
Abstract
Introduction
Homocysteine (Hcy) is considered a newly identified risk factor for osteoporosis. Nevertheless, the underlying mechanism of folate (FA), a key factor in the metabolism of Hcy, in protection against osteoblast dysfunction remains unclear. The purpose of this study was to investigate the mechanism by which FA attenuates Hcy-induced osteoblast damage.
Materials and methods
The Hcy-induced MC3T3-E1 cells were treated with different concentrations of FA. Cell morphology, cell density, cell proliferation ability, alkaline phosphatase (ALP) activity and mineralization capacity were observed and determined; the gene expression of B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (BAX) and ERS-associated factors, including glucose-regulated protein 78 (GRP-78), activating transcription factor 4 (ATF-4) and growth arrest and DNA damage inducible gene 153 (CHOP/GADD153), were assessed by RT-PCR; and protein levels of GRP-78 and ATF-4 were analyzed by western blotting.
Results
Hcy suppressed the proliferation, differentiation and mineralization ability of MC3T3-E1 cells in a concentration-dependent manner and activated the ERS signaling pathway. After intervention with different concentrations of FA, the cell viability and density, ALP activity, number of mineralized nodules, calcium content and Bcl-2 gene expression were all significantly increased, whereas the gene expression of GRP-78, CHOP/GADD153, ATF-4 and Bax was markedly downregulated, and protein levels of GRP-78 and ATF-4 were also markedly decreased.
Conclusion
The adverse effects of Hcy on osteoblast differentiation are dose dependent. FA not only protects against osteoblasts apoptosis but also has a direct osteogenic effect on Hcy-induced osteoblasts, which could be partially mediated by inhibition of the PERK-activated ERS pathway.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Homocysteine (Hcy) is a sulfur-containing amino acid formed as a key intermediate in the methionine cycle. Hcy levels vary considerably among individuals due to genetic background, medication and diet. Elevated plasma concentrations of Hcy, known as hyperhomocysteinemia (HHcy), have been proven to be closely related to several chronic diseases, including atherosclerotic cardiovascular disease [1], Alzheimer's disease [2], senile macular degeneration [3] and cancer [4]. Moreover, several studies have previously shown that Hcy promotes osteoclastic bone resorption, inhibits osteoblastic bone formation and induces osteoporosis via endoplasmic reticulum stress (ERS), mitochondrial injury and other pathways [5, 6]. In addition, Hcy also affects the bone reconstruction process and increases the risk of fracture by upregulating intracellular reactive oxygen species and activating matrix metalloproteinases [7].
Folate (FA) is a key cofactor in the metabolism of the Hcy methylation pathway. Consequently, impairment of the methylation process due to FA deficiency results in the accumulation of Hcy. A preliminary study found that the plasma Hcy concentration of ovariectomized rats was significantly increased, while bone mineral density and alkaline phosphatase (ALP) activity were markedly decreased. However, FA administration markedly reduces levels of Hcy, increases bone ALP activity and enhances bone mineral density in an osteoporosis model of ovariectomized rats [8], but the specific mechanism has not been well described.
Based on existing data, we hypothesized that FA plays a bone-protective role by alleviating Hcy-induced ERS and improving the proliferation and differentiation of osteoblasts. Consequently, we assessed the effect of FA on the proliferation and differentiation of osteoblasts induced by Hcy, as well as the expression of apoptosis-related genes B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (BAX) in osteoblasts. Then, we investigated the gene and protein expression of key factors of the ERS pathway, including glucose-regulated protein 78 (GRP-78), activating transcription factor 4 (ATF-4) and growth arrest and DNA damage inducible gene 153 (CHOP/GADD153). The present study explored the possible bone protection mechanism of FA at the cellular and molecular levels, and provides a theoretical basis for the prevention and treatment of osteoporosis.
