
Abstract
The gas nitrous oxide (N2O) is generated in a variety of abiotic, biotic, and anthropogenic processes and it has recently been under scrutiny for its role as a greenhouse gas. A single enzyme, nitrous oxide reductase, is known to reduce N2O to uncritical N2, in a two-electron reduction process that is catalyzed at two unusual metal centers containing copper. Nitrous oxide reductase is a bacterial metalloprotein from the metabolic pathway of denitrification, and it forms a 130 kDa homodimer in which the two metal sites CuA and CuZ from opposing monomers are brought into close contact to form the active site of the enzyme. CuA is a binuclear, valence-delocalized cluster that accepts and transfers a single electron. The CuA site of nitrous oxide reductase is highly similar to that of respiratory heme-copper oxidases, but in the denitrification enzyme the site additionally undergoes a conformational change on a ligand that is suggested to function as a gate for electron transfer from an external donor protein. CuZ, the tetranuclear active center of nitrous oxide reductase, is isolated under mild and anoxic conditions as a unique [4Cu:2S] cluster. It is easily desulfurylated to yield a [4Cu:S] state termed CuZ * that is functionally distinct. The CuZ form of the cluster is catalytically active, while CuZ * is inactive as isolated in the [3Cu1+:1Cu2+] state. However, only CuZ * can be reduced to an all-cuprous state by sodium dithionite, yielding a form that shows higher activities than CuZ. As the possibility of a similar reductive activation in the periplasm is unconfirmed, the mechanism and the actual functional state of the enzyme remain under debate. Using enzyme from anoxic preparations with CuZ in the [4Cu:2S] state, N2O was shown to bind between the CuA and CuZ sites, suggesting direct electron transfer from CuA to the substrate after its activation by CuZ.
Please cite as: Met. Ions Life Sci. 14 (2014) 177–210
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction: The Biogeochemical Nitrogen Cycle
The bulk elements carbon, oxygen, hydrogen, nitrogen, and sulfur are the fundamental building blocks for all classes of biomolecules. They are abundant in our environment and were readily available during evolution to be combined into molecules of increasing complexity that form the basic classes of biological macromolecules: carbohydrates, lipids, amino acids, and nucleic acids. Water is an obvious and omnipresent source both for oxygen and hydrogen, and nature has devised various different pathways for the assimilation of carbon, the most prominent being the light-driven fixation of CO2 in the Calvin cycle during photosynthesis. The remaining elements, nitrogen and sulfur, are present in a series of different modifications and oxidation states that are chemically or enzymatically interconverted in global, biogeochemical cycles [1]. For nitrogen, this cycle spans eight oxidation levels ranging from + V in nitrate, NO −3 , to –III in ammonia, NH3 [2, 3], and only the latter can be incorporated into amino acids via the reactions of glutamine synthase or glutamate dehydrogenase. Nitrate undergoes a two-electron reduction to nitrite, NO −2 , catalyzed by a molybdenum-containing nitrate reductase, and nitrite in turn constitutes the central metabolic hub of the nitrogen cycle (Figure 1).

The biogeochemical nitrogen cycle. A network of reactions catalyzed by metal-containing proteins connects the different modifications of the element. The pathway of denitrification (highlighted in green) is a four-step metabolism to reduce nitrate (NO −3 ) via nitrite (NO −2 ), nitric oxide (NO), and nitrous oxide (N2O) to dinitrogen (N2). Nitrous oxide reductase catalyzes the final step of denitrification, and although the reaction is thermodynamically highly favored, a high activation energy barrier conveys substantial kinetic stability to N2O.
From here, the reductive ammonification pathway bridges a step of six electrons with a single enzyme that exists in two distinct versions. Assimilatory nitrite reductase is a siroheme-dependent metalloprotein that in bacteria and plants is employed for the production of ammonium to be incorporated into biomolecules, consequently using NADPH as an electron donor. In contrast, the dissimilatory variant of nitrite reductase is a multiheme c enzyme that obtains electrons from the pool of menaquinol in the membrane, coupling its oxidation to the generation of a proton motive force for energy conservation. The enzyme, NrfA, contains five covalently attached heme groups per monomer and forms functional dimers [4]. The reversal of nitrate ammonification is realized in the pathway of nitrification, the sole oxygen-dependent process in the nitrogen cycle. Here, ammonium is oxidized to hydroxylamine by ammonia monooxygenase, in a process with strong parallels to methane oxidation to methanol by methane monooxygenase. Hydroxylamine is then converted to nitrite by an octaheme hydroxylamine dehydrogenase, followed by the two-electron oxidation to nitrate by a molybdenum-dependent nitrite oxidase. An alternative route for ammonium oxidation exists as an anaerobic process, in which ammonium is comproportionated with nitric oxide, NO, to yield hydrazine, N2H4, by hydrazine hydrolase. The product is then oxidized to N2 by an octaheme hydrazine dehydrogenase, and this anammox process – while being the latest discovery in the nitrogen cycle – is now known to predominate in many habitats, giving it a significant influence on the global nitrogen balance [5]. Nevertheless, the highest metabolic flux through the nitrogen cycle occurs along the remaining reductive pathway, leading in four enzymatic steps from nitrate via nitrite, nitric oxide and nitrous oxide, N2O, to the stable end product dinitrogen, N2. Each step is energy-conserving through the generation of a proton motive force, and in summary this denitrification pathway is the most energetically favorable process known to operate in the absence of molecular oxygen [2, 6, 7]. Its final two intermediates, N2O and N2, play a particular role in the nitrogen cycle. Both are stable, inert gases, and for each nature seems to have evolved only a single enzyme able to catalyze a reductive conversion. For N2, this is the assimilatory enzyme nitrogenase, a complex iron-sulfur enzyme and the agent of biological nitrogen fixation [8]. N2O, in contrast, is converted to N2 and H2O by the copper enzyme nitrous oxide reductase (N2OR) (equation 1), the topic of the present chapter.
N2OR is unusual in various ways, as it contains two non-canonical metal centers that allow reacting a highly inert substrate molecule. Due to the limited stability of this enzyme, N2O is a common byproduct of this microbial metabolism and gives rise to substantial environmental problems (see Section 2.4).
2 Nitrous Oxide: Environmental Effects and Atmospheric Chemistry
The colorless, slightly sweet-smelling gas nitrous oxide was first described by Joseph Priestley in 1774 as ‘dephlogisticated nitrous air’ in his “Experiments and Observations on Different Kinds of Air” that also encompassed the first description of O2, NH3, and NO [9]. It was found to be a useful anesthetic and also to cause a certain light-headedness that was soon popularized through laughing gas parties and traveling shows [10]. Today the gas is still in use as a mild sedative in dental surgery and in obstetrics, where the application in medical-grade purity does no longer give rise to fits of uncontrolled happiness. For major surgical procedures, nitrous oxide is used as a carrier gas for more potent agents such as halothane.
2.1 Chemical Properties of Dinitrogen Monoxide
The N2O molecule is isoelectronic to CO2 and shares several of its physicochemical properties. From rotational spectroscopy measurements, the bond lengths in N2O gas were determined to be 1.1282 Å for the N–N bond and 1.1842 Å for the N–O bond. These bonds are notably shorter than the respective average values of 1.25 Å and 1.21 Å for the corresponding double bonds, and this can be ascribed to the existence of two resonance structures (Figure 2). The central nitrogen carries a positive partial charge in both structures, resulting in an orbital contraction that helps to rationalize the shortened bond lengths.

The N2O molecule. The linear molecule shows N–N and N–O bond lengths that are shorter than those expected for double bonds. This is due to resonance stabilization that also conveys a slightly positive partial charge to the central nitrogen atom, while both terminal atoms carry a slightly negative partial charge. The resulting small dipole moment of N2O substantially contributes to its solubility in polar solvents such as water.
The two resonance structures carry opposing charges on their terminal atoms, resulting in a low dipole moment of 0.161 D in spite of the terminal oxygen atom. N2O has an electronic configuration of (1σ)2(2σ)2(3σ)2(4σ)2(5σ)2(6σ)2 (1π)4(7σ)2(2π)4, so that the highest occupied π orbitals, 1π and 2π, each are a degenerate pair of allyl-type orbitals. Calculations have suggested the 2π orbital to be slightly N–N bonding and N–O antibonding, in agreement with the shorter N–N distance. The LUMO is an antibonding 3π molecular orbital, whose π character is strengthened by the fact that oxygen has a higher electronegativity than nitrogen [10]. The N2O molecule exhibits three fundamental absorptions with superimposed rotational fine structure in infrared spectroscopy, an asymmetric stretch (Σ+) at ν 1 = 1284.91 cm–1, a bend (Π) at ν 2 = 588.77 cm–1, and a symmetric stretch (Σ+) at ν 3 = 2223.76 cm–1. Both stretches (ν 1 and ν 3) also appear in the Raman spectrum, and these properties provided initial proof that the linear structure of the molecule was asymmetric. Nitrous oxide liquefies at 183.7 K and solidifies at 170.8 K. It is well soluble in water with a mole fraction of 4.4⋅10–4 at 298 K, so that solvated nitrous oxide is readily accessible as an enzyme substrate [11]. Hydration of N2O in water could potentially yield hyponitrous acid (H2N2O2), a weak acid with pK 1 = 7.2 and pK 2 = 11.5, but the equilibrium for this reaction is far on the side of N2O, which is consequently the dominant species in aqueous solution.
2.2 Nitrous Oxide, the Greenhouse Effect, and Ozone Depletion
In the atmosphere, nitrous oxide appears as a trace gas with a current concentration of about 325 ppb, and it can be traced through climate history by analyzing air entrapped in polar ice cores. Interestingly, the present concentration is higher than at any time during the last 45,000 years [12]. From low levels during the last glacial period (the Würm ice age in the European Alpine region) the atmospheric concentration of N2O increased to about 275 ppb and remained largely constant for the last 10,000 years. It started to rise in the 19th century, concomitant with the onset of industrialization, and is increasing since, at an average annual rate of 0.8 ppbv [13]. Due to its chemical stability, the atmospheric half-life of N2O is estimated to be 120 years, so that any increase in production will lead to an atmospheric accumulation that persists for centuries to come [14].
