its value in the B complex is almost triple that in the Al complex.

Deformation densities of the pair-wise orbital interactions in the dyads at the GGA-BPE-D3/QZ2P//MP2/aug-cc-pVTZ level. The associated orbital interaction energies are given in kcal mol–1. The color code of the charge flow is red–blue and the isovalue is 0.005 au
An AIM analysis can be used to predict the presence of an interaction and to estimate its strength and type. A bond critical point (BCP) is present between the Z and N atoms, and its electron density, Laplacian, and total energy density are given in Table 1. For a given Z···N interaction, the electron density also varies with the hybridization of R, being larger from sp3 to sp2 to sp. The value of Laplacian is positive for all complexes, indicating the electron density is depleted at the Z···N BCP. However, the sign of energy density varies dependent on the nature of the triel atom. That is, it is positive in the Al complex but is negative in the B complex. According to the proposition of Koch and Popelier [61], the B···N interaction has a partially covalent character, while the Al···N interaction is basically electrostatic in nature.
In order to further explore the nature of the triel bond, we divided the interaction energy into five parts in Table 2: electrostatic energy (Eele), exchange energy (Eex), repulsion energy (Erep), polarization energy (Epol), and dispersion energy (Edisp). Eele is defined as the Coulomb energy between the two unperturbed partners, Eex is chiefly caused by the overlap of molecular orbitals, Erep mainly originates from Pauli repulsion, Epol is defined as the energy lowering due to the intramolecular relaxation of each molecule’s ALMOs in the field of all other molecules in the system, and Edisp is derived via a supermolecule approach using size-consistent correlation methods, such as MP2 and CCSD(T). The exchange energy is much larger than the electrostatic energy in the B complex, while both terms are almost equal in the Al complex. Because the repulsion energy depends on the exchange energy and they cancel each other, we do not focus on both terms. Considering the B···N interaction or the Al···N interaction, three attractive terms (Eele, Epol, and Edisp) are more negative from electrostatic to polarization to dispersion, as shown in Fig. 4. The electrostatic energy is about twice that of the polarization energy in the Al···N interaction; thus, this triel bond is electrostatically dominated. For the B···N interaction, electrostatic and polarization energies are comparable; thus, this interaction is mainly determined by a combination of electrostatic and polarization. In the B complexes, the relatively larger Epol means that the shape of the orbitals has undergone a large change, which is consistent with the deformation of the RBH2 molecule. Both Eele and Epol are more negative from sp3 to sp2 to sp, while Edisp has a slight change with the hybridization. Each term in the B complex is greater than that of the Al complex and their largest difference is found for Epol, indicating the boron atom is more easily polarized. Politzer et al. [62] pointed out that noncovalent interactions are Coulombic, with inclusion of polarization and dispersion, and thus the above energy decomposition analysis is necessary.

Dependence of energy components on the hybridization of carbon atom adjoined to the triel atom
Solvent effect
It is interesting to study the strength of triel bonding in solvents; thus, we placed the six complexes above into two solvents of heptane and DMSO. Table 3 presents the changes of binding distance, charge transfer, and electron density in the solvent relative to in the gas phase. The binding distance is shortened and charge transfer and electron density are increased, indicating that the triel bond is strengthened in solvents. This is because the B and Al atoms are easily polarized by solvents and thus engage in a stronger triel bond. Moreover, these changes are prominent with the increase of solvent polarity. Although the shortening of the binding distance is greater in the Al complex than that in the B analogue, both charge transfer and electron density have a larger increase in the latter complex. As a result, the B···N treil bond is greatly enhanced in the solvent relative to the Al···N treil bond. Here the binding distance does not truly reflect the strength of the triel bond in some cases, which supports the conclusion that close contacts between atoms are not always a reliable indicator of the actual interactions [63].
In both solvents, the binding distance shortens and electron density increases from sp3 to sp2 to sp for either the B complex or the Al complex, indicating that the strength of triel bond is stronger following the same order, which is different from that in the gas phase. Obviously, solvation has an important effect on the strength of the triel bond. With enhancement of the triel bond, its type is also varied. The energy density is from one positive value in the gas phase to a negative value in the solvent, becoming a partially covalent interaction.
Influence of F substitution
In this section, CH2=CHZH2···NH3 (Z = B, Al) was chosen to study the influence of F substituents on the strength of triel bonding, where different sites of H atoms in CH2=CHZH2 are replaced by F substituents. Figure 5 shows the optimized structures of F derivatives of CH2=CHZH2···NH3 (Z = B, Al). For simplicity, the F derivatives of CH2=CHZH2···NH3 are denoted as nF-Z, where Z denotes a triel atom (B or Al) and n is the number of F substituents (1–3). If one F substitution happens at the C position adjoined with the –ZH2 group, NH3 is inclined to approach the triel atom along the side of this F substitution since it is favorable for the attractive interaction between the F atom and the NH group. Otherwise, NH3 attacks the triel atom from the opposite side in 1F-Z-b and 2F-Z-a, which is due to the presence of attractive interactions between the F atom at the terminal C atom and NH group. There is one exception in 1F-Z-a, where its conformation is like that in CH2=CHZH2···NH3. Clearly, the C–C–Z–H dihedral angle has a prominent change in most complexes excluding 1F-Z-a relative to the unsubstituted complex. It is noted that we tried to make the conformations of F derivatives of CH2=CHZH2···NH3 (Z = B, Al) uniform, but they change again into the ones shown in Fig. 5.