Materials and methods
Cell culture and treatment
MC3T3-E1 cells, which are preosteoblast cells with osteoblast differentiation ability, were used for the in vitro experiments and obtained from the Chinese Academy of Medical Sciences. The cells were cultured in low-glucose DMEM (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA) and 0.5% penicillin and streptomycin (Sigma, USA) in a humidified incubator with 5% CO2 at 37 °C. To induce differentiation, cells were seeded into 6-well culture plates and allowed to reach confluence. The media was then replaced with serum-free and low-glucose DMEM containing different concentrations of Hcy (0, 10–5, 10–4, 10–3 and 10–2 mol/L, Sigma, USA) and cultured to specific times and assessed for the effect of Hcy on osteogenic differentiation. Based on the experimental results, the optimal inhibitory Hcy concentration of 10–3 mol/L was selected for subsequent experiments. Twenty-four hours after culture in serum-free low-glucose DMEM containing 10–3 mol/L Hcy, MC3T3-E1 cells were treated with various concentrations of FA (0, 10–7, 10–6, 10–5 and 10–4 mol/L, Yuanye Biology, Shanghai) to induce osteoblast differentiation (Fig. 1). All experimental manipulations were approved by the Ethics Committee of The First Hospital of Lanzhou University, Gansu, China.

Experimental flowchart
Determination of cell viability
Cell viability was determined using the LIVE/DEADTM Viability Kit (Thermo Fisher Scientific). Live cells are labeled with Calcein AM, which is hydrolyzed into a bright green fluorescent product after entering the cell. When the membrane integrity of dying or dead cells is compromised, ethidium homodimer-1 can enter the cell and bind to nucleic acids, exhibiting bright red fluorescence. Cells were cultured in 96-well plates at a density of 1×104 cells/well for 48 h and then incubated with LIVE/DEAD reagents for 10 min at room temperature. Stained cells were observed under a fluorescence microscope (Olympus BX51 Microscope), and twenty representative images were acquired to compute cell density and the live cell percentage. Sampling areas were manually determined after visually assessing the entire sample region.
MTT assay
MC3T3-E1 cells (1 × 104 cells/well) were seeded into 96-well plates and incubated overnight at 37 °C. After a 24-h incubation, the cells were treated with various concentrations of Hcy and/or FA for 48 h. Then, 100 µL MTT reagent (5 mg/mL) was added to each well and incubated for another 4 h. After the culture medium was removed, 150 μL of dimethyl sulfoxide (Sigma, USA) was added to each well, and the plates were shaken for 10 min. Optical densities (ODs) of cell supernatants were measured at 570 nm using an ultraviolet spectrophotometer (Shanghai Tianmei Instrument Company, Shanghai, China). Cell activity is expressed as the percentage of the value of normal cells.
Alkaline phosphatase (ALP) activity
MC3T3-E1 cells were plated into 6-well plates at a density of 5 × 105 cells/well for 72 h. Then, the cells were washed three times with PBS, dissolved in 0.5 mL of Tris–HCl buffer (pH 7.6) containing 0.1% Triton X-100 and scraped into distilled water. ALP activity was measured using an ALP assay kit (Nanjing Jiancheng Biotechnology Co. Ltd., Nanjing, China) in accordance with the manufacturer’s instructions. Absorbance was measured at 490 nm using a RT-6000 Enzyme Label Analyzer (Shenzhen Leidu Life Technology Co. Ltd., Shenzhen, China).