The problematic aspect of this increase is that N2O is a potent greenhouse gas that absorbs sunlight reflected from the Earth’s surface with approximately the 300-fold efficiency of CO2 [14, 15]. The control and reduction of N2O emissions thus should be an imminent aspect of global climate policies, but while attempts are undertaken to regulate and limit CO2 emissions, the impact of nitrous oxide has long gone unnoticed. Only in 2009, the gas has been designated the dominant anthropogenic emission of the 21st century [16], and public awareness is rising. Most critically, nitrous oxide not only acts as a greenhouse gas, but is also involved in complex atmospheric chemistry.
N2O is the predominant source of stratospheric NO x that react with and deplete the reservoir of ozone, O3. However, in conjunction with the notorious ozone-depleting chlorofluorocarbons (CFC) the effect of NO x may even be beneficial, as chlorine oxide radicals generated through the decomposition of CFCs react with NO and NO2 in the lower stratosphere to form stable compounds such as ClONO2 that do not react with ozone [10, 17].
Today the international treaties to limit the production of CFCs have shown substantial effects, but at the same time this brings back N2O as a major sink for ozone that will play a key role in the foreseeable future [16]. Nitrous oxide is among the greenhouse gases considered in the second phase of the Kyoto Protocol to the United Nations Framework Convention on Climate Change, together with CO2, CH4, SF6, and the CFCs. This treaty covers the commitment period 2013–2020, but remains to be ratified by most countries, with a decision by the European Parliament projected for 2015 [18].
2.3 Abiotic and Biotic Sources
Nitrous oxide is primarily produced in the course of a variety of biological processes, in particular bacterial denitrification (see Section 2.4), but in recent times a number of abiotic routes for the generation of substantial quantities of the gas were found. The nitrate-dependent oxidation of Fe(II) was recognized as a central geochemical process, but while for most minerals the reduced nitrogen species generated here are nitrite or ammonium, the mineral siderite (FeCO3) was described to yield N2O as a main product [19]. More recently, a study investigating a brine pond in Antarctica revealed that several Fe(II) components of the mineral dolerite could also generate N2O in large quantities [20].
On a global scale, such processes nevertheless are dwarfed by the biological generation of nitrous oxide through the pathways of nitrification and denitrification in the nitrogen cycle [6, 21]. Approximately 40 % of the global nitrous oxide emissions originate directly or indirectly from human activities (US Environmental Protection Agency, http://epa.gov/climatechange/ghgemissions/gases/n2o.html). Herein, 70 % are related to agricultural soil management, with a minor fraction of approximately 5 % being due to the breakdown of nitrogen compounds in the manure and urine of livestock, while the bulk is due to the use of nitrogen fertilizers in industrial crop production (see Section 2.4). About 10 % of anthropogenic N2O release are attributed to combustion engines in industry and transportation, and another 10 % to industrial and chemical production processes. A major factor hereby is the release of N2O as a side product in the synthesis of adipic acid, one of the two components required for the production of nylon [22].
2.4 Bacterial Denitrification
In the metabolic pathway of denitrification, N2O is generated through the reductive coupling of two molecules of nitric oxide (NO) by the membrane-integral nitric oxide reductase. It is released to the periplasmic space in Gram-negative bacteria, where a soluble nitrous oxide reductase catalyzes the two-electron reduction to N2 (see equation 1 in Section 1), using electrons that originate from menaquinol. Energetically, denitrification is the most efficient respiratory pathway in the absence of molecular oxygen as a terminal electron acceptor, and consequently denitrifiers thrive in environments with low oxygen levels and abundant supply of nitrate [7]. Coincidentally, these conditions are met quite precisely in modern industrial agriculture, where the growth-limiting element nitrogen is provided as a fertilizer, commonly in the form of nitrate salts. Soil denitrifiers compete with the assimilating crops for this nutrient, to the effect that approximately half of the fertilizer dispersed on the fields is returned to the N2 from which it was produced in the Haber-Bosch process [3].
With a current global fertilizer production of approximately 160 Mt per year, the overall metabolic flux through the denitrification pathway is therefore substantial. The second condition for denitrification, an anoxic or microoxic environment, is met quite readily as well, as the availability of O2 diminishes drastically already millimeters or centimeters into the rather stratified soil of a regularly watered field. Nitrate reduction increases sharply at this oxycline, where traces of O2 may be encountered, but can generally be tolerated. The weakest link in the chain of denitrificatory enzymes, however, is the one catalyzing the final step from N2O to N2, nitrous oxide reductase. Its copper centers show the highest sensitivity, and while the reaction of the enzyme is presumably coupled to the generation of a proton motive force [23], the organism can tolerate its failure, in particular because the gaseous substrate N2O can be released and will not accumulate to inhibit the preceding NO reductase. The result is a direct correlation of the increasing use of nitrogen fertilizers in agriculture and the rise of atmospheric N2O released through incomplete denitrification. While agriculture thus is just one of the factors contributing to the rise of atmospheric levels of nitrous oxide, it is the one most directly related to human population growth with a direct link to the ongoing industrialization of highly populated countries such as India or China [16].
3 Nitrous Oxide Reductase
The enzymatic conversion of N2O into the stable products N2 and water (equation 1) is carried out by copper-containing nitrous oxide reductase. After noticing a general requirement for copper for N2O reduction by Alcaligenes faecalis [24], the enzyme itself was identified in a dedicated search for copper-containing enzymes in Pseudomonads [25], with the first ortholog to be characterized being the one from Pseudomonas stutzeri strain ZoBell (originally P. perfectomarina) [26].
A variety of other orthologs was characterized over the years and detailed insight was gained concerning the two metal sites, but it was only in the year 2000 that a first crystal structure became available for the enzyme from Pseudomonas nautica [27, 28] (now Marinobacter hydrocarbonoclasticus) [29] and in 2003 for Paracoccus denitrificans [27, 30]. Structural data was later presented for N2O reductase from Achromobacter cycloclastes [31], and more recently for the originally studied enzyme from P. stutzeri [32]. Gaining an unambiguous picture of the function and properties of N2O reductase has been complicated by the fact that the enzyme has been isolated in various forms that differ in content and state of the active centers, as well as in their activity.
3.1 Anatomy of an Unusual Copper Enzyme
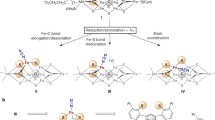
The enzyme N2O reductase is the product of the nosZ gene, an open reading frame encoding a protein of approximately 640 amino acid residues that is located in the bacterial periplasmic space. Variations to this organization were reported for the denitrifying hyperthermophilic crenarchaeon Pyrobaculum aerophilum [33], and for few genera of bacteria including Thiomicrospira denitrificans and Wolinella succinogenes that contain an additional cytochrome c domain in their nosZ gene [23]. The two domains of the canonical NosZ protein fold into an N-terminal, seven-bladed β-propeller, and a C-terminal β-sandwich that attains the cupredoxin fold commonly observed in mononuclear type-I copper proteins [34]. Accordingly, each individual domain has a distinct metal center embedded, and the interaction of two chains in the dimeric enzyme assures that they are located in an appropriate orientation to afford a functional interaction (Figure 3).

Structure of the copper enzyme N2O reductase (PDB ID 3SBR). (a) N2OR is the product of the nosZ gene, and the peptide chain is organized into two distinct domains that harbor one of the metal centers each. The N-terminal domain attains a seven-bladed β-propeller fold that coordinates the tetranuclear active site CuZ through seven histidine residues at its hub. The C-terminal domain shows a cupredoxin fold similar to type-I copper proteins and binds the dinuclear mixed-valent CuA center. (b) The distance between the two metals centers within a single polypeptide chain is above 40 Å, but the enzyme forms stable homodimers in a head-to-tail arrangements that brings the CuA site of one monomer within a distance of 10 Å from the CuZ site of the other and vice versa.
NosZ proteins form stable homodimers with a strict head-to-tail arrangement, where the N-terminal domain of one monomer interacts with the C-terminal domain of the other and vice versa. In consequence, the metal centers within a single protein chain are more than 40 Å apart in the NosZ dimer, while the distance between the centers in different chains amounts to a mere 10 Å, placing it well within the range required for efficient electron transfer [35]. Both metal centers of N2O reductase exclusively employ copper, and both exhibit a degree of specialization that is only observed here. The cupredoxin domain is the site of the binuclear CuA center, an electron transfer site with a characteristic spectroscopic signature that is also found in many respiratory oxidases. Its function in N2O reductase is to mediate single-electron transfer from an external electron donor, commonly a c-type cytochrome or a cupredoxin, to the second metal site where substrate reduction occurs. This second site is a tetranuclear Cu center termed CuZ that so far has only been found in N2O reductase. Its architecture and exact composition have been a matter of debate for many years (see Section 3.3).
3.1.1 Distinct Forms of the Enzyme
The observed differences in operon architecture are in at least one case directly related to the structure of the NosZ protein itself. Nos operons that lack the nosR gene and instead contain nosG, nosH, and further periplasmic cytochromes, typically contain a larger open reading frame for nosZ that includes a third domain with the α-helical secondary structure and the typical CXXCH heme binding motif of c-type cytochromes. This has been analyzed in W. succinogenes [23, 36], and it is commonly interpreted to originate from a fusion event of the reductase enzyme with a soluble electron donor. A cytochrome c 552 of similar architecture serves as electron donor to the enzyme of M. hydrocarbonoclasticus [37], while for other NosZ orthologs the type-I copper protein pseudoazurin was described to also fulfill this function [38]. This functional variability in the architecture of electron transfer chains is not unprecedented within the pathway of denitrification and a very similar case was described for cytochrome cd 1 nitrite reductase [39].
As an additional layer of complexity, NosZ itself was found to occur in a series of forms that are distinct in terms of spectroscopy and reactivity [40]. When isolated in the absence of dioxygen, preparations had a deep purple color, with high catalytic activity and approximately 8 mol of Cu per mol of dimeric enzyme of P. stutzeri [41, 42]. This was designated form I of N2O reductase, and it is considered to be the physiologically relevant active form [43], although the copper content at the time was significantly underestimated. When isolated in the presence of dioxygen, the enzyme showed reduced catalytic activity and a different optical spectrum.
