
Abstract
The structures and energies of copper-doped small silicon clusters CuSi n (n = 4–10) and their anions were investigated systematically using CCSD(T)/aug-cc-pVTZ-DK//MP2/6-31G(2df,p), G4//MP2/6-31G(2df,p), and the B3LYP/6-311+G* basis set. The performance of the methods used for the prediction of energetic and thermodynamic properties was evaluated. Comparing experimental [Xu et al. (2012) J Chem Phys 136:104308] and theoretical calculations, it was concluded that the CCSD(T) results are very accurate and exhibit the best performance; the mean absolute deviation from experimental data was 0.043 eV. The excellent agreement of vertical detachment energy (VDE) between experimental results and CCSD(T) calculations indicates that the ground state structures of CuSi n − (n = 4–10) presented in this paper are reliable. For CuSi10, assigning 2.90±0.08 eV to the experimental adiabatic electron affinity (AEA) and 3.90±0.08 eV to the VDE is more reasonable than to 3.46±0.08 eV and 3.62±0.08 eV, respectively, based on the CCSD(T) calculations and the previous photoelectron spectrum of CuSi10 − (Xu et al., op. cit.). The AEAs of CuSi n (n = 4–10), excluding CuSi7, are in excellent agreement with experimental data, showing that the ground state structures of CuSi n (n = 4–6, 8–10) reported in this paper are reliable. CuSi10 is suggested to be the smallest endohedral ground state structure. However, adding an additional electron to CuSi10 pulls out the Cu atom from the center location, forming an exohedral ground state structure of CuSi10 −. The charge transfer and dissociation energy of Cu from CuSi n and their anions determined to examine the nature of bonding and their relative stabilities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Silicon-based clusters have been investigated both experimentally and theoretically because they enable not only miniaturization of electronic devices but also exert control over a wide variety of properties by changing size, shape, and composition [1–10]. In particular, doping a transition metal atom into silicon clusters can alter the structure of the silicon cage, reduce the chemical activity of the surface dangling bonds and enhance stability [11–20]. Materials of Cu-doped silicon possess excellent semiconductor properties so copper is a common trace impurity used in the manufacture of silicon devices as integrated circuits and solar cells. Copper plays key roles in the silicon-based semiconductor material. Exploring fundamental properties, such as the ground and low-lying electronic state of neutral and charged Cu-doped silicon clusters, charge transfer, electron affinities (EA), and dissociation energies (DE), can provide valuable information not only for designing and synthesizing of novel functional cluster-assembled materials but also for improving microelectronic devices.
A large number of experiment studies on transition-metal-doped silicon clusters have been presented since Beck [1] reported the observation of stable silicon cluster-metal atom compounds formed by a chemical reaction in a supersonic jet expansion. For copper-doped silicon cluster, Beck [21] studied the photoionization mass spectrum of CuSin with the technique of laser vaporization supersonic expansion and found that CuSi8 + and CuSi11 + clusters are relatively less stable than neighboring cluster CuSi6 +, CuSi7 +, CuSi9 +, and CuSi10 +. In the light of observations of laser photodissociation of mass-selected CuSi7 + and CuSi10 + at 355 nm, Duncan and co-workers [22] concluded that both CuSi7 + and CuSi10 + are exohedral structures and that Cu–Si bonding is weaker than Si–Si bonding. In terms of investigation of the reflection time-of-flight mass spectrometric stability of CuSi n +, Neukermans et al. [23] found that CuSi6 +, CuSi7 +, and CuSi10 + clusters are relatively abundant. Janssens et al. [24] carried out an experiment using argon physisorption as a structural probe for endohedrally doped silicon clusters and concluded that, starting from n = 12, the CuSi n + clusters are completely caged structures with the Cu atom stuffed in the interior site. Fielicke and coworkers [25, 26] tested the far-infrared vibrational spectra of CuSi n + (n = 6–11) employing an argon-tagging infrared multiple photon dissociation technique. Recently, Xu et al. [27] measured the EAs of CuSi n (n = 4–18) using photoelectron spectroscopy.
From a theoretical point of view, various density functional theories (DFT) can be used to study the structures and properties of small CuSi n clusters. For example, Dkhissi [28] studied the geometries and electronic properties of CuSi n (n = 1–4) employing OLYP, OPW91, OB95, VSXC, PBE0, and B3LYP, and evaluated the performance of these approaches. Hagelberg and coworkers [29–31] studied the geometric, energetic, electronic, and bonding properties of CuSi n (n = 1–6) with B3LYP, FNDMC (fixed-node diffusion Monte Carlo), and CASSCF methods. Facelli and coworkers [32] searched for the ground-state structure of CuSi n (n = 4,6,8,10,12) using MGA (modified genetic algorithms) followed by the B3PW91 scheme. For mid-sized CuSi n clusters, most theoretical calculations have been performed by means of the B3LYP scheme. For instance, in the range of n = 9–14, the geometries and properties of CuSi n were investigated by Lan et al. [33], Hossain et al. [34], He et al. [35, 36], Zdetsis et al. [37, 38], Gueorguiev et al. [39], Guo et al. [8], See et al. [40], and Hagelberg and coworkers [41–43]. Chuang et al. [44] investigated the electronic structures of CuSi n (n = 1–16) using first principles calculations. In addition to these, the geometries and properties of the anion CuSi n − (n = 1–18) were recently studied using B3LYP schemes [27, 45].
Although DFT methods are widely used, the quantitative performance of DFT in determining the energetic and thermodynamic properties of transition metal-containing species is not satisfactory. For instance, the best performing DFT method is the B97-1 functional, the mean absolute deviation of which from experimental data for the prediction of gas phase enthalpies of formation for a large set of 3d transition-metal-containing molecules is 7.2 kcal mol−1 [46]. Due to competing low-lying excited states, the existence of spin-orbit coupling, strong relativistic effects, and increased electron correlation, the systematic improvement of DFT methods to accurately predict the energetic properties of transition metal-containing species faces many difficulties [47].
The objective of the present study was to apply the G4 and CCSD(T) methods to the determination of the ground-state structures, EA, DE, relative stabilities, and growth patterns of neutral and anionic CuSin(0,−1) (n = 4–10) clusters. To facilitate comparison, the B3LYP scheme was also adopted. The performance of these methods for the prediction of energetic and thermodynamic properties was evaluated. In order to examine the effect of the doped Cu atom on the silicon clusters, the ground state structures of the bare silicon clusters were also calculated using identical methods.