The optimized structures of F derivatives of CH2=CHZH2···NH3 (Z = B, Al).
Owing to the electron-withdrawing property, the F substitution results in an increase of the positive MEP on the π-hole of the triel atom (Table 4). Generally, the triel atom has a larger MEP with an increase in F number. Moreover, the F substitution at the CH position causes a larger increase in the positive MEP than that at the CH2 position. In addition, the F substitution has the same effect on the π-hole of both B and Al atoms.
Relative to CH2=CHZH2···NH3, the Z···N distance is shortened owing to the F substitution. However, no good linear relationship is found between the Z···N distance and the positive MEP on the triel atom. For example, 1F-Al-b has the shorter Al···N distance but the smaller positive MEP on the Al atom than 1F-Al-a. Such inconsistency may be due to the cooperativity between the triel bond and the F···H secondary interaction as well as the conformation difference. Similarly, the interaction energy is increased owing to the F substitution, and the increase of interaction energy shows some inconsistencies with the positive MEP on the triel atom. In addition, the stronger triel bond is partly attributed to the increase of charge transfer. Thus, we think that the triel bond should be understood with a combination of electrostatic and orbital interactions.
Conclusions
We performed theoretical calculations for the complexes of CH3ZH2···NH3, CH2CHZH2···NH3, and CHCZH2···NH3 (Z = B and Al) in view of the geometrics, energies, charge transfer, orbital interactions, and AIM analyses. The main conclusions are summed up as follows:
- (1)
The positive MEP on the π-hole of the triel atom in RZH2 is related to the carbon hybridization of alkyl group R. It is larger in the order of sp2 < sp < sp3 for the B atom but sp2 < sp3 < sp for the Al atom. This is a combinative result of the electronegativity of R and the conjugative effect.
- (2)
The triel bond is stronger in the sequence of sp2 < sp3 < sp in the B complex but sp2 ≈ sp3 < sp in the Al complex. Obviously, some inconsistencies between the interaction energy and the positive MEP on the π-hole of the triel atom are found for both B and Al complexes. These inconsistencies are partly attributed to strong orbital interaction and large polarization energy.
- (3)
Solvation not only reinforces the triel bond but affects its nature as well. The interaction strength of both B and Al complexes follows the same order: sp3 < sp2 < sp. The triel bond in the Al complex varies from an electrostatic interaction in the gas phase to a partially covalent interaction in solvent.
- (4)
The F substitution in the triel donor also strengths the triel bond and its enhancing effect is related to the number, position, and conformation of F substitution. The F substitution causes a prominent change in the configuration of the complex.
References
Schneider HJ (2009) Angew Chem Int Ed 48:3924–3977
Scheiner S (1997) Hydrogen bonding: a theoretical perspecitive. Oxford University Press, New York
Politzer P, Murray JS, Clark T (2010) Phys Chem Chem Phys 12:7748–7757
Politzer P, Murray JS, Clark T (2013) Phys Chem Chem Phys 15:11178–11189
Politzer P, Murray JS (2013) Chem Phys Chem 14:278–294
Guo X, Liu YW, Li QZ, Li WZ, Cheng JB (2015) Chem Phys Let 620:7–12
Wei YX, Li QZ, Scheiner S (2018) Chem Phys Chem 19:736–743
Grabowski SJ (2014) Chem Phys Chem 15:2985–2993
Xu HL, Cheng JB, Yang X, Liu ZB, Li WZ, Li QZ (2017) Chem Phys Chem 18:2442–2450
Dong WB, Yang X, Cheng JB, Li WZ, Li QZ (2018) J Fluorine Chem 207:38–44
Zierkiewicz W, Michalczyk M, Scheiner S (2018) Molecules 23:1416
Nziko PV, Scheiner S (2016) Phys Chem Chem Phys 18:3581–3590
Esrafili MD, Mohammadian-Sabet F (2016) Struct Chem 27:1157–1164
Bauzá A, Frontera A (2016) Chem Phys Chem 17:3181–3186
Egli M, Gessner RV (1995) Proc Natl Acad Sci USA 92:180–184