In this form II, the 7-line hyperfine pattern in the EPR spectrum of CuA (Figure 4) was generally less well resolved in the g || region, and at the time this was ascribed to an underlying signal of a distinct form of CuZ, termed CuZ * [40, 43]. As such, CuZ was thus associated with an active state, CuZ * with an inactive state of the center. Anoxic reduction of CuA with a 10-fold molar excess of dithionite changed the color of the protein to blue (form III), leading to a catalytically inactive state dominated by a single charge-transfer band with a maximum at 650 nm, and an axial S = ½ EPR signal without a resolved hyperfine structure in the g || region [40]. With CuA in the reduced state, the spectroscopic features of form III solely originate from the CuZ center, but while a distinction was made for the EPR data, it was not initially recognized that the UV/vis signatures reveal the identity and ratio of CuZ versus CuZ * that reflect on the structure/function relationship of the enzyme’s active site (Figure 5) [44].

Electron paramagnetic resonance spectrum of N2O reductase. In a continuous-wave EPR experiment at X-band (ν = 9.4 GHz) recorded at 10 K, N2OR shows a characteristic axial signal dominated by the contribution from the CuA site. Both the g || and the g ⊥ region of the EPR spectrum are split into a complex hyperfine pattern that was best resolved in the purple form I of the enzyme where the second metal site, CuZ, was in the [4Cu:2S] configuration. The seven-line pattern is well resolved in the first harmonic (solid), and the second harmonic (dashed) shows that both g regions are split with an intensity ratio of 1:2:3:4:3:2:1 that is explained by two coupled copper nuclei (63,65Cu nuclear spin I = 3/2) in a fully valence-delocalized arrangement.

Electron excitation spectra of N2O reductase. (a) The spectrum of purple N2OR (form I) shows maxima at 538 nm and 795 nm and can be deconvoluted into individual bands by a multi-Gaussian fit. (b) The individual contributions of the CuA and CuZ centers can be reconstructed from the fit, so that the properties of the two sites can be studied in isolation. In addition, the reduction with sodium ascorbate will only reduce CuA, and the CuZ contribution is readily accessible experimentally. (c) In N2OR form I, the contribution of the CuZ site can be modelled by two bands with maxima at 552 nm and 650 nm. Further reduction with sodium dithionite will only deplete the transition at 552 nm, while the remaining band persists. (d) In form II N2OR, CuZ is present in the [4Cu:S] CuZ * state, with only a single band with a maximum at 650 nm from the tetranuclear cluster. Contrary to form I, this state can be further reduced with sodium dithionite, yielding an enzyme that is all-Cu(I) and thus colorless.
A fourth form of N2O reductase was described as a ‘reconstituted’ enzyme, where Cu was first removed to generate the apo-protein that could subsequently be replenished with copper. This was achieved by anoxic dialysis against KCN, followed by the addition of Cu(en)2SO4 [40]. This procedure led to an enzyme with a lower copper content of approximately 4 Cu per homodimer of N2OR that had the spectroscopic features of an intact CuA site, but with CuZ fully absent. Besides providing evidence that the assembly of CuZ required further maturation factors, this procedure also gave access to a form of the enzyme in which one of the metal centers could be studied without interference from the other. The same effect was reached through a variant strain, P. stutzeri MK402, in which the biogenesis of CuZ was prevented through a Tn5 insertion, leading to an enzyme containing exclusively CuA, termed form V [40].
The Cu content of different preparations of nitrous oxide reductase was generally between 7 and 8 Cu per dimer, but the exogenous addition of copper salts to the growth medium yielded a highly active form of the enzyme with an increased Cu content of 10–12/dimer [45, 46]. This could be rationalized from the crystal structures that became available at the same time, and showed the CuZ site to be an entirely novel, tetranuclear copper center, with four metal ions arranged around a central ligand and coordinated to a total of seven histidine residues at the hub of the N-terminal β-propeller domain [27, 28, 30]. Both relevant forms of CuZ, the CuZ site of N2OR form I and the CuZ * site of form II, contain the same number of copper ions within the cluster, and even the original assumption that CuZ * represents an oxidatively damaged form of CuZ was recently questioned by the finding that an enzyme with the typical features of N2OR form I could be isolated under oxic conditions from M. hydrocarbonoclasticus [47].
3.1.2 Three-Dimensional Structures
Initial advances towards structural data for the metal sites of nitrous oxide reductase was made for the CuA site in respiratory cytochrome c oxidase or in engineered cupredoxins as detailed below, but no information was available for the active site of the denitrificatory enzyme, the CuZ center. In the initial description of the ortholog from P. nautica (M. hydrocarbonoclasticus) [29, 48], CuZ was identified as a novel tetranuclear site coordinated by seven histidines, with a central oxygen ligand [28, 49, 50]. This structure was obtained from a preparation corresponding to form III of N2O reductase, with CuA in the reduced state and the distinct blue color of an absorption band with a maximum at 600 nm that consequently had to originate from CuZ. This assignment raised immediate criticism from the spectroscopic community, as an environment consisting exclusively of imidazole nitrogens and an oxygen lacked the soft, donating ligand required to yield the prominent ligand → metal charge-transfer transition (LMCT) represented by the blue color of the preparation. Indeed, a re-determination of the M. hydrocarbonoclasticus N2OR structure, combined with the structure of the enzyme from Paracoccus denitrificans at 1.6 Å resolution soon thereafter clarified that the central oxygen in CuZ was indeed a sulfido species that likely was misinterpreted due to partial occupancy [27, 30].
Both structures were obtained as a blue form III of the enzyme and interpreted to show the CuZ * form of the active center. In 2006, the enzyme from A. cycloclastes was crystallized and refined to 1.7 Å resolution, showing for the first time an enzyme in form II, which differed from the previous structures in having the CuA site in the oxidized state [31]. Note, however, that the copper sites of nitrous oxide reductase are likely prone to swift photoreduction upon exposure to X-rays, so that spectroscopic monitoring of crystals will be required before unambiguous assignments of oxidation state can be made. The most recent structural data on N2OR came from P. stutzeri, the organism where the enzyme was originally discovered. Using anoxically isolated protein, purple crystals of form I N2OR were obtained [51], and the resulting structure showed several relevant features, including the CuZ state of the active site cluster [32].
3.2 CuA: More than an Electron Transfer Center
Beside its role as an electron transfer site in nitrous oxide reductase, further interest in the CuA site arose from the fact that its spectroscopic features indicated a possible structural and electronic similarity to one of the copper sites in respiratory cytochrome c oxidase [52]. Due to a better resolution of the observed spectroscopic features, nitrous oxide reductase eventually proved to be highly instrumental to understand the structure and properties of this site, but agreement was only reached after lengthy debates with the advent of several crystal structures [53]. Yet, only the most recent analyses of the denitrificatory enzyme have revealed intriguing properties that after all do set the CuA site in N2OR apart from the one in cytochrome c oxidase and that help to explain the improved resolution in EPR spectra of the latter [32, 44].
3.2.1 Spectroscopic Properties of CuA
Copper-containing proteins are frequently classified within a scheme of three basic types according to a proposal by Malkin and Malmström [54]. When the enzyme nitrous oxide reductase was initially isolated from P. stutzeri in different form as described above [43], it was readily apparent that this novel protein would not fit into the established scheme. For the purple form I of the enzyme, EPR studies at X-band showed a distinct axial signal with g || = 2.18 and g ⊥ = 2.03, with characteristic hyperfine splittings with seven lines in a 1:2:3:4:3:2:1 intensity ratio that were not consistent with a mononuclear copper site (63/65Cu nuclear spin I = 3/2) (Figure 4) [43]. The g || region featured seven equidistant lines with A || = 3.83 mT, and a similar pattern was observed in the g ⊥ region with A ⊥ = 2.8 mT. This hyperfine structure disappeared upon reduction of the protein in the absence of dioxygen to yield to a broad featureless signal with g values at 2.18 and 2.06. Notably, the pink form II of the enzyme that is characterized by an intact CuA site, but a CuZ site in the CuZ * state, showed a similar spectrum to form I, but with an inferior resolution for the hyperfine pattern in both the g || and g ⊥ regions.
Based on these and further studies, Kroneck and coworkers concluded that CuA in nitrous oxide reductase was a valence-delocalized [Cu1.5+:Cu1.5+] binuclear center, and that the same was true for the corresponding site in cytochrome c oxidase [52]. This claim was soon contested by the proponents of a mononuclear CuA site in the respiratory complex [55], largely under the impression of the available spectroscopic data for the respiratory enzyme. While the additional copper center there was found to show an unusual g || value of 2.18 and lacked the four-line hyperfine pattern typical for type-I and type-II copper sites unless it was chemically denatured, the characteristic seven-line pattern (Figure 4) was never observed clearly. In retrospect this was ascribed to the additional presence of heme groups in cytochrome c oxidase [53, 56], but more recent data has provided an alternative explanation (see Section 3.2.3).
In the absence of a crystal structure, Cu K-edge EXAFS was highly valuable for obtaining initial information on the architecture of CuA. A study on the solubilized cupredoxin domain of subunit II in cytochrome c oxidase from Bacillus subtilis indicated the presence of two coppers, each of which was coordinated to a single histidine and two cysteines [57]. In addition, a direct metal-metal interaction was observed at a distance of 2.5 Å. In a follow-up, the authors compared the CuA domains of the oxidases of B. subtilis and Thermus thermophilus in different redox states, and obtained a Cu-Cu distance of 2.43 Å for the valence-delocalized oxidized state, versus 2.51 Å for the fully-reduced [Cu1+:Cu1+] state [58].