Computational methods
The three different methods used were as follows: (1) Becke’s three-parameter [48] and Lee-Yang-Parr’s gradient-corrected correlation functional [49] (B3LYP) and 6-311+G* basis sets were used for structural optimization and frequency calculations. (2) The G4 [50] method was carried out to evaluate energies. In this method the equilibrium geometries were computed at the MP2/6-31G(2df,p) level instead of the B3LYP/6-31G(2df,p) in original G4 theory. The MP2/6-31G(2df,p) method was employed only for structural optimization. Frequency calculations were performed at the MP2/6-31G(d) level of theory. Note that the 6-31G(d) basis sets for Cu were modified by Mitin, Baker, and Pulay [51], and the “2df” polarization functions for the Cu atom are “2fg” basis sets, which are based on the modified 6-31G(d) basis sets as detailed in [50]. (3) The CCSD(T) schemes [52, 53] and aug-cc-pVTZ-DK basis sets [54] were used to perform single-point energy calculations at MP2/6-31G(2df,p) geometries. It is well known that relativistic effects are very important for heavy metals. Thus, the single-point energy computations use Douglas-Kroll basis sets and employ the spin-free, one-electron Douglas-Kroll-Hess Hamiltonian [55–58]. Before MP2/6-31G(2df,p) optimization, the harmonic frequency analysis at MP2/6-31G(d) level of theory was performed to guarantee that the optimized structures are local minima. The MP2/6-31G(d) frequencies were then scaled by a factor of 0.9434 [59] to give the zero-point vibrational energy (ZPVE) for the G4 and the CCSD(T) methods. All calculations were performed by means of the Gaussian 09 package [60].
To search for the lowest-energy structure, a large number of isomers need to be studied. Accordingly, in the optimization process of cluster structures, we considered a great number of isomers, which can be classified into the following four types: one is the “substitutional structure”, which can be regarded as being derived from the lowest-energy structure of Sin+1 (and/or Sin+1 −) by replacing a Si atom with a Cu atom. The second is the “attaching structure”, in which the Cu atom is bound to different positions on the surface, edge or apex of the lowest-energy structure of Si n (and/or Si n −). The third type is the “evolving structure”, in which the Si atom is bound to various positions on the surface, edge or apex of the lowest energetic structure of CuSi n (and/or CuSi n −). The remaining geometries were designed by us and are named the “fourth type”. Starting with these structures, we obtained as many of the refined low-lying structures as possible at the B3LYP/6-311+G* and G4 level. Then, we refined the energies of the selected low-energy isomers using CCSD(T) methods. In addition, the spin multiplicities of the singlet, doublet, triplet, and quartet state were taken into account for n ≤ 3 species because the ground states of Si and Si2 are triplet, and singlet and triplet compete with each other for the ground state of Si3. From the above description, we can conclude that the structures reported in this article represent true global minima.
Results and discussion
The total energies of the selected structures of CuSi n (n = 1–10) and their anions are listed in Tables S1–S8 in the Supplementary Material.
CuSi1-3 and their anions
The ground-state structures of neutral CuSi1-3 and their anions are shown in Fig. 1. The lowest-energy structure of CuSi (Fig. 1a) is predicted to be the 2Π ground state at the CCSD(T)/aug-cc-pVTZ-DK//MP2/6-31G(2df,p), the G4//MP2/6-31G(2df,p), and the B3LYP/6-311+G* level of theory. This result is the same as the previous theoretical and experimental study [61–67]. The frequencies at the MP2/6-31G(d) level are 345 cm−1 (scaled by 0.9434), which is in agreement with both experimental values of 360 ± 20 cm−1 [67] and 330 ± 15 cm−1 [68]. This also indicates that using the MP2/6-31G(d) frequency scaled by a factor of 0.9434 as the ZPVE correction to obtain the total energy at 0 K is reasonable. The B3LYP/6-311+G* frequencies (324 cm−1) agree well with the experimental value of 330±15 cm−1 [68], but are smaller than another experimental value by 36 cm−1. The MP2/6-31G(2df,p) bond lengths of 2.176 Å are in accord with the results of relativistic DK CASSCF/CASPT2 benchmark calculations of 2.199 Å [64]. The B3LYP/6-311+G* bond distances are 2.250 Å. We noted that an experimental value of 2.298 Å for the geometry of 2Σ state was presented in 1995 [69]. The lowest-energy structure of CuSi2 (Fig. 1b) is evaluated to be the 2A1 ground state. This result is in agreement with those of Dkhissi [28] and Xiao et al. [30], but in contradiction to the previous study [65], in which the lowest-energy geometry was calculated to be the 2B1 state. Energetically, the 2B1 state is less stable than the 2A1 state by 0.03, 0.21, and 0.15 eV at the B3LYP, the G4, and the CCSD(T) level, respectively. Neutral CuSi3 is predicted to have a 2B2 ground state (Fig. 1c) with a low-lying 2A1 excited state at the G4 and the CCSD(T) level. The energy differences between 2B2 and 2A1 state are 0.13 and 0.22 eV, respectively. At the B3LYP level, the structure of the 2B2 state is a saddle point on the potential energy surface due to having an imaginary b 2 frequency of 278 cm−1. Following the mode b 2, it collapses to a geometry with C s symmetry and 2A′ state, which is more stable in energy (by 0.18 eV) than that of the 2A1 state. All the results for CuSi3 are different from previous studies [28, 30] in which the ground-state structures predicted by OLYP, OPW91, OB95, VSXC, PBE0, and B3LYP schemes were the 2A1 state.