Mineralization assay
Alizarin red staining (ARS) was performed to determine the mineralization of MC3T3-E1 cells after 28 days of culture. Cells were washed twice with phosphate-buffered saline (PBS) and fixed in 95% ethanol for 10 min, and 1% alizarin red (Sigma, USA) was added and incubated for 30 min and then decolorized with 50% ethanol. A microscope (Olympus, Tokyo, Japan) was used to determine the number of mineralized nodules with a 10 mm coordinate network. To detect the calcium content in mineralized nodules, the absorbance was then measured at 570 nm [9]. The calcium content of the cell layer was normalized to the total DNA content.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from cell monolayers using TRIzol reagent (TaKaRa, Tokyo, Japan). cDNA was synthesized using the Prime Script™ RT reagent Kit according to the manufacturer’s instructions (TaKaRa, Tokyo, Japan). Then, cDNA was used for RT-PCR assays conducted on an ABI 7300 FAST Real-Time PCR system (ABI, USA) using SYBR Premix Ex Taq TM (TaKaRa, Tokyo, Japan). A 40-cycle thermal program was used, consisting of predenaturation at 95 °C for 30 s, denaturation at 95 °C for 5 s and annealing at 60 °C for 31 s. Each experiment was performed in triplicate. The relative mRNA expression was normalized to β-actin and calculated using the 2−∆∆CT method. The sequences of RT-PCR primers are shown in Table 1.
Western blotting
Western blotting was performed to assess the protein expression of GRP-78 and ATF-4. Briefly, protein concentration was measured using the BCA Protein Assay Kit (Solarbio). Equal protein concentrations were loaded, separated by SDS-PAGE (Solarbio), and transferred to PVDF membranes (Solarbio), which were blocked and incubated with a primary antibody at 4 °C overnight and then with secondary antibodies for 2 h at room temperature. Immunoreactivity was detected using an enhanced chemiluminescence system (Millipore). Quantification of western blots was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data are shown as the mean ± standard deviation (SD) and were analyzed using one-way analysis of variance (ANOVA) with GraphPad Prism 7.0. P values less than 0.05 were considered statistically significant.
Results
FA improves the morphology of MC3T3-E1 cells induced by Hcy
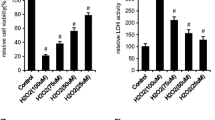
Representative fluorescence images of MC3T3-E1 cells labeled with live/dead dye solution are shown in Fig. 2, where live cells are labeled in green and dead cells in red. After 48 h in culture, the cell population was uniformly distributed and established frequent cell-to-cell connections, and the typical morphology of MC3T3-E1 cells, such as polygonal, triangular and spindle-shaped cells, is shown in green. The effects of Hcy and FA on cell density and cell survival were further explored, and the results showed that after treatment of the cells with different concentrations of Hcy, a reduced density of cells and a lower percentage of live cells were observed and dead cells were appeared in Hcy-treated cells at concentrations of 10–3 and 10–2 mol/L (Fig. 2a, c, e). However, after adding FA to interfere with Hcy-induced MC3T3-E1 cells, the live cells gradually appeared denser and more fused, and dead cells gradually disappeared as the FA concentration increased (Fig. 2b). As shown in Fig. 2d, f, quantitative analysis also showed that the percentage of live cells increased from 21.8 to 93.2 and the density of cells increased from 6.5 × 104 to 10.5 × 104 cells cm−2 with the increasing FA concentration (0–10–4 mol/L).

Representative fluorescent images of MC3T3 cells with live cells stained as green and dead cells as red. a The morphology of MC3T3-E1 cells induced by Hcy. b The morphology of Hcy-induced MC3T3-E1 cells treated with FA. c, d The percentage of live cells. e, f The number of cells per cm2 was calculated (× 104/cm2). Data are represented as the mean ± SD. ***P < 0.001
FA stimulates cell proliferation in MC3T3-E1 cells induced by Hcy
The effects of Hcy on MC3T3-E1 cell proliferation were assessed using an MTT assay. We found that the cell proliferation ability of MC3T3-E1 cells exposed to different concentrations of Hcy (0, 10–5, 10–4, 10–3 and 10–2 mol/L) was significantly decreased (Fig. 3a). Treatment of MC3T3-E1 cells with different concentrations of FA (0, 10–7, 10–6, 10–5 and 10–4 mol/L) resulted in increased cell proliferation ability, suggesting a stimulatory effect on cell proliferation in response to FA. Meanwhile, the stimulatory effect of FA was dose dependent (Fig. 3b). The optimal stimulatory effect on cell proliferation was observed in response to 10–4 mol/L FA treatment.