3.2.2 Three-Dimensional Structure(s) of CuA
While the CuA site in N2O reductase had proved valuable for understanding the electronic properties of the center, structural information was first provided along different lines. Extensive efforts to solve the three-dimensional structure of respiratory cytochrome c oxidase were undertaken by several groups, until the prokaryotic ortholog from Paracoccus denitrificans was solved to 2.8 Å resolution in 1995 [59], followed shortly thereafter by the eukaryotic enzyme from bovine heart mitochondria [60, 61]. The same arrangement was also found in a solubilized form of subunit II of cytochrome bo 3 oxidase from Escherichia coli [62]. These structures unequivocally settled the debate concerning the number of metal ions in CuA, and they underlined that the ligand environment in CuA is highly conserved.
The two Cu ions of CuA show a distorted tetrahedral coordination environment that represents the classical ‘entatic’ state between a square-planar geometry with hard ligands (preferred by Cu(II)) and a tetrahedral geometry with soft ligands (preferred by Cu(I)) [63]. In CuA, the two cysteine ligands are shared in a μ2-bridging fashion, and each ion in addition has a histidine ligand (Figure 6). The coordination environment is complemented in each case by a further ligand that differs between the two metals. For Cu1, this ligand is the oxygen atom of a backbone carbonyl group, while for Cu2 the first coordination sphere is completed by the sulfur of a methionine, at a slightly longer distance. The site thus is of high – if not perfect – symmetry, and shows the features of electronic delocalization, in particular the characteristic seven-line hyperfine pattern.

The electron transfer site CuA of N2O reductase. Stereo representations of the two conformations observed in structures of N2OR. (a) In purple N2OR from P. stutzeri, as isolated, residue His583 was in most cases not a ligand to Cu1 of the CuA site resulting in a shift of the copper ion into the plane formed by the two cysteine residues, Cys618 and Cys622, and Met629 (PDB ID 3SBQ). The experimental data is consistent with a sharpening of the EPR hyperfine structure in this conformation. (b) In all instances with substrate bound to CuZ, the side chain of His583 was flipped back to coordinate Cu1 of the CuA center, linking the metal cluster to the protein surface via residue Asp576 to allow for electron transfer from an external donor (PDB ID 3SBR).
Based on the crystal structures, an excellent biomimetic model complex was presented soon thereafter [64]. While the complex was centrosymmetric and therefore had perfectly identical geometries for both metals, the observed seven-line hyperfine pattern in EPR spectroscopy showed a hyperfine splitting of 4.99 mT for A || and 3.63 mT for A ⊥, notably larger than for the protein. This was concomitant with a Cu-Cu distance in the model complex of 2.92 Å that was significantly longer than the 2.5 Å determined by EXAFS spectroscopy for CuA [57, 65]. The short distance between both metals in this center is commonly interpreted as one of the rare cases of a direct metal-metal interaction in Biology [57]. In the structure of P. denitrificans N2OR determined at 1.6 Å resolution, the Cu-Cu distance was 2.51 Å [30], but was significantly longer at 2.63 Å in the purple form I structure from P. stutzeri [32]. For this enzyme, 63Cu- or 65Cu-enrichment as well as 15 N-labeling had allowed for a significantly improved resolution for EPR spectroscopy that then formed the basis for molecular orbital calculations on a structural core unit consisting of Cu ions with two μ2-bridging sulfides and a terminal amine ligand on each metal [66]. The obtained values agreed very well with the experimental data, and subsequent ENDOR studies helped to refine the picture further (see Section 5.1) [67, 68].
The CuA center is located in a domain that shows a conserved tertiary structure commonly termed the cupredoxin fold [69, 70]. Cupredoxins are mononuclear copper proteins (‘type-I copper proteins’) that coordinate a single copper ion in a rigid binding site at the periphery of a 100–140 aa peptide with a characteristic β-barrel forming a Greek-key motif [34]. In cupredoxins, the metal is coordinated by a cysteine that allows for a LMCT, giving rise to an intense absorption maximum around 600 nm that conveys the typical color of these ‘blue copper proteins’. Further ligands are two histidines and a fourth ligand that commonly is a methionine, but can also be a different amino acid that modulates the midpoint potential of the Cu+/Cu2+ redox pair [71]. Within the protein chain, one of the histidine ligands is found in the first third of the sequence, while the other three ligands cluster in a single loop near the C-terminal that connects the last two β strands.
Interestingly, the very same architecture is found both in cytochrome c oxidase and nitrous oxide reductase, where a cupredoxin domain holds the CuA site. In P. stutzeri N2O reductase, the ligands to CuA are His583 in the N-terminal part of the cupredoxin domain, and Cys618, Trp620, Cys622, His626, and Met629 that all form part of the terminal loop between β strands 8 and 9 (Figures 3 and 6). This loop is longer than in a typical type-I copper protein, and in fact Sanders-Loehr, Canters, and coworkers could show that a simple insertion of a CuA-binding loop into a cupredoxin such as amicyanin led to the assembly of an intact CuA center in a valence-delocalized state [72, 73]. The copper-binding loop of the CuA site in nitrous oxide reductase faces the second metal site, CuZ, and two residues, Phe621 and Met627 in P. stutzeri, form part of the substrate binding pocket, as detailed below.
3.2.3 Unexpected Flexibility: CuA in P. stutzeri Nitrous Oxide Reductase
In the structure of purple N2O reductase from P. stutzeri, the CuA site differed not only in the consistently longer metal-metal bond when compared to earlier structure determinations, but also in the conformation of one of the coordinating ligands to a copper, His583 [32]. While all earlier structure determinations of proteins containing a CuA site – natively or through engineering of the CuA-binding loop – invariably showed the conserved ligand environment described above, His583 of P. stutzeri N2O reductase was found in two different conformations, with its Nδ atom either coordinating Cu2 of the CuA site, or forming a hydrogen bond to the hydroxyl group of nearby residue Ser550 (Figure 6). The two different conformations are related through a rotation of the imidazole moiety of the side chain of His583 by approximately 135° [32]. With its Nε nitrogen, His583 retains a hydrogen bond to the β-carboxy group of Asp576, an aspartate residue located at the surface of the enzyme, in an area that was postulated as a possible docking site for an electron transfer partner [37]. The arrangement observed in the crystal structures thus gave rise to the hypothesis that His583 can act as a gate for electron transfer from an external electron donor to the CuA site of N2O reductase. If a direct coordination of His583 to Cu2 was indeed a prerequisite for reduction of CuA, then the rotation of the histidine side chain towards Ser550 would serve this conduit and prevent the enzyme from executing its catalytic function [32, 44].
However, in order to function as an efficient gate the flip of the imidazole moiety should be linked to a triggering event within the catalytic cycle. In various structures of N2OR form I, His583 was observed in either conformation, flipped in or out with respect to Cu2, indicating a significant degree of structural flexibility on the part of the reductase. Notably, in several crystal structure determinations the conformation of His583 differed between the two monomers of the same enzyme dimer. This is not straightforward to rationalize in a crystal structure that always represents the macroscopic conformational average over all unit cells, and it may imply a slight asymmetry of the dimeric arrangement that affects the crystallization process. No such asymmetry has been described to date, and it will be interesting to conduct an in-depth analysis based on high-quality structural data.
To date His583 (or the corresponding residue in other CuA domains) was so far only found in a flipped-out state in the purple form I of N2OR (Figure 6). While Ser550 is a conserved residue in N2O reductase, it is absent in heme-copper oxidases, where no flip of a histidine ligand to CuA seems to occur. In N2O reductase, the purple form I has the second metal site, CuZ, in a [4Cu:2S] conformation, so that there must be structural changes in the vicinity of both metal sites that enable His583 to abandon its coordination to Cu2 of CuA. The exact determinants of these changes remain to be identified, but their functional relevance is underlined by the finding that the one case where His583 was always found flipped towards CuA – thus enabling electron transfer from an external partner – was when substrate was bound to the nearby CuZ site (see Section 5.3).
3.3 The Tetranuclear CuZ Center
In contrast to CuA, the second metal site in N2O reductase, CuZ, has no precedence in any other known enzyme, and its exact architecture and relevant states remain to be fully understood. As the addition of the strong reductant sodium dithionite, Na2S2O4, produced a blue species with an absorption maximum around 640 nm and a distinct EPR spectrum, the presence of a second copper site in N2OR was suspected early on [26]. A copper site that was not reduced by dithionite seemed highly unusual, and as copper determinations at the time yielded on average 8 Cu per enzyme dimer [41, 42], the site – now designated CuZ – was considered to be a second dinuclear center with a sulfide ligand that gave rise to the characteristic charge-transfer band [74]. The EPR signature of CuZ, with g || = 2.18 and g ⊥ = 2.06, was distinct from the signature of CuA (Figure 4) and remained after the latter was reduced to an all-Cu(I), diamagnetic state [43]. This form of CuZ was described as an S = ½ resting state of the cluster [75]. In retrospect, many studies on CuZ suffered from the intrinsic structural and electronic heterogeneity found in CuZ, and only with the advent of three-dimensional structures for different forms of nitrous oxide reductase the picture has become clearer.
A recurring theme in literature on N2OR is on the different states of the CuZ site. After the initial description of two forms, a catalytically active CuZ and a catalytically inactive CuZ * [74, 76, 77], more recent data have led to a more differentiated (and complicated) picture. Ascorbate reduces the CuA site to a colorless and EPR-silent [Cu+:Cu+] state, so that CuZ can be straightforwardly analyzed without influence from the other cluster. Here, the difference between both forms of CuZ is readily apparent. In electron excitation spectroscopy, CuZ * shows the single LMCT band at 640 nm described above, while the CuZ center is characterized by two distinct bands at 550 nm and 650 nm. Further reduction of CuZ with sodium dithionite depletes the absorption at 550 nm, but notably a complete reduction of the site is not achieved and the band at 650 nm is retained even after prolonged incubation with strong reductants. In contrast, dithionite in combination with the redox mediator methyl viologen can reduce CuZ * to an all-cuprous, colorless state. After reduction of CuA, both CuZ and CuZ * appear blue in color, and according to the original assignment both species would be classified as form III N2OR. Nevertheless the key to understanding the reactivity of nitrous oxide reductase most likely lies exactly within the distinction of these states.