The ground-state structures of CuSi1-3 and their anions optimized at the MP2/6-31G(2df,p) level. Bond lengths in Ångstroms. Only silicon atoms are numbered
For the anion, the lowest-energy structures of CuSi− (Fig. 1d) were evaluated to be in the 3Σ− ground state at all three levels. This result is the same as that of previous theoretical studies [61, 63, 64, 66, 67]. The MP2/6-31G(2df,p) bond distance of 2.174 Å is in agreement with the CASSCF/CASPT2+DK values of 2.164 Å [64, 69]. The lowest-energy structures of CuSi2 − and CuSi3 − are an isosceles triangles (Fig. 1e) and rhombus geometry (Fig. 1f), respectively, with the 1A1 ground state, in agreement with a previous theoretical study by Li et al. [44]
CuSi4 and its anion
The geometries of neutral CuSi4 and their anions optimized with MP2/6-31G(2df,p) scheme are plotted in Fig. 2. Six isomers for CuSi4 are reported. Isomers 2a, 2b, 2c, and 2f can be regarded as attaching a Cu atom to the surface, edge, and apex of the ground-state rhombic structure of Si4 [2, 70–73], respectively. The C 2v -symmetry 2a isomer of the 2B2 state is predicted to be the ground-state structure at the G4 and the CCSD(T) levels. Energetically, it is more stable than 2b by 0.15 and 0.06 eV, than 2c by 0.38 and 0.20 eV, and than 2f by 0.70 and 0.54 eV at the G4 and the CCSD(T) levels, respectively. The C s -symmetry isomer 2d of the 2A″ state can be regarded as being derived not only from the trigonal bipyramid of Si5 [70–73] by replacing an axial Si atom with a Cu atom but also from the ground-state tetrahedral structure of CuSi3 by facing-capping with a Cu atom. It is less stable than that of 2a by 0.26 and 0.21 eV in energy at the G4 and CCSD(T) levels, respectively. The C s-symmetry isomer 2e can be regarded as attaching a Cu atom to an edge of the ground-state tetrahedral structure CuSi3. It is less stable than the 2a structure by 0.17 and 0.27 eV in energy at the G4 and the CCSD(T) levels, respectively.

The geometries of CuSi4 and their anions optimized at the MP2/6-31G(2df,p) level. Bond lengths in Ångstroms. Only silicon atoms are numbered
At the B3LYP level, the C 2v -symmetry structure 2a is a saddle point due to having an imaginary b 2 frequency of 56 cm−1. It undergoes Jahn-Teller distortion to give a C s -symmetry isomer of the 2A′ state, which was predicted to be a ground-state structure in previous studies [29–31]. However, it, 2b, and 2c are nearly isoenergetic, namely the Cu atom moves almost freely on the rhombus Si4 frame.
Five minima for anion CuSi4 − are presented. The C s symmetry 2g isomer of the 1A′ state, corresponding to neutral 2d, is predicted to be the ground state structure at the G4 and CCSD(T) levels. This result is the same as the recent experimental study conducted by Xu et al. [27]. Geometries 2h (corresponding to neutral 2b and 2c) and 2k (corresponding to neutral 2f) are also in C s symmetry with 1A′ state. Energetically, they are higher than that of 2g by 0.16 and 0.13 eV and 0.60 and 0.43 eV at the G4 and the CCSD(T) levels, respectively. The C 1-symmetry 2i isomer, corresponding to neutral 2e, is less stable than the ground-state structure 2g by 0.18 and 0.24 eV at the G4 and the CCSD(T) levels, respectively. When the geometry of neutral 2a obtains an additional electron, we can obtain a C 2v -symmetry anionic isomer with the 1A1 electronic state. However, it is unstable due to having imaginaries. It undergoes Jahn–Teller distortion to give the C 2-symmetry 2j isomer of 1A state. Energetically, it is less stable than the 2g by 0.35 eV at the G4 and the CCSD(T) levels.
At the B3LYP level, isomer 2h is more stable than the 2g by 0.16 eV as reported previously [27, 41, 44]. The isomer 2j is unstable because of having an imaginary b frequency of 45 cm−1. Following the mode b, it collapses finally to isomer 2h. In short, B3LYP results differ from these of G4 and CCSD(T) levels.
CuSi5 and its anion
The geometries of neutral CuSi5 and their anions optimized with the MP2/6-31G(2df,p) scheme are plotted in Fig. 3. Two isomers for neutral are reported. One (3a) has C 2v symmetry with 2B1 ground state at the G4 and the CCSD(T) levels, which can be regarded as being derived not only from the ground state trigonal bipyramid of Si5 [70–73] by face capping with a Cu atom (“attaching structure”) but also from the ground state tetragonal bipyramid of Si6 [2, 70–73] by replacing a Si atom with a Cu atom (“substitutional structure”). And another (3b) is C s symmetry with 2A′ state and belongs to the “attaching structure”. Energetically, it is less stable than the ground state structure 3a by 1.34 and 0.94 eV at the G4 and the CCSD(T) levels, respectively. At the B3LYP level, the C 2v symmetry structure 3a of 2B1 is unstable due to having an imaginary b 1 frequency of 45 cm−1. It undergoes Jahn-Teller distortion to give the C s-symmetry isomer of 2A′ state. This result is the same as the outcome reported previously by Xiao et al. [30].

The geometries of CuSi5 and their anions optimized at the MP2/6-31G(2df,p) level. Bond lengths in Ångstroms. Only silicon atoms are numbered
For negatively charged ions, two isomers are also presented. Adding an additional electron to the neutral 3a structure, we can obtain a C 2v -symmetry anionic isomer with 1A1 state. It is unstable due to having an imaginary b 1 frequency. Following the mode b 1, it collapses to the isomer (3c) with C s symmetry and 1A′ ground state. The C 3v -symmetry geometry 3d of 1A1 state is higher in energy than the 3c structure by 0.52, 1.13, and 0.87 eV at the B3LYP, the G4, and the CCSD(T) levels, respectively.
CuSi6 and its anion
The geometries of neutral CuSi6 and their anions optimized with MP2/6-31G(2df,p) scheme are shown in Fig. 4. Six minima for neutral are presented. The C 2v -symmetry 4a isomer of 2B2 state is predicted to be the ground state at the G4 and the CCSD(T) levels, which can be regarded as being derived from the ground-state pentagonal bipyramid of Si7 [70–73] by replacing a horizontal Si atom with a Cu atom. Isomers 4b, 4c, 4e and 4f can be regarded as attaching a Cu atom to the surface, edge or apex of the lowest-energy structure of Si6 (for Si6, tetragonal bipyramid, face-capped trigonal bipyramid, and edge-capped trigonal bipyramid compete with each other for the ground-state structure (see refs. [70–73]). They possess C s symmetry with the exception of the 4f isomer, which have C 4v symmetry. The electronic state is 2A′′, 2A′, 2A′ and 2A1, respectively. Energetically, they are less stable than the ground state 4a by 0.16 (and 0.30), 0.25 (and 0.50), 0.57 (and 0.85), and 0.85 (and 1.24) eV at the CCSD(T) (and G4) level, respectively. The C s symmetry isomer 4d of 2A′ state is an “evolving structure”, namely it can be regarded as being derived from the ground state of CuSi5 by attaching a Si atom. It is less stable in energy than ground state 4a by 0.33 and 0.27 eV at the G4 and the CCSD(T) levels, respectively.