Cell proliferation was evaluated using MTT assay. a The ability of cell proliferation in MC3T3-E1 cells with different concentrations of HCY (0, 10–5, 10–4, 10–3 and 10–2 mol/L). b The ability of cell proliferation in MC3T3-E1 cells after treated with different concentrations of FA (0, 10–7, 10–6, 10–5 and 10–4 mol/L). Data are represented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
FA promotes osteogenic differentiation in MC3T3-E1 cells induced by Hcy
Osteoblast differentiation is the most essential step for bone formation. To elucidate the effect of FA on the process of osteoblast differentiation, ALP activity was investigated after cells had been cultured in Hcy-containing medium for 3 days with or without FA. We found that the ALP activity decreased dramatically with increasing concentrations of Hcy (0–10–2 mol/L). However, MC3T3-E1 cells treated with FA exhibited a gradual increase in ALP activity (Fig. 4a, b).

The osteogenic differentiation in MC3T3-E1 cells. a Effect of Hcy on ALP activity. b Effect of FA on ALP activity in Hcy-induced MC3T3-E1 cells. c Effect of Hcy on the mineralization of MC3T3-E1 cells (100 ×). d Effect of FA on the mineralization of MC3T3-E1 cells induced by Hcy (100 ×). e–h Quantification of mineralized nodules number and calcium contents were performed in Hcy-induced cells threated with or without FA. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
Mineralization of these cells was investigated using Alizarin red staining, and the total number of nodules per culture dish was quantified macroscopically. In regular culture medium without Hcy, an average of 100 nodules were counted per well. However, when different concentrations of Hcy were added to the medium, the number of mineralized nodules gradually decreased in a concentration-dependent manner as the Hcy concentrations increased (Fig. 4c, e). To determine whether the addition of FA modified the effects of Hcy, FA (10−7–10−4 mol/L) was added to the culture medium after 24 h of Hcy exposure, which showed that FA stimulated the number of nodules formed in a dose-dependent manner (Fig. 4d, f). In addition, calcium deposition in the mineralized nodules was also assessed using Alizarin red staining. Similarly, the calcium content was significantly inhibited by Hcy (Fig. 4g), whereas FA significantly stimulated calcium deposition in all Hcy-containing media in a dose-dependent manner, with maximal stimulation being reached at a FA concentration of 10–4 mol/L (Fig. 4h). In summary, FA treatment attenuates the deleterious effects of Hcy on osteogenic differentiation.
FA reduces apoptosis induced by Hcy in MC3T3-E1 cells
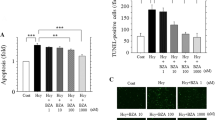
We first examined gene expression levels of the apoptosis-related proteins Bcl-2 and Bax in cells with Hcy administration. The data showed that Bax expression was increased and Bcl-2 expression was decreased in cells exposed to Hcy (Fig. 5a), indicating that Hcy promoted apoptosis of osteoblasts. Prolonged or severe ERS induces apoptosis. To examine whether FA ameliorates Hcy-induced apoptosis in osteoblasts, MC3T3-E1 cells were treated with different concentrations of FA after a 24-h incubation with 10–3 mol/L Hcy. The gene expression levels of Bcl-2 were increased while those of Bax were decreased as the FA concentration increased (Fig. 5b). Taken together, these results demonstrate that FA significantly ameliorates Hcy-induced apoptosis in MC3T3-E1 cells.

FA protects MC3T3-E1 cells against apoptosis induced by Hcy. a The gene expression levels of Bcl-2 and Bax in cells interfered with Hcy for 24 h. b The expression levels of Bax and Bcl-2 with different concentrations of FA after a 24-h incubation with Hcy at a concentration of 10−3 mol/L. Data are represented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
FA reduces ERS induced by Hcy in osteoblast cells
In this study, the ERS response was assessed by