3.3.1 Structural Data on CuZ
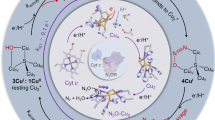
After the initial clarification of the central ligand of CuZ to be a partially occupied sulfide rather than on oxide [27, 28], the CuZ centers of M. hydrocarbonoclasticus [27], P. denitrificans [30], and A. cycloclastes [31] were consistently described as [4Cu:μ-S] centers [44, 78]. The picture was amended with the structure of purple form I N2OR from P. stutzeri that showed a [4Cu:2S] configuration (Figure 7) and revealed that this was associated to the CuZ state, while the single-sulfide species [4Cu:μ-S] represented CuZ * [32, 44]. The tetranuclear site seems to be rather susceptible to partial degradation during the isolation procedure of the enzyme, and while the four copper ions are stably retained, it is in the presence of the additional sulfide ion bridging atoms Cu1 and Cu4 where the differences manifest.


CuZ, the catalytic center of N2O reductase. (a) The tetranuclear CuZ site is located at the hub of the N-terminal β-propeller domain. It constitutes a unique [4Cu:2S] cluster coordinated to seven histidine residues. (b) The spectroscopically and functionally distinct forms CuZ (above) and CuZ* (below) differ in the presence of the second sulfur atom in the cluster. While CuZ undergoes a redox transition from a [2Cu+:2Cu2+] state to a [3Cu+:1Cu2+] and cannot be further reduced, CuZ* is isolated as [3Cu+:1Cu2+] and can be further reduced to a [4Cu+] state by sodium dithionite and methyl viologen. (c) The variation of CuZ structures from different crystal structures of nitrous oxide reductases. The first structures (PDB ID 1QNI, 1FWX) showed mixtures of CuZ and CuZ*, where a partially occupied sulfur atom between Cu1 and Cu4 was interpreted as a bridging water or hydroxo species. A form II structure (PDB ID 2IWF) then had two water ligands at the Cu1–Cu4 edge, but would bind the inhibitor iodide in a bridging fashion (PDB ID 1IWK). Only in the purple form I structure (PDB 3SBR), CuZ was in the complete [4Cu:2S] state.
Degradation of the site in the CuZ state leads to formation of CuZ *, and the relative abundance of both states tends to vary from one protein batch to another. In addition, the reverse process, a conversion of CuZ * to CuZ, had not been achieved in vitro, and it is assumed that this is only possible with the help of additional maturation factors during the biogenesis of the enzyme [11]. As a consequence, the electron density maps obtained from X-ray diffraction data are prone to showing an average of different forms of the cluster, and this refers in particular to mixtures of the CuZ and CuZ * states. The magnitude of a peak in an electron density map primarily reflects the number of electrons at a given position, and in addition the value obtained represents an average of all unit cells in the entire crystal. For a 1:1 mixture of CuZ and CuZ * within a crystal, this means that the electron density maximum at the position of atom SZ2, where both forms of the cluster differ, will have half the magnitude of a fully occupied CuZ state. In number of electrons this corresponds to only 8 of the 16 electrons expected for the element sulfur, but it also corresponds to a fully occupied oxygen atom, and in the original structures it was indeed modeled as such. An oxygen atom in the model will satisfy the observed electron density so that no conspicuous residual peaks will be visible in F o–F c difference electron density maps. Oxygen was modeled in this position in the structures of M. hydrocarbonoclasticus and P. denitrificans, but in both cases the Cu–‘O’ bond lengths refined to 2.3 Å (PDB ID 1QNI, 1FWX) [27, 30], falling far more in the expected range of a Cu–S bond [44]. The current interpretation of the existing CuZ structures (Figure 7) thus suggests that CuZ exists as a [4Cu:2S] cluster, while CuZ * only lacks the bridging SZ2, but does not contain a water ligand connecting Cu1 and Cu4. The situation differed slightly in the structure of the A. cycloclastes enzyme, where two distinct electron density maxima were interpreted as water ligands at reasonable bond distances of 2.0–2.1 Å (PDB ID 2IWF), or where iodide as an inhibitor bound between Cu1 and Cu4 in a bridging manner (PDB ID 2IWK) [31]. This enzyme had the spectroscopic properties of a form II N2OR with CuZ in the CuZ * state, but it did unambiguously show exogenous ligands binding to the center for the first time.
3.3.2 States of CuZ and Catalytic Properties
Detailed activity assays for N2O reductase were first presented for the P. stutzeri enzyme [43]. As detailed above, the observed activities were highest for the anoxically isolated form I of the enzyme that we now understand to contain the intact [4Cu:2S] CuZ cluster (Figure 7). Subsequently, Snyder and Hollocher published activities for a ‘purple’ form of P. denitrificans N2OR that were two orders of magnitude higher, but the precise state of the enzyme preparation was not clearly stated [79]. More recent activity data on the form II preparation of M. hydrocarbonoclasticus N2OR provided further insights [50]. Here, the protein was isolated with the tetranuclear site in the CuZ * state, and the observed activity of 0.01 U N2O was far below the one reported for other orthologs. However, the enzyme could be activated by complete reduction of CuZ * to an all-Cu(I) form using sodium dithionite in conjunction with methyl viologen as a redox mediator (see Section 3.3). In this state, the activity of the enzyme was determined to 160 U N2O, and the conclusion drawn was that CuZ * must be the active form of the CuZ cluster and that reductive activation of the enzyme is a necessary requirement for catalytic activity [47, 78]. In this mechanism, the activation of the enzyme still is far slower than the observed catalytic turnover, and it is not straightforward to reconcile fast oxidation of CuZ * during N2O reduction with the observed, slow activation of the cluster with dithionite [44].
A possible solution for the issue came with the identification of a further state of the CuZ site that was proposed to be an additional catalytic intermediate. This species was characterized by an additional absorption maximum at 680 nm and termed CuZ 0 [80]. Solomon and coworkers identified CuZ 0 as a [3Cu+:1Cu2+] form of the cluster that was, however, distinct from the isoelectronic CuZ * and that can be rapidly reduced back to the all-Cu(I) state of the site [81]. For the mechanism of nitrous oxide reduction, this suggested that within the catalytic cycle, the fully reduced state was required to bind and activate the kinetically inert N2O molecule, but that the catalytic one-electron oxidation of the tetranuclear cluster would lead to CuZ 0 rather than the CuZ * resting state, allowing for a quick reductive reactivation of the site to keep up with the observed rate of catalysis. At the same time, a single-turnover experiment, where activated and reduced N2OR was incubated with its substrate in the absence of further reductant, yielded the enzyme in the CuZ * resting state, while N2O had obtained one electron each from CuA and CuZ to yield the products N2 and H2O [81]. Note that this mechanistic proposal strictly depends on the availability of the all-Cu(I) state of CuZ, which only seems to be accessible with the [4Cu:S] CuZ * site, while in the [4Cu:2S] form, CuZ, even extended incubation with sodium dithionite and viologens will not reduce all four copper ions [44, 47]. This may be explained with the presence of the additional sulfur donor ligand that forms highly covalent bonds with Cu1 and Cu4 of the cluster and stabilizes the more oxidized state [34, 81]. The activity reported for the purple form I N2OR from P. stutzeri would thus either be due to a residual fraction of CuZ * that was reductively activated, or it would reflect the catalytic potential of the CuZ itself that, however, was too low to explain the activities observed in vivo [34]. In this mechanism, N2O was still suggested to bind to the exposed edge of CuZ * formed by Cu1 and Cu4 in the absence of a bridging sulfur [81–83], and the binding mode observed experimentally [32] was proposed to reflect the lower activity of the purple form I of N2OR [81].
From a physiological point of view, however, this mechanistic proposal encounters two major problems. First, the reductive activation of N2OR to generate the all-Cu(I) state of CuZ * that is a prerequisite for catalysis can only be achieved at very low redox potentials in vitro, and it additionally required the presence of an abiological redox mediator. Both components will not be readily available in the oxidizing environment of the bacterial periplasm, and to date there is no mechanism known to drive such low-potential redox chemistry outside the cytoplasm in an in vivo situation [44]. Second, the most careful isolations of N2OR, in particular those in the absence of dioxygen, commonly yield a high CuZ/CuZ * ratio, i.e., a high proportion of the tetranuclear site in the [4Cu:2S] state [32, 40, 47]. It thus seems safe to assume that the CuZ * center is only generated from CuZ through the loss of a sulfur. N2OR is isolated from its natural host and from cells that have turned over N2O in a physiological context. If indeed the [4Cu:2S] state was an inactive one, this would imply that the bacteria have a way to remove the S2 sulfide anion from CuZ for reductive activation and catalysis, but to subsequently replenish the center to its [4Cu:2S] form that then can be isolated. The biogenesis of CuZ is not fully understood to date (see Section 4.3), but it has long been known that there is no straightforward way to re-introduce sulfur into a defective CuZ center.
4 Biogenesis and Assembly of Nitrous Oxide Reductase
Denitrification is a respiratory metabolism that relies on the generation of a proton motive force across the cytoplasmic membrane. All its enzymatic steps are located in the periplasm (in the case of Gram-negative denitrifiers), and the required metalloenzymes therefore must be assembled in this compartment that in several aspects differs from the ‘inside’ of the cell, the cytoplasm. First, the assembly of metal centers in biomolecules is never an unspecific process. This is particularly true for the relatively rare and highly toxic copper that is tightly chaperoned at all times in a physiological context [84]. Denitrifiers thus require not only the different enzymes, but in most cases also a complex biogenesis pathway that assembles the active sites post-translationally. Second, the general currency of energy for the cell, ATP, is not available in the periplasm, so that any energy-consuming step in said pathways must be fueled in a different way.
4.1 The nos Operon
The structural gene for N2O reductase is termed nosZ [85] (Figure 8). In most instances, the derived NosZ protein shows the two-domain architecture mentioned above (Figure 3), but in some cases an additional domain encoded for a c-type cytochrome that presumably represents a fused electron transfer partner (see Section 3.1) [23, 78]. This may not represent a fundamental difference in the functionality of the enzymes, but it has substantial consequences for the required machinery for N2OR biogenesis. Two-domain NosZ proteins are exported via the twin-arginine translocation (Tat) pathway, where the entire protein domain can cross the cytoplasmic membrane in a folded or partially folded state [86]. However, if a c-type cytochrome domain is included, as in W. succinogenes NosZ, its maturation, i.e., the covalent attachment of the heme group to the protein chain [87], is strictly dependent on the Sec translocon that transports proteins in their unfolded state [88]. In the latter case, all assembly steps of the two copper sites of N2O reductase thus have to take place in the periplasm, after the attachment of the heme group to the cytochrome domain, while Tat-dependent export of the more canonical two-domain NosZ would in principle allow to assemble the CuA and CuZ fully or in part in the cytoplasm. Although it is presently assumed that the key steps of metal center biogenesis do occur in the periplasm in both variants of the nos operon, the operon architecture itself is different [23].