The geometries of CuSi6 and their anions optimized at the MP2/6-31G(2df,p) level. Bond lengths in Ångstroms. Only silicon atoms are numbered
Previous studies have shown that isomers 4a and 4b compete with each other for the ground state structure [29–32, 41]. At present, isomers 4a, 4b, 4c and 4d compete with each other for the ground state structure at the B3LYP level. Isomers 4b and 4c are more stable than 4a by 0.08 and 0.06 eV, respectively, and isomer 4d is less stable than 4a by 0.06 eV in energy at the B3LYP level. It is obvious that the B3LYP results differ from those of G4 and CCSD(T).
Five minima for anion CuSi6 − are presented. The C 2v symmetry geometry 4g of 1A1 state, corresponding to neutral 4a, is predicted to be the ground state structure at the G4 and CCSD(T) levels. The C s symmetry isomer 4h of 1A′ state, C 4v symmetry isomer 4i of 1A1 state, and C 3v symmetry isomer 4k of 1A1 state (corresponding to neutral 4e) is less stable than that of 4g by 0.30 (and 0.51), 0.38 (and 0.78), and 0.64 (and 1.03) eV in energy at the CCSD(T) (and the G4) levels of theory, respectively. The C s symmetry isomer 4j of 1A′ state, corresponding neutral 4b and/or 4d, is less stable in energy than that of 4g by 0.41 and 0.41 eV at the G4 and the CCSD(T) levels, respectively.
As noted in a previous study [27], many isomers for anion CuSi6 − are degenerate in energy at the B3LYP level. For example, isomer 4a is less stable than structures 4b and 4c by only 0.04 and 0.01 eV, respectively, and more stable than the isomer 4d by only 0.07 eV.
CuSi7 and its anion
The geometries of neutral CuSi7 clusters and their anions optimized with the MP2/6-31G(2df,p) scheme are shown in Fig. 5. Five minima for neutral clusters are presented. Both C 2v symmetry 5a isomer of 2B1 state and C 5v symmetry 5c isomer of 2A1 state can be regarded as attaching a Cu atom to the edge and vertex of the ground state pentagonal bipyramid of Si7 [70–73], respectively. Energetically, the 5a structure is more stable than that of 5c by 0.37 and 0.32 eV at the G4 and the CCSD(T) level, respectively. The C s symmetry structure 5b of 2A′ state, analogous to the ground state structure of BeSi7 [5], belongs to a “fourth type”. At the CCSD(T) level, it is less stable in energy than that of 5a by 0.22 eV, but more stable by 0.18 eV at the G4 level. The C s symmetry structure 5d of 2A′ can be regarded as being derived from the ground state distorted bicapped octahedron of Si8 [71, 72, 74, 75] by replacing a Si atom with a Cu atom. It is less stable than that of 5a by 0.16 and 0.45 eV in energy at the G4 and the CCSD(T) levels, respectively. The C s symmetry structure 5e of 2A′ state is a “fourth type”. It is less stable in energy than that of 5a by 0.48 eV at the CCSD(T) level, but more stable by 0.12 eV at the G4 level.

The geometries of CuSi7 and their anions optimized at the MP2/6-31G(2df,p) level. Bond lengths in Ångstroms. Only silicon atoms are numbered
At the B3LYP level, the lowest-energy structure is predicted to be 5a geometry. It is more stable in energy than the 5b, 5c, 5d, and 5e by 0.13, 0.12, 0.20, and 0.24 eV, respectively. The B3LYP result is similar to that of the CCSD(T), and different from that of the G4.
Five minima for anion CuSi7 − are reported. The C s symmetry structure 5f of 1A′ state is predicted to be the ground state. It corresponds to neutral 5e. That is, the ground state geometry of anion CuSi7 − differs from its neutral ground state geometry. The isomers 5g (corresponding to neutral 5b) and 5h (corresponding to neutral 5d) also share C s symmetry with 1A′ state. Energetically, they are less stable than isomer 5f by 0.15 (0.35) and 0.15 (0.46) eV at the CCSD(T) (the G4) levels, respectively. The C 5v symmetry isomer 5i of 1A1 state is less stable than 5f by 0.21 and 0.90 eV in energy at the CCSD(T) and the G4 levels, respectively. The C 2v symmetry isomer 5j of 1A1 state is less stable than 5f by 0.54 and 1.23 eV in energy at the CCSD(T) and the G4 levels, respectively. The energetic order of G4 is the same as the result of the CCSD(T) for negatively charged ion CuSi7 −.
At the B3LYP level, isomers 5f and 5i are nearly degenerate in energy. The 5f structure is more stable in energy than 5i by only 0.01 eV. The C 5v symmetry isomer 5i cannot be the ground-state structure of anion CuSi7 − because its simulated DOS spectrum does not match the experimental spectrum [27]. The isomers 5g, 5h, and 5j are less stable in energy than the 5f by 0.26, 0.13, and 0.54 eV at the B3LYP level, respectively.
CuSi8 and its anion
The geometries of neutral CuSi8 and their anions optimized with the MP2/6-31G(2df,p) scheme are shown in Fig. 6. Five minima for neutral are presented. The ground state structure of neutral CuSi8 is predicted to be the 6a geometry with C 1 symmetry, which can be regarded as replacing a Si atom of the ground state bicapped pentagonal bipyramid of Si9 [72, 75] with a Cu atom. The C s symmetry 6b of 2A'' state can be regarded as attaching a Si atom to the ground state structure of CuSi7, namely an “evolving structure”. The C s symmetry 6c of 2A'' state can be regarded as attaching a Cu atom to the edge of the ground state distorted bicapped octahedron of Si8 [71, 72, 74, 75]. The C s symmetry 6d of 2A′ state can be viewed as attaching a Si atom to the ground state structure of anion CuSi7 −. The D 2h symmetry 6e Cu-body-centered cubic structure of 2B1u state, similar to the ground state structure of BeSi8 [5], belongs to the “fourth type”. Energetically, the 6a structures are more stable than those of 6b, 6c, 6d, and 6e by 0.39 (0.28), 0.41 (0.82), 0.75 (0.42), and 1.66 (0.84) eV at the CCSD(T) (the G4) level, respectively.

The geometries of CuSi8 and their anions optimized at the MP2/6-31G(2df,p) level. Only silicon atoms are numbered
At the B3LYP level, isomers 6a and 6c are nearly degenerate in energy. The 6a structure is more stable in energy than that of 6c by only 0.01 eV. Isomers 6b and 6d are less stable in energy than 6a by 0.26 and 0.57 eV, respectively. The endohedral 6e at the B3LYP level is a saddle point on the potential surface due to having imaginary frequency. It undergoes Jahn-Teller distortion to finally yield 6a geometry.