Hof F, Scofield DM, Schweizer WB, Diederich F (2004) Angew Chem Int Ed 43:5056–5059
Zhou P, Zou JW, Tian FF, Shang ZC (2009) J Chem Inf Model 49:2344–2355
Fischer FR, Wood PA, Allen FH, Diederich F (2008) Proc Natl Acad Sci USA 105:17290–17294
Kamer KJ, Choudhary A, Raines RT (2013) J Org Chem 78:2099–2103
Cormanich RA, Rittner R, O’Hagan D, Bühl M (2016) J Comput Chem 37:25–33
Grabowski SJ (2015) Chem Phys Chem 16:1470–1479
Leopold KR, Canagaratna M (1997) Acc Chem Res 30:57–64
Gao L, Zeng YL, Zhang XY, Meng LP (2016) J Comput Chem 37:1321–1327
Phillips JA, Giesen DJ, Wells NP, Halfen JA, Knutson CC, Wrass JP (2005) J Phys Chem A 36:8199–8208
Hase Y (2007) Spectrochim Acta A 68:734–738
Müller J, Ruschewitz U, Indris O, Hartwig H, Stahl W (1999) J Am Chem Soc 121:4647–4652
Phillips JA, Halfen JA, Wrass JP, Knutson CC, Cramer CJ (2006) Inorg Chem 45:722–731
Liu MX, Zhuo HY, Li QZ, Li WZ, Cheng JB (2016) J Mol Model 22:10
Esrafili MD, Mohammadian-Sabet F (2016) Mol Phys 114:1528–1538
Zhang JR, Wei YX, Li WZ, Cheng JB, Li QZ (2018) Appl Organometal Chem 32:e4367
Dong WB, Wang YQ, Yang X, Cheng JB, Li QZ (2018) J Mol Graphics Model 84:118–124
Zhang JR, Li WZ, Cheng JB, Liu ZB, Li QZ (2018) RSC Adv 8:26580–26588
Yourdkhani S, Korona T, Hadipour NL (2015) J Comput Chem 36:2412–2428
Grabowski SJ (2018) J Comput Chem 39:472–480
Bhunya S, Malakar T, Ganguly G, Paul A (2016) ACS Catal 6:7907–7934
Hamilton CW, Baker RT, Staubitz A, Manners I (2009) Chem Soc Rev 38:279–293
Grabowski SJ (2015) Molecules 20:11297–11316
Grabowski SJ (2017) Struct Chem 28:1163–1171
Jabłoński M (2018) J Comput Chem 39:1177–1191
Fiacco DL, Leopold KR (2003) J Phys Chem A 107:2808–2814
Bauzá A, Frontera A (2017) Theor Chem Acc 136:37
An XL, Liu HP, Li QZ, Gong BA, Cheng JB (2008) J Phys Chem A 112:5258–5263
Scheiner S, Grabowski SJ, Kar T (2001) J Phys Chem A 105:10607–10612
Alkorta I, Rozas I, Elguero J (2000) J Fluorine Chem 101:233–238
Zou JW, Jiang YJ, Guo M, Hu GX, Zhang B, Liu HC, Yu QC (2005) Chem Eur J 11:740–751
Li QZ, Jing B, Li R, Liu ZB, Li WZ, Luan F, Cheng JB, Gong BA, Sun JZ (2011) Phys Chem Chem Phys 13:2266–2271
Robertson CC, Wright JS, Carrington EJ, Perutz RN, Hunter CA, Brammer L (2017) Chem Sci 8:5392–5398
Shen D, Su PF, Wu W (2018) Phys Chem Chem Phys 20:26126–26139
Hansen AS, Du L, Kjaergaard HG (2014) Phys Chem Chem Phys 16:22882–22891
Riley KE, Murray JS, Fanfrlík J, Řezáč J, Solá RJ, Concha MC, Ramos FM, Politzer P (2011) J Mol Model 17:3309–3318
Boys SF, Bernardi FD (1970) Mol Phys 19:553–556
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Scalmani G, Cossi M, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene MLX, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VGDS, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Gonzalez C, Wong MW, Ittsburgh PA, Pople JA (2009) Gaussian 09, revision A02. Gaussian Inc, Wallingford.
Bader RFW (2000) AIM Program, v. 2.0, McMaster University, Hamilton, Canada
Bulat FA, Toro-Labbe A, Brinck T, Murray JS, Politzer P (2010) J Mol Model 16:1679–1691
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Su PF, Li H (2009) J Chem Phys 131:014102
Schmidt MW, Baldridge KK, Boalz JA, Elbert ST, Gorden MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993) J Comput Chem 14:1347–1363
SCM, ADF, Release 2008.01; Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, Available at: http://www.scm.com
Politzer P, Murray JS, Clark T, Resnati G (2017) Phys Chem Chem Phys 19:32166–32178
Batsanov SS (2001) Inorg Mater 37:871–885
Koch U, Popelier P (1995) J Phys Chem 99:9747–9754
Politzer P, Murray JS, Clark T (2015) J Mol Model 21:52
Politzer P, Murray JS (2017) Crystals 7:212
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, Z., Li, Y. Triel bonds in RZH2···NH3: hybridization, solvation, and substitution. J Mol Model 25, 219 (2019). https://doi.org/10.1007/s00894-019-4089-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-019-4089-1