The nos operon of Pseudomonas stutzeri. The nosZ gene (Z) is the structural gene for N2O reductase, while the integral membrane protein NosR, encoded by the preceding gene (R), is required both for the biogenesis of NosZ and for electron transfer to the reductase. NosF (F) and NosY (Y) form an ABC transporter that is presumed to shuttle sulfur into the periplasm, in a form that can be incorporated into CuZ with the help of the nosD gene (D) product. NosL (L) is a putative copper chaperone that provides Cu(I) for the assembly of both metal sites.
The W. succinogenes nos operon consists of twelve open reading frames, the first of which encodes the larger version of NosZ with the additional cytochrome domain. The operon in addition encodes for two periplasmic assembly factors, NosD and NosL, and ABC Transporter, NosFY, and contains three further open reading frames (orfs) of unknown function. It also holds a putative menaquinol oxidase, NosGH, and two periplasmic cytochromes, NosC1 and NosC2, with a putative role in electron transfer to the enzyme [23]. In contrast, P. stutzeri possesses a smaller nos cluster that is organized into three transcriptional units (Figure 8) [89]. It commences with the monocistronic nosR gene, encoding a complex, cofactor-containing membrane protein required both for NosZ maturation and for N2O reduction [90].
Following nosR, the nosZ gene encoding the actual enzyme is a second monocistronic transcription unit that includes an N-terminal leader peptide with the typical twin-arginine motif for Tat-dependent translocation. The following nosD operon is a four-gene transcription unit that holds the factors required for periplasmic maturation of CuZ (and possibly CuA) [89]. All four genes, nosDFYL, are also present in the W. succinogenes operon, indicating that this is a general maturation machinery, while NosGH, NosC1 and NosC2 may combine to play the complex role of the NosR protein in P. stutzeri. As the main focus of the present text is on P. stutzeri N2OR, we will restrict the discussion of maturation factors to this version of the operon (Figure 8).
4.2 Protein Maturation and CuA Insertion
In P. stutzeri, the nosZ gene is translated in the cytoplasm, so that its folded – or partially folded – protein product is exported via the Tat pathway. It is currently not known whether NosZ is exported as a monomer or a dimer, and whether it reaches the periplasm as a metal-free apo protein or already has pre-assembled copper sites included. The presence and location of the known maturation factors, however, strongly indicates that the major part of the assembly process occurs in the extracellular compartment. Here, the less intricate CuA site is assembled along a different route, as deletions of any of the genes nosDFYL will leave the binuclear site unaffected, while the resulting protein is fully deplete of NosZ [11]. In a metal-depleted NosZ protein, CuA can be readily reconstituted by the addition of Cu(II) salts, while CuZ cannot [43], and it is assumed that in vivo the CuA site employs a copper chaperone, such as ScoA, that is also required for the assembly of heme-copper oxidases [11]. This is in line with the concept of rack-induced metal binding by cupredoxin domains and the facile reconstitution of the binuclear CuA site has also been observed in model proteins with an engineered CuA-binding loop [72, 73].
4.3 Assembly of CuZ
The ABC transporter NosFY and the periplasmic proteins NosD and NosL that form part of all known gene clusters encoding a functional N2O reductase system are required for assembly of the CuZ active site. This site is substantially more complex than CuA (Figures 6 and 7), and consequently its biogenesis is more intricate, to the point that in vitro reconstruction without the help of additional factors has not been achieved [7]. The CuZ center consists of two essential building blocks, the four copper ions and the two sulfide ions, that must be provided along different routes.
As mentioned previously, Cu trafficking is a highly regulated process in all organisms, and specialized chaperones are required to provide the metal, commonly in the Cu(I) state [84]. For N2OR, the NosL protein was suggested for this role [11, 78, 91]. An NMR structure of NosL is available that shows the apo-state of the protein, but contains cysteine residues that may well serve as ligands for Cu(I) [92]. NosFY is an ABC transporter that shuttles a necessary assembly intermediate from the cytoplasm to the periplasm, but its actual cargo is unknown to date [89]. It is, however, homologous to the Atm1 type of ABC transporters that are involved in iron-sulfur cluster biogenesis. Two structures of Atm1 orthologs have become available most recently and imply that the transported species may be a glutathione persulfide [93, 94]. In conjunction with NosFY, NosD is thought to serve as an assembly chaperone or a periplasmic binding protein for the species transported by NosFY. The emerging picture thus is that NosL is the copper donor at least for the assembly of CuZ in N2OR, while NosD obtains an activated sulfide from the cytoplasm, transported via NosFY, and shuttles it to the enzyme. The process is undoubtedly more complex than this, as the addition of any combination of sulfides or thiols and Cu(I) or Cu(II) salts in vitro is not sufficient to achieve even a partial reconstitution of CuZ.
5 Activation of Nitrous Oxide: The Workings of Nitrous Oxide Reductase
The reaction of nitrous oxide reductase requires two electrons per N2O molecule (equation 1) that are provided by a soluble single-electron donor such as cytochrome c 552 and reach the enzyme via an electron input site close to the CuA center. The structural analysis of purple N2OR I from P. stutzeri not only revealed novel structural features of the metal sites, but it also allowed to demonstrate the mode of substrate binding by pressurization of crystals with N2O gas prior to flash-cooling and diffraction data collection [32]. In contrast, earlier studies based on the [4Cu:μ-S] structure of CuZ * and the iodide-inhibited state of the site observed in A. cycloclastes N2OR [31] (Figure 7) led to a model where nitrous oxide bound to the cluster edge formed by atoms Cu1 and Cu4 that in the CuZ state was occupied by the second sulfide [78, 82, 83]. While we have discussed the electronic and functional implications above, this distinction will be relevant for mechanistic considerations in the context of the architecture of the enzyme’s active site.
5.1 Substrate Access
While the substrate N2O is gaseous at ambient temperatures, it is well soluble in water and overall slightly polar, albeit far less so than the surrounding solvent, water. The structure of nitrous oxide reductase shows a feature that is present in an overwhelming majority of gas-converting enzymes, even for gas molecules as small as H2 [95]. In N2OR, a hydrophobic (i.e., devoid of ordered solvent molecules) channel leads from the protein surface to the substrate binding site observed between CuA and CuZ in the P. stutzeri enzyme [32].
The channel is formed by the interface of the two monomers that form the tight N2OR dimer, and it terminates in a small vestibule located right at the tetranuclear site (Figure 9). This is the only visible direct connection from the protein surface to the metal sites, and although, at present, an unambiguous conformation of the functionality of this channel by obstruction through site-directed mutagenesis has not been made, the overall arrangement is highly suggestive. In this position, however, the substrate channel does not help to make a distinction concerning the relevance of the substrate binding modes predicted by X-ray crystallography versus theory [34, 44]. While the hydrophobic channel indeed leads right up to a gate formed by two bulky amino acid side chains (Phe621 and Met627 in P. stutzeri) on the face of CuZ that is directed towards CuA, the additional space gained through the loss of the second sulfide, S2, in the CuZ * state would allow the substrate to access the Cu1-Cu4 edge of the cluster as well.

Substrate access and N2O binding at the active site of N2O reductase. (a) Stereo representation of the molecular surface at the interface between the cupredoxin domain (blue) and the β-propeller domain with the active site situated below. Substrate access is provided via a hydrophic channel along the domain interface, leading to a vestibule close to CuZ. The access to the N2O binding site on CuZ is controlled by residues Phe621 and Met627. (b) Observed binding of N2O between CuA and CuZ in gas-pressurized crystals of P. stutzeri N2OR form I (PDB ID 3SBR). A difference electron density map for the substrate is shown below a transparent van-der-Waals surface. Upon N2O binding, residue His583 at CuA was invariably found to coordinate to the nearby copper ion (Figure 6). The positioning of the substrate between the two metal sites suggests that electron transfer might occur directly from CuA to N2O, rather than involving a reduction of CuZ.
5.2 Gated Electron Transfer
An unexpected finding in the structure of P. stutzeri N2OR was the flexibility of His583, a ligand to Cu2 of the CuA site (Figure 6) [32]. Based on the available structural data the 135° flip of the imidazole side chain of this histidine residue cannot be related to the redox state of the site, and both conformations seem accessible. However, all structures of gas-pressurized crystals of the P. stutzeri enzyme showed the ligand to be coordinated to the metal ion, as did all earlier observations, where the tetranuclear site was in the CuZ * state. The flipped-out state of His583 was thus only observed in form I of N2OR, i.e., with an intact [4Cu:2S] cluster, and in the absence of the substrate N2O. As His583 in both its conformations remains hydrogen-bonded to residue Asp576 that is presumed to be the electron entry site from an external electron transfer protein [37], the on-off coordination to CuA was suggested to form a conformational gate for intramolecular electron transfer, with the consequence that electron flow will only be permitted after the substrate is bound at CuZ [32, 44]. Obviously, this intricate coupling mechanism is tightly linked to the physiological activity of the enzyme, based on a reduced electron transfer protein as a redox partner. Using small-molecule reductants such as ascorbate or sodium dithionite, complete reduction of CuA is swift, but there is no way of controlling at which points the reductants will interact with the enzyme. The question of gated electron transfer in nitrous oxide reductase thus touches the distinction between reactivities observed in vitro and those occurring in the actual physiological environment of the bacterial periplasm.