Five minima for anion CuSi8 − are reported. The C s symmetry structure 6f of 1A′ state, corresponding to neutral 6a, is predicted to be the ground state. The isomers 6g (corresponding to neutral 6c), 6h (corresponding to neutral 6d), and 6i (corresponding to neutral 6b) are also C s symmetry with 1A′ state. Energetically, these isomers are less stable than that of 6f by 0.32 (0.83), 0.48 (0.23), and 0.82 (0.68) eV at the CCSD(T) (the G4) level, respectively. The endohedral octahedron 6j with O h symmetry and 1A1g state is less stable than that of 5f by 1.13 and 0.30 eV in energy at the CCSD(T) and the G4 levels, respectively.
At the B3LYP level, the “attaching structure” 6g is calculated to be the lowest-energy geometry as previously reported by Xu et al. [27]. It is more stable than the structures of 6f, 6h, and 6j by 0.08, 0.48, and 1.52 eV in energy, respectively. The 6i isomer at the B3LYP level is a saddle point on the potential surface due to having an imaginary a'' frequency of 64 cm−1. Following the mode a'', it collapses down to 6f geometry.
CuSi9 and its anion
The structures of neutral CuSi9 and their anions optimized with MP2/6-31G(2df,p) scheme are shown in Fig. 7. Three minima for neutral are reported. The C s symmetry 7a of 2A′ state can be regarded as being derived from the ground state tetracapped trigonal prismatic structure of Si10 [72, 74, 75] by replacing a Si atom with a Cu atom. The C s symmetry 7b structure of 2A′ state, Cu atom semi-encapsulated in distorted tricapped trigonal prism of Si9, is predicted to be the ground-state structure. The C 2 symmetry 7c isomer of 2A state is an encapsulated structure. At the CCSD(T) level, the 7a structures are more stable than the 7b and 7c isomers by 0.38 and 0.80 eV in energy, respectively. At the G4 level, they are nearly degenerate in energy. The 7a structure is less stable than the 7b isomer by only 0.03 eV, and more stable than the 7c isomer by 0.07 eV. At the B3LYP level, the energetic orders are the same as those predicted with the CCSD(T) scheme. The 7a structures are more stable than the 7b and 7c isomers by 0.45 and 0.63 eV, respectively.

The geometries of CuSi9 and their anions optimized at the MP2/6-31G(2df,p) level. Only silicon atoms are numbered
For these anions, three minima are also reported. At the CCSD(T) and the B3LYP level, the C s symmetry 7d structure of 1A′ state is predicted to be the ground state. Energetically, it is more stable than that of the 7e isomer with C 3v symmetry and 1A1 state by 0.05 and 0.23 eV, and than that of the 7f isomer with C s symmetry and 1A′ state by 0.61 and 0.74 eV, respectively. The G4 results differ from those of the CCSD(T) and B3LYP levels . At the G4 level, the 7d structure is less stable than the 7e and 7f isomer by 0.55 and 0.32 eV in energy, respectively. It is noted that our B3LYP results differ from the B3LYP outcomes reported previously [27, 33], in which the ground state structures were evaluated to be 7e and 7f isomers, respectively.
CuSi10 and its anion
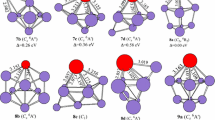
The structures of neutral CuSi10 clusters and their anions are shown in Fig. 8. Earlier experimental work [21] suggests relatively high stabilities for CuSi10 but low stabilities for CuSi n with n greater than or less than 10. The most likely ground state structure of CuSi10 is thus a cage-like structure with a Cu atom stuffed into the interior site. King [76] employed topological models and demonstrated that the cage-like structure is a dual polyhedron of a square antiprism type. Previous ab initio computations on CuSi10 clusters [32–34, 37, 41] showed that the ground state structure is not a dual polyhedron with a square antiprism, and is not even an endohedral structure. Our results are similar to those reported by King [76]. Two isomers for neutral are reported. The D 4d symmetry 8a structure, with a Cu atom encapsulated in a bicapped antitetragonal prism of Si10, is predicted to be the 2A1 ground state. The C s symmetry 8b geometry of 2A′ state is an exohedral isomer, which can be regarded as being derived from the lowest energy structure of Si11 [77, 78] by replacing a Si atom with a Cu atom. Energetically, it is less stable than that of 8a by 0.37 and 1.71 eV at the CCSD(T) level and the G4 level, respectively, but more stable by 0.30 eV at the B3LYP level.

The isomers of CuSi10 clusters and their anions obtained at the MP2/6-31G(2df,p) level. Only silicon atoms are numbered
For negatively charged ions, two isomers are also presented. The exohedral 8c geometry with C s symmetry and 1A′ state, corresponding to neutral 8b, is predicted to be the ground state. It is more stable in energy than the endohedral 8d isomer with D 4d symmetry and 1A1 electronic state by 0.11 and 0.66 eV at the CCSD(T) and the B3LYP levels, respectively, but more stable by 0.95 eV at the G4 level. It should be noted that, when adding an additional electron to the ground state structure of neutral CuSi10, the Cu atom in the center is pulled out by this additional electron and then the exohedral ground state structure of anion CuSi10 − is formed. It is observed that the exohedral 8c structures are different from those reported by Xu et al. [27]. At the B3LYP level, the 8c structure and the isomer reported by Xu et al. [27] is nearly degenerate in energy; the 8c structures are less stable than those reported by Xu et al. [27] by only 0.08 eV. However, using the structure reported by Xu et al. [27] as an optimized initial geometry, the 8d structure is finally obtained with the MP2/6-31G(2df,p) method. That is to say that the MP2 method tends to form endohedral structures, while the density functional method tends to generate exohedral geometry.
From the aforementioned discussion, we conclude that (1) the ground state encapsulating structures of neutral CuSi n starts with n = 10, while the ground state structure of anion CuSi10 − is exohedral in structure, which is different from its neutral structure. In addition to CuSi10 −, the ground state structures of CuSi4 − and CuSi7 − are also different from their neutral ground state structures. (2) The ground state structures evaluated by the G4 scheme are the same as the results of CCSD(T) with n ≤ 6. The energetic order of the G4 is also, by and large, similar to that of CCSD(T) for CuSi n with n ≤ 6. However, starting from n = 7, the ground state structures and energetic orders predicted by the G4 scheme differ from those predicted by the CCSD(T). This indicates that the G4 scheme may be suitable only for small clusters. Our initial idea was firstly to use the G4 method to optimize geometries and determine the ground state of CuSi n and their anions. And then to use the CCSD(T)/aug-cc-pVTZ-DK schemes to perform calculations of single-point energies and properties of ground state structures in order to save computation time [it is well known that the computation time required for CCSD(T) is expensive]. Unfortunately, this idea is wrong. (3) At the B3LYP, many isomers are sometimes nearly degenerate in energy. As a result, it is difficult to determine the ground state structures. On the other hand, the B3LYP method tends to form an exohedral structure, while the ground state structures of transition-metal-doped silicon clusters tend to become cage-like structures.
Electron affinities
The adiabatic electron affinities (AEAs) [defined as the difference of total energies in the manner AEA = E(optimized neutral) – E(optimized anion)] and vertical detachment energies (VDEs) (defined as the difference of total energies in the manner VDE = [E(neutral at optimized anion geometry) – E(optimized anion)] of CuSin clusters are calculated at the CCSD(T) level. These values are listed in Table 1 along with the experimental values. From Table 1, we can conclude that the theoretical VDEs predicted by the CCSD(T)/aug-cc-pvTZ-DK scheme are in excellent agreement with experimental values (taken from [27]). The average absolute deviations from experiment are only about 0.043 eV. The largest deviations are 0.12 eV. This also indicates that the ground state structures of CuSi n − (n = 4–10) presented in this paper may be correct. The theoretical AEAs of CuSi n (n = 4–10) with the exception of n = 7 are also in excellent agreement with experimental values. The average absolute deviations from experiment are about 0.042 eV, and the largest errors are around 0.08 eV. This indicates that the ground state structures of CuSi n (n = 4–10) excluding n = 7 reported in this paper may be correct. Although anion photoelectron spectroscopy is a powerful experimental technique for measuring AEAs and, in principle, is the most accurate method to determine the AEAs of the corresponding neutral species, it is difficult to determine accurate AEAs if the recorded spectroscopy shows a featureless long and very rounded tail, with no clear onset. The spectroscopy of CuSi10 − is one such example as described by the authors themselves [27]. Consequently, theoretical calculations are necessary to help determine accurate AEAs. In the light of the CCSD(T) calculation, the previous photoelectron spectrum (taken from [27]) of CuSi10 − was reassigned. The experimental AEAs and VDEs of CuSi10 were measured to be 2.90±0.08 eV and 3.90±0.08 eV, respectively, rather than 3.46±0.08 eV and 3.62±0.08 eV [27]. For CuSi7, the theoretical AEA is predicted to be 2.64 eV, which is smaller than the experimental value by 0.26 eV. It is well known that the photoelectron spectroscopy of the anion represents the transitions from the ground state of the anion to the ground or excited states of their corresponding neutral. If a transition from the ground state of CuSi7 − (5f structure) to the 5b structure of CuSi7 occurs, the theoretical AEA with ZPVE correction is calculated to be 2.90 eV, which is equal to the experimental value [27]. That is, the photoelectron spectrum favors the 5b structure as the ground state, while the CCSD(T) calculation favors the 5a structure as the ground state. In this case, we cannot confirm if the ground state structure of CuSi7 is 5a or 5b. Further experimental studies such as determining Raman or infrared spectra would be necessary to determine the ground state structure of CuSi7.
Dissociation energies
The dissociation energies (DEs; defined as the energy required in the reaction CuSi n →Cu+Si n for CuSi n and CuSi n −→Cu+Si n − for CuSi n −) of CuSi n and their anions were evaluated and are listed in Table 2. From the DEs, the stability of Cu atom bonding to silicon clusters can be found. The higher values of these DEs indicate that the cluster bonding of a Cu atom is stable. A better way of comparing the local relative stability of various size clusters is by means of the incremental binding energies. To facilitate comparison, Fig. 9 sketches the DEs of the Cu atom from the ground-state structures of CuSi n and their anions as a function of cluster size. From Fig. 9, we can see that (1) the CuSi n clusters where n = 4, 7, and 9 are less stable than for n = 2, 5, and 8 because the DEs are local minima for n = 4, 7, and 9 and local maxima for n = 2, 5, and 8. (2) The CuSi n − anion with n = 4 and 7 is less stable than with n = 2, 5, 8, and 9. (3) The DEs of neutral clusters are smaller than those of their anions. This can be readily explained. Neutral CuSi n clusters possess an open shell electronic structure. When they gain an electron, the electronic structures change to a closed shell (except for CuSi n − species—its ground state is triplet), where the electronic repulsions are minimized according to the Pauli exclusion principle [79]. Especially for n = 9, the relative stability changes from poorest to best. The maximum AEA of CuSi9 among all these clusters (see Table 2) also illustrates this point of view.

Dissociation energy (eV) with ZPVE corrections for the reaction CuSi n →Cu+Si n and CuSi n −→Cu+Si n − versus the number of atoms n for Cu-doped silicon clusters
Charge transfer
To further understand the interaction between silicon clusters and the Cu atom, natural population analysis (NPA) were performed with the MP2(full)/GTlarge//MP2/6-31G(2df,p) method. The NPA valence configurations and charges on the Cu atoms are listed in Table 3. From Table 3, we can see that (1) the valence configuration is 3d9.58–9.714s0.43–0.654p0.20–0.884d0.13–0.14 for Cu in CuSi n species with n = 2–9. Obviously, the 3d shell (and 4d shell) of Cu in the clusters is nearly unchanged [the configuration of the free Cu atom is (core)4s1.013d9.824p0.034d0.14 at the MP2(full)/GTlarge//MP2/6-31G(2df,p) level]. The charge transfer takes place mainly from 4s to 4p orbitals, leading to strong hybridization between 4s and 4p orbitals. (2) The calculated charges of the Cu atom in CuSi5, CuSi6 and CuSi8 species are nearly zero, indicating no charge transfer from Cu to the silicon clusters. Thus, the bonding in CuSi5, CuSi6 and CuSi8 species is covalent in nature. In CuSi2, CuSi3, CuSi4, CuSi7 and CuSi9, the Cu atom has a charge of 0.22, 0.30, 0.30, 0.47, and −0.36 a.u., respectively, indicating mixed ionic and covalent bonding between Cu atom and silicon clusters. It is interesting to note that, in the CuSi9 cluster, the charge transfers from Si atoms to the Cu atom, which indicates that the Cu atom acts as an electron acceptor, not a donor. On the other hand, the ground state structure of CuSi9 is exohedral structure. Both result in the poor relative stability of CuSi9. This result is consistent with that of DE analysis. (3) The valence configuration of Cu in CuSi is very close to the configuration of the free Cu atom, namely charge transfer occurs not only between Cu 4s and Cu 4p, but also between the Cu atom and the Si atom, i.e., there is weak interaction between the Cu and Si atoms in CuSi species. (4) The calculated charge of the Cu atom in the CuSi10 cluster is −2.81 a.u. Thus, the bonding in CuSi10 is ionic in nature although charge transfers from Si s and p orbitals to Cu 4p, 4d and/or 4f orbitals; thus, the Cu atom acts as an electron acceptor. The cage-like framework stabilizes the ground state structure of CuSi10. (5) In the case of anion CuSi n −clusters (n = 1–10) with the exception of n = 7, the majority of the extra charge on the electron was found to be localized to silicon clusters, especially for CuSi10 −, in which the charge of the Cu atom is positive. Compared with neutral CuSi9, not only is the extra electron’s charge localized in the silicon clusters, but also the electron back-donation from the Cu atom to the silicon cluster is induced and strengthens the bond between Cu and the silicon cluster. In CuSi7 −, the extra electron’s charge is localized on the Cu 4p orbitals.