5.3 Activation of Nitrous Oxide
The reduction of N2O to N2 by nitrous oxide reductase (equation 1) takes place at the tetranuclear site, CuZ. The N2O molecule observed in the P. stutzeri X-ray structure was not directly coordinated to any of the metal ions of the cluster, but rather it was fixed by hydrogen bonds to the oxygen atom, originating from His626, a ligand to CuA, and a structural water molecule (Figure 9) [32]. This structure did not represent a coordination complex in the classical sense, but rather revealed a substrate-binding site in close proximity to the metal centers. This binding event is presumed to be the trigger for His583 to flip back towards the CuA center and enable electron transfer to the substrate. Nevertheless, the suggestive implication that derives from this mode of N2O binding is that the substrate is located in between the two metal sites, making it unlikely that in this arrangement electron transfer from CuA would proceed first into CuZ and then to the substrate, but rather directly from CuA to an activated state of N2O that is coordinated to CuZ.
5.4 The Fate of the Products
The substrate N2O is a weakly polar molecule with a positive partial charge on the central nitrogen atom and according negative partial charges on the terminal N and O (Figure 2). It is well suited to access the active site of N2O reductase via a largely hydrophobic access channel. After reduction by two electrons, the gaseous, apolar product N2 will be able to exit the enzyme along the same route. However, the oxygen atom of N2O remains as a polar water molecule, and the hydrophobic substrate channel seems to disfavor the passage of water.
In the structures of N2O reductase, water molecules are found in close proximity to the metal sites, and indeed the position held by the oxygen atom of N2O in the substrate complex structure (Figure 9) is commonly occupied by water if substrate is absent. From here, a network of coordinated water molecules, interacting through hydrogen bonds, stretches through the protein matrix. This arrangement suggests that after the reduction of N2O, the fate of the two product molecules differs, and while N2 will exit through the original substrate channel, the water molecule will leave the active site in a different way, to move through a hydrophilic cavity and exit the protein at a remote site. In many enzymes, such optimizations serve to prevent product inhibition and accelerate the reaction rate, but in the present case it seems more likely that this is a mechanism to accommodate the very different physicochemical properties of the two reaction products.
6 General Conclusions
Nitrous oxide reductase is a complex and fascinating metalloenzyme that catalyzes the conversion of an inert gas of high chemical interest and ecological relevance. Its copper sites have features that so far are unique in bioinorganic chemistry, and the seemingly straightforward two-electron redox process it catalyzes is of sufficient complexity to defy a final elucidation.
At present, the model of the action of N2O reductase brought forward by Solomon, Moura, and coworkers integrates the majority of the available experimental data and presents the outlines of a mechanism in which the simultaneous coordination of N2O to two copper ions within CuZ * is required to overcome the activation energy barrier for substrate reduction [34, 81]. It places the purple form I of the enzyme on a catalytic sideline, but this in particular raises the question why all biochemical evidence then points to CuZ * being a degradation product of the intact [4Cu:2S] CuZ site, and why this doubly-sulfurated form I of N2OR can readily be isolated from bacteria that have actively turned over N2O during denitrificatory growth, using the exact protein that was isolated after harvesting of the biomass. It therefore is imminent to address the issue of assembly and maturation of the CuZ center and generate structural pictures – ideally verified by single-crystal spectroscopy – that align the different states of the enzyme with the spectroscopy data at the highest resolution possible.
Moreover, it will also be essential to determine whether a full-reduced, all-Cu(I) state of CuZ can be generated in vivo and which mechanisms might be in place to deliver the required low-potential electrons to a periplasmic enzyme. No such process was observed to date and it seems quite difficult to envision indeed, but one of the great aspects of science surely is that very few things, really, seem to be impossible in nature. Astounding progress has been made in recent years, and the field seems all set to eventually provide some of those famed ‘final answers’ regarding the biological reduction of nitrous oxide.
References
P. M. H. Kroneck, in Biogeochemical Cycles of Elements, Vol. 43 of Metal Ions in Biological Systems, Eds A. Sigel, H. Sigel, R. K. O. Sigel, Taylor & Francis, Boca Raton, USA, 2005, pp. 1–7.
O. Einsle, P. M. H. Kroneck, Biol. Chem. 2004, 385, 875–883.
D. E. Canfield, A. N. Glazer, P. G. Falkowski, Science 2010, 330, 192–196.
O. Einsle, Methods Enzymol. 2011, 496, 399–422.
B. Kartal, W. J. Maalcke, N. M. de Almeida, I. Cirpus, J. Gloerich, W. Geerts, H. J. M. O. den Camp, H. R. Harhangi, E. M. Janssen-Megens, K. J. Francoijs, H. G. Stunnenberg, J. T. Keltjens, M. S. M. Jetten, M. Strous, Nature 2011, 479, 127–132.
R. Knowles, Microbiol. Rev. 1982, 46, 43–70.
W. G. Zumft, Microbiol. Mol. Biol. Rev. 1997, 61, 533–616.
D. C. Rees, F. A. Tezcan, C. A. Haynes, M. Y. Walton, S. Andrade, O. Einsle, J. B. Howard, Philos. Trans. R. Soc. A 2005, 363, 971–984.
J. Priestley, Experiments and Observations on Different Kinds of Air, J. Johnson, London, 1774.
W. C. Trogler, Coord. Chem. Rev. 1999, 187, 303–327.
W. G. Zumft, P. M. H. Kroneck, Adv. Microb. Physiol. 2007, 52, 107–225.
M. Leuenberger, U. Siegenthaler, Nature 1992, 360, 449–451.
W. C. Trogler, J. Chem. Educ. 1995, 72, 973–976.
J. T. Houghton, Climate Change 1996: The Science of Climate Change, Cambridge University Press, Cambridge, 1996.
O. Badr, S. D. Probert, Appl. Energ. 1993, 44, 197–231.
A. R. Ravishankara, J. S. Daniel, R. W. Portmann, Science 2009, 326, 123–125.
P. O. Wennberg, R. C. Cohen, R. M. Stimpfle, J. P. Koplow, J. G. Anderson, R. J. Salawitch, D. W. Fahey, E. L. Woodbridge, E. R. Keim, R. S. Gao, C. R. Webster, R. D. May, D. W. Toohey, L. M. Avallone, M. H. Proffitt, M. Loewenstein, J. R. Podolske, K. R. Chan, S. C. Wofsy, Science 1994, 266, 398–404.
European Commission, in “Climate action: Commission proposes ratification of second phase of Kyoto Protocol”, 2013.
S. Rakshit, C. J. Matocha, M. S. Coyne, Soil Sci. Soc. Am. J. 2008, 72, 1070–1077.
V. A. Samarkin, M. T. Madigan, M. W. Bowles, K. L. Casciotti, J. C. Priscu, C. P. Mckay, S. B. Joye, Nat. Geosci. 2010, 3, 341–344.
G. A. Kowalchuk, J. R. Stephen, Annu. Rev. Microbiol. 2001, 55, 485–529.
R. A. Reimer, C. S. Slaten, M. Seapan, M. W. Lower, P. E. Tomlinson, Environ. Prog. 1994, 13, 134–137.
J. Simon, O. Einsle, P. M. H. Kroneck, W. G. Zumft, FEBS Lett. 2004, 569, 7–12.
H. Iwasaki, T. Saigo, T. Matsubara, Plant Cell Physiol 1980, 21, 1573–1584.
T. Matsubara, W. G. Zumft, Arch. Microbiol. 1982, 132, 322–328.
W. G. Zumft, T. Matsubara, FEBS Lett. 1982, 148, 107–112.
K. Brown, K. Djinovic-Carugo, T. Haltia, I. Cabrito, M. Saraste, J. J. G. Moura, I. Moura, M. Tegoni, C. Cambillau, J. Biol. Chem. 2000, 275, 41133–41136.
K. Brown, M. Tegoni, M. Prudêncio, A. S. Pereira, S. Besson, J. J. G. Moura, I. Moura, C. Cambillau, Nat. Struct. Biol. 2000, 7, 191–195.
C. Sproer, E. Lang, P. Hobeck, J. Burghardt, E. Stackebrandt, B. J. Tindall, Int. J. Syst. Bacteriol. 1998, 48, 1445–1448.
T. Haltia, K. Brown, M. Tegoni, C. Cambillau, M. Saraste, K. Mattila, K. Djinovic-Carugo, Biochem. J. 2003, 369, 77–88.
K. Paraskevopoulos, S. V. Antonyuk, R. G. Sawers, R. R. Eady, S. S. Hasnain, J. Mol. Biol. 2006, 362, 55–65.
A. Pomowski, W. G. Zumft, P. M. H. Kroneck, O. Einsle, Nature 2011, 477, 234–237.
P. Völkl, R. Huber, E. Drobner, R. Rachel, S. Burggraf, A. Trincone, K. O. Stetter, Appl. Environ. Microbiol. 1993, 59, 2918–2926.
E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt, L. Tian, Chem. Rev. 2014, 114, 3659–3853.
C. C. Page, C. C. Moser, X. X. Chen, P. L. Dutton, Nature 1999, 402, 47–52.
S. Teraguchi, T. C. Hollocher, J. Biol. Chem. 1989, 264, 1972–1979.
S. Dell’Acqua, S. R. Pauleta, E. Monzani, A. S. Pereira, L. Casella, J. J. G. Moura, I. Moura, Biochemistry 2008, 47, 10852–10862.
K. Mattila, T. Haltia, Proteins: Struct. Funct. Bioinform. 2005, 59, 708–722.
I. V. Pearson, M. D. Page, R. J. M. van Spanning, S. J. Ferguson, J. Bacteriol. 2003, 185, 6308–6315.
J. Riester, W. G. Zumft, P. M. H. Kroneck, Eur. J. Biochem. 1989, 178, 751–762.
W. G. Zumft, P. M. H. Kroneck, Adv. Inorg. Chem. 1996, 11, 193–221.
P. Wunsch, H. Körner, F. Neese, R. J. M. van Spanning, P. M. H. Kroneck, W. G. Zumft, FEBS Lett. 2005, 579, 4605–4609.
C. L. Coyle, W. G. Zumft, P. M. H. Kroneck, H. Körner, W. Jakob, Eur. J. Biochem. 1985, 153, 459–467.
A. Wüst, L. Schneider, A. Pomowski, W. G. Zumft, P. M. H. Kroneck, O. Einsle, Biol. Chem. 2012, 393, 1067–1077.
J. M. Charnock, A. Dreusch, H. Körner, F. Neese, J. Nelson, A. Kannt, H. Michel, C. D. Garner, P. M. H. Kroneck, W. G. Zumft, Eur. J. Biochem. 2000, 267, 1368–1381.