Conclusions
The structures and energies of copper-doped small silicon clusters CuSi n (n = 4–10) and their anions were studied systematically by means of the CCSD(T)/aug-cc-pVTZ-DK//MP2/6-31G(2df,p), the G4//MP2/6-31G(2df,p), and the B3LYP/6-311+G* methods. The results revealed that (1) starting from n = 10, the Cu atom in CuSi n falls completely into the center of the Si outer frame, forming a metal-encapsulated ground state structure, but not for the corresponding anion. When adding an additional electron to the ground state structure of neutral CuSi10, the Cu atom in the center location is pulled out by this additional electron, forming the exohedral ground state structure of anion CuSi10 −. (2) The ground state structure and energy difference predicted by the G4 and the B3LYP method is different from that of the CCSD(T) scheme. Both the energetics and the structural features calculated by the G4 and the B3LYP approaches are unreliable for transition-metal-doped silicon clusters. (3) The reliable VDEs of CuSi n are predicted to be 2.65 eV for CuSi4, 3.18 eV for CuSi5, 2.65 eV for CuSi6, 3.27 eV for CuSi7, 3.18 eV for CuSi8, 3.56 eV for CuSi9, and 3.82 eV for CuSi10. They are in excellent agreement with experimental data taken from [27]. The average absolute deviations from experimental data are only 0.043 eV. The excellent agreement between the previously measured photoelectron spectroscopy and the CCSD(T) theoretical simulations indicate that the ground state structures of CuSi n − (n = 4–10) presented in this paper are reliable. (4) In light of the CCSD(T) calculation and the previous photoelectron spectrum of CuSi10 −, assigning 2.90±0.08 eV and 3.90±0.08 eV to the experimental AEAs and VDEs is more reasonable than to 3.46±0.08 eV and 3.62±0.08 eV, respectively (see [27]). The AEAs of CuSi n were evaluated to be 2.38 eV for CuSi4, 2.83 eV for CuSi5, 2.51 eV for CuSi6, 2.64 eV for CuSi7, 3.11 eV for CuSi8, 3.21 eV for CuSi9, and 2.90 eV for CuSi10. With the exception of CuSi7, these are in excellent agreement with experimental data [27]. The average absolute deviations from experimental data are only 0.042 eV. The excellent agreement between experimental data and CCSD(T) theoretical values show that the ground state structures of CuSi n (n = 4–6,8–10) presented in this paper are reliable. (5) From the DEs of Cu from CuSi n clusters and their anions and the analysis of charge transfer, we can reasonably infer that CuSi n clusters with n = 4, 7, and 9 are less stable than with n = 2, 5, and 8, and anion CuSi n − with n = 4 and 7 are less stable than with n = 2, 5, 8, and 9.
References
Beck SM (1987) J Chem Phys 87:4233–4234
Honea EC, Ogura A, Peale DR, Félix C, Murray CA, Raghavachari K, Sprenger WO, Jarrold MF, Brown WL (1999) J Chem Phys 110:12161–12172
Guo LJ, Zhao GF, Gu YZ, Liu X, Zeng Z (2008) Phys Rev B 77:195417-1–195417-8
Koyasu K, Atobe J, Furuse S, Nakajima A (2008) J Chem Phys 129:214301-1–214301-7
Fan HW, Yang JC, Lu W, Ning HM, Zhang QC (2010) J Phys Chem A 114:1218–1223
Koyasu K, Atobe J, Akutsu M, Mitsui M, Nakajima A (2007) J Phys Chem A 111:42–49
Hiura H, Miyazaki T, Kanayama T (2001) Phys Rev Lett 86:1733–1736
Han JG, Hagelberg F (2009) J Comput Theor Nanosci 6:257–269
Wang J, Ma QM, Xu RP, Liu Y, Li YC (2009) Phys Lett A 373:2869–2875
Lu J, Yang JC, Xing ZF, Ning HM (2014) J Theor Comput Chem 13:1450038-1–1450038-24
Lu J, Yang JC, Kang YL, Ning HM (2014) J Mol Model 20:2114-1–2114-12
Guo LJ, Liu X, Zhao GF (2007) Chem Phys 126:234704-1–234704-7
Xu HG, Zhang ZG, Feng Y, Yuan JY, Zhao YC, Zheng WJ (2010) Chem Phys Lett 487:204–208
Kong XY, Xu HG, Zheng WJ (2012) J Chem Phys 137:064307-1–064307-9
Li JR, Wang GH, Yao CH, Mu YW, Wan JG, Han M (2009) J Chem Phys 130:164514-1–164514-9
Khanna SN, Rao BK, Jena P, Nayak SK (2003) Chem Phys Lett 373:433–438
Ma L, Zhao JJ, Wang JG, Lu QL, Zhu LZ, Wang GH (2005) Chem Phys Lett 411:279–284
Ren ZY, Li F, Guo P, Han JG (2005) J Mol Struct: THEOCHEM 718:165–173
Kumar V, Kawazoe Y (2001) Phys Rev Lett 87:045503-1–045503-4
Kumar V, Kawazoe Y (2002) Phys Rev B 65:073404-1–073404-4
Steven MB (1989) J Chem Phys 90:6306–6312
Jaeger JB, Jaeger TD, Duncan MA (2006) J Phys Chem A 110:9310–9314
Neukermans S, Wang X, Veldeman N, Janssens E, Silverans RE, Lievens P (2006) Int J Mass Spectrom 252:145–150
Janssens E, Gruene P, Meijer G, Woste L, Lievens P, Fielicke A (2007) Phys Rev Lett 99:063401-1–063401-4
Gruene P, Fielicke A, Meijer G, Janssens E, Ngan VT, Nguyen MT, Lievens P (2008) Chem Phys Chem 9:703–706
Ngan VT, Gruene P, Claes P, Janssens E, Fielicke A, Nguyen MT, Lievens P (2010) J Am Chem Soc 132:15589–15602
Xu HG, Wu MM, Zhang ZG, Yuan JY, Sun Q, Zheng WJ (2012) J Chem Phys 136:104308-1–104308-10
Dkhissi A (2008) Int J Quantum Chem 108:996–1003
Ovcharenko IV, Lester WA Jr, Xiao C, Hagelberg F (2001) J Chem Phys 114:9028–9032
Xiao CY, Abraham A, Quinn R, Hagelberg F, Lester WA Jr (2002) J Phys Chem A 106:11380–11393
Xiao C, Hagelberg F (2000) J Mol Struct: THEOCHEM 529:241–257
Ona O, Bazterra VE, Caputo MC, Ferraro MB, Fuentealba P, Facelli JC (2004) J Mol Struct: THEOCHEM 681:149–155
Lan YZ, Feng YL (2009) Phys Rev A 79:033201-1–033201-9
Hossain D, Pittman CU, Gwaltney SR (2008) Chem Phys Lett 451:93–97
He JG, Wu KC, Sa RJ, Li QH, Wei YQ (2010) Chem Phys Lett 490:132–137
He JG, Wu KC, Liu CP, Sa RJ (2009) Chem Phys Lett 483:30–34
Zdetsis AD (2007) Phys Rev B 75:085409-1–085409-10
Zdetsis AD, Koukaras EN, Garoufalis CS (2009) J Math Chem 46:971–980
Gueorguiev GK, Pacheco JM, Stafstrom S, Hultman L (2006) Thin Solid Films 515:1192–1196
Sen P, Mitas L (2003) Phys Rev B 68:1554041-1–1554041-4
Xiao CY, Hagelberg F, Lester WA (2002) Phys Rev B 66:075425-1–075425-23
Xiao C, Hagelberg F, Ovcharenko I, Lester WA Jr (2001) J Mol Struct: THEOCHEM 549:181–192
Hagelberg F, Yanov I, Leszczynski J (1999) J Mol Struct: THEOCHEM 487:183–192
Chuang FC, Hsu CC, Hsieh YY, Albao MA (2010) Chin J Phys 48:82–102
Li GL, Ma WL, Gao AM, Chen HY, Finlow D, Li QS (2012) J Theor Comput Chem 11:185–196
Jiang WY, Laury ML, Powell M, Wilson AK (2012) J Chem Theor Comput 8:4102–4111
Jiang WY, DeYonker NJ, Determan JJ, Wilson AK (2012) J Phys Chem A 116:870–885
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Mayhall NJ, Raghavachari K, Redfern PC, Curtiss LA (2009) J Phys Chem A 113:5170–5175
Mitin AV, Baker J, Pulay P (2003) J Chem Phys 118:7775–7782
Raghavachari K, Trucks GW, Pople JA, Gordon MH (1989) Chem Phys Lett 157:479–483
Watts JD, Gauss J, Bartlett RJ (1993) J Chem Phys 98:8718–8733
Balabanov NB, Peterson KA (2005) J Chem Phys 123:064107-1–064107-15
Douglas M, Kroll NM (1974) Ann Phys (NY) 82:89–155
Hess BA (1985) Phys Rev A 32:756–763
Hess BA (1986) Phys Rev A 33:3742–3748
Jansen G, Hess BA (1989) Phys Rev A 39:6016–6017
Scott AP, Radom L (1996) J Phys Chem 100:16502–16513
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al (2010) Gaussian 09 Revision C.01. Gaussian, Inc, Wallingford
Wu ZJ, Su ZM (2006) J Chem Phys 124:184306-1–184306-15
Tomonari M, Mochizuk YJ, Tanaka K (1999) Theor Chem Acc 101:332–335
Boldyrev AI, Simons J, Scherer JJ, Paul JB, Collie CP, Saykally RJ (1998) J Chem Phys 108:5728–5732
Turski P, Barysz M (1999) J Chem Phys 111:2973–2977
Plass W, Stoll H, Preuss H, Savin A (1995) J Mol Struct: THEOCHEM 339:67–81
Tsipis AC, Gkarmpounis DN (2012) J Comput Chem 33:2318–2331
Lefebvre Y, Schamps J (2000) J Mol Spectrosc 201:128–133
Scherer JJ, Paul JB, Collier CP, Saykally RJ (1995) J Chem Phys 102:5190–5199
Turski P, Barysz M (2000) J Chem Phys 113:4654–4661
Li S, Van Zee RJ, Weltner W Jr, Raghavachari K (1995) Chem Phys Lett 243:275–280
Hao DS, Liu JR, Yang JC (2008) J Phys Chem A 112:10113–10119
Yang JC, Xu WG, Xiao WS (2005) J Mol Struct: THEOCHEM 719:89–102
Raghavachari K (1986) J Chem Phys 84:5672–5686
Raghavachari K (1991) J Chem Phys 94:3670–3678
Vasiliev I, Ogut S, Chelikowsky JR (1997) Phys Rev Lett 78:4805–4808
King RB (1991) Z Phys D 18:189–191
Zhu XL, Zeng XC (2003) J Chem Phys 118:3558–3570
Ning HM, Fan HW, Yang JC (2011) Comput Theor Chem 976:141–147
Lee HM, Ge M, Sahu BR, Tarakeshwar P, Kim KS (2003) J Phys Chem B 107:9994–10005
Acknowledgment
This work was supported by the National Natural Science Foundation of China (Grant No. 21263010).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 21 kb)
Rights and permissions
About this article

Cite this article
Lin, L., Yang, J. Small copper-doped silicon clusters CuSin (n = 4–10) and their anions: structures, thermochemistry, and electron affinities. J Mol Model 21, 155 (2015). https://doi.org/10.1007/s00894-015-2702-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-015-2702-5