T. Rasmussen, B. C. Berks, J. Sanders-Loehr, D. M. Dooley, W. G. Zumft, A. J. Thomson, Biochemistry 2000, 39, 12753–12756.
S. Dell’Acqua, S. R. Pauleta, J. J. Moura, I. Moura, Philos. Trans. R. Soc. B 2012, 367, 1204–1212.
M. J. Gauthier, B. Lafay, R. Christen, L. Fernandez, M. Acquaviva, P. Bonin, J. C. Bertrand, Int. J. Syst. Bacteriol. 1992, 42, 568–576.
M. Prudêncio, A. S. Pereira, P. Tavares, S. Besson, I. Cabrito, K. Brown, B. Samyn, B. Devreese, J. Van Beeumen, F. Rusnak, G. Fauque, J. J. G. Moura, M. Tegoni, C. Cambillau, I. Moura, Biochemistry 2000, 39, 3899–3907.
M. Prudêncio, A. S. Pereira, P. Tavares, S. Besson, I. Moura, J. Inorg. Biochem. 1999, 74, 267–267.
A. Pomowski, W. G. Zumft, P. M. H. Kroneck, O. Einsle, Acta Crystallogr. Sect. F Cryst. Comm. 2010, 66, 1541–1543.
P. M. H. Kroneck, W. A. Antholine, J. Riester, W. G. Zumft, FEBS Lett. 1988, 242, 70–74.
H. Beinert, Eur. J. Biochem. 1997, 245, 521–532.
R. Malkin, B. G. Malmström, Adv. Enzymol. Relat. Subj. Biochem. 1970, 33, 177–244.
P. M. Li, B. G. Malmström, S. I. Chan, FEBS Lett. 1989, 248, 210–211.
P. M. H. Kroneck, W. A. Antholine, J. Riester, W. G. Zumft, FEBS Lett. 1989, 248, 212–213.
N. J. Blackburn, M. E. Barr, W. H. Woodruff, J. Vanderooost, S. de Vries, Biochemistry 1994, 33, 10401–10407.
N. J. Blackburn, S. deVries, M. E. Barr, R. P. Houser, W. B. Tolman, D. Sanders, J. A. Fee, J. Am. Chem. Soc. 1997, 119, 6135–6143.
S. Iwata, C. Ostermeier, B. Ludwig, H. Michel, Nature 1995, 376, 660–669.
T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, S. Yoshikawa, Science 1995, 269, 1069–1074.
T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, S. Yoshikawa, Science 1996, 272, 1136–1144.
M. Wilmanns, P. Lappalainen, M. Kelly, E. Sauer-Eriksson, M. Saraste, Proc. Natl. Acad. Sci. USA 1995, 92, 11955–11959.
B. L. Vallee, R. J. P. Williams, Proc. Natl. Acad. Sci. USA 1968, 59, 498–505.
R. P. Houser, V. G. Young, W. B. Tolman, J. Am. Chem. Soc. 1996, 118, 2101–2102.
G. Henkel, A. Müller, S. Weissgräber, G. Buse, T. Soulimane, G. C. M. Steffens, H. F. Nolting, Angew. Chem. Int. Ed. 1995, 34, 1488–1492.
F. Neese, W. G. Zumft, W. E. Antholine, P. M. H. Kroneck, J. Am. Chem. Soc. 1996, 118, 8692–8699.
F. Neese, R. Kappl, J. Hüttermann, W. G. Zumft, P. M. H. Kroneck, J. Biol. Inorg. Chem. 1998, 3, 53–67.
B. Epel, C. S. Slutter, F. Neese, P. M. H. Kroneck, W. G. Zumft, I. Pecht, O. Farver, Y. Lu, D. Goldfarb, J. Am. Chem. Soc. 2002, 124, 8152–8162.
P. Wittung, B. Kallebring, B. G. Malmström, FEBS Lett. 1994, 349, 286–288.
M. Saraste, Q. Rev. Biophys. 1990, 23, 331–366.
E. T. Adman, Adv. Protein Chem. 1991, 42, 145–197.
C. R. Andrew, P. Lappalainen, M. Saraste, M. T. Hay, Y. Lu, C. Dennison, G. W. Canters, J. A. Fee, C. E. Slutter, N. Nakamura, J. Sanders-Loehr, J. Am. Chem. Soc. 1995, 117, 10759–10760.
C. Dennison, E. Vijgenboom, S. de Vries, J. Vanderoost, G. W. Canters, FEBS Lett. 1995, 365, 92–94.
J. A. Farrar, A. J. Thomson, M. R. Cheesman, D. M. Dooley, W. G. Zumft, FEBS Lett. 1991, 294, 11–15.
P. Chen, S. I. Gorelsky, S. Ghosh, E. I. Solomon, Angew. Chem. Int. Edit. 2004, 43, 4132–4140.
M. L. Alvarez, J. Y. Ai, W. G. Zumft, J. Sanders-Loehr, D. M. Dooley, J. Am. Chem. Soc. 2001, 123, 576–587.
T. Rasmussen, B. C. Berks, J. N. Butt, A. J. Thomson, Biochem. J. 2002, 364, 807–815.
S. R. Pauleta, S. Dell’Acqua, I. Moura, Coord. Chem. Rev. 2013, 257, 332–349.
S. W. Snyder, T. C. Hollocher, J. Biol. Chem. 1987, 262, 6515–6525.
S. Dell’Acqua, S. R. Pauleta, P. M. Paes de Sousa, E. Monzani, L. Casella, J. J. Moura, I. Moura, J. Biol. Inorg. Chem. 2010, 15, 967–976.
E. M. Johnston, S. Dell’Acqua, S. Ramos, S. R. Pauleta, I. Moura, E. I. Solomon, J. Am. Chem. Soc. 2014, 136, 614–617.
S. Ghosh, S. I. Gorelsky, P. Chen, I. Cabrito, J. J. G. Moura, I. Moura, E. I. Solomon, J. Am. Chem. Soc. 2003, 125, 15708–15709.
S. I. Gorelsky, S. Ghosh, E. I. Solomon, J. Am. Chem. Soc. 2006, 128, 278–290.
T. V. O’Halloran, R. A. Pufhal, G. Munson, D. Huffman, Biochemistry 1996, 35, 110–110.
A. Viebrock, W. G. Zumft, J. Bacteriol. 1988, 170, 4658–4668.
T. Palmer, B. C. Berks, Nat. Rev. Microbiol. 2012, 10, 483–496.
R. Kranz, R. Lill, B. Goldman, G. Bonnard, S. Merchant, Mol. Microbiol. 1998, 29, 383–396.
V. A. M. Gold, F. Duong, I. Collinson, Mol. Membr. Biol. 2007, 24, 387–394.
U. Honisch, W. G. Zumft, J. Bacteriol. 2003, 185, 1895–1902.
P. Wunsch, W. G. Zumft, J. Bacteriol. 2005, 187, 1992–2001.
M. A. McGuirl, J. A. Bollinger, N. Cosper, R. A. Scott, D. M. Dooley, J. Biol. Inorg. Chem. 2001, 6, 189–195.
L. M. Taubner, M. A. McGuirl, D. M. Dooley, V. Copie, Biochemistry 2006, 45, 12240–12252.
J. Y. Lee, J. G. Yang, D. Zhitnitsky, O. Lewinson, D. C. Rees, Science 2014, 343, 1133–1136.
V. Srinivasan, A. J. Pierik, R. Lill, Science 2014, 343, 1137–1140.
F. Leroux, S. Dementin, B. Burlatt, L. Cournac, A. Volbeda, S. Champ, L. Martin, B. Guigliarelli, P. Bertrand, J. Fontecilla-Camps, M. Rousset, C. Leger, Proc. Natl. Acad. Sci. USA 2008, 105, 11188–11193.
Acknowledgment
The authors thank Peter Kroneck, Walter Zumft, Jörg Simon, Sofia Pauleta, and Isabel Moura for stimulating discussions. This work was supported by Deutsche Forschungsgemeinschaft, Deutscher Akademischer Austauschdienst, the BIOSS Centre for Biological Signalling Studies, and the European Research Council.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Abbreviations and Definitions
Abbreviations and Definitions
- aa:
-
amino acid
- anammox:
-
anaerobic ammonia oxidation
- ATP:
-
adenosine 5′-triphosphate
- CFC:
-
chlorofluorocarbon
- DNRA:
-
dissimilatory nitrate reduction to ammonia
- en:
-
ethylenediamine (= 1,2-diaminoethane)
- EPR:
-
electron paramagnetic resonance
- ENDOR:
-
electron nuclear double resonance
- EXAFS:
-
extended X-ray absorption fine structure
- LMCT:
-
ligand-to-metal charge transfer
- LUMO:
-
lowest unoccupied molecular orbital
- NADPH:
-
nicotinamide adenine dinucleotide phosphate (reduced)
- NMR:
-
nuclear magnetic resonance
- N2OR:
-
nitrous oxide reductase
- NO x :
-
atmospheric nitrogen oxides (NO x =NO + NO2)
- orf:
-
open reading frame
- ppbv:
-
parts per billion by volume
- Tat:
-
twin-arginine translocation
- U:
-
unit of enzymatic activity (μmol (substrate) · min–1 · mg–1 (protein))
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Schneider, L.K., Wüst, A., Pomowski, A., Zhang, L., Einsle, O. (2014). No Laughing Matter: The Unmaking of the Greenhouse Gas Dinitrogen Monoxide by Nitrous Oxide Reductase. In: Kroneck, P., Torres, M. (eds) The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment. Metal Ions in Life Sciences, vol 14. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9269-1_8
Download citation
DOI: https://doi.org/10.1007/978-94-017-9269-1_8
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9268-4
Online ISBN: 978-94-017-9269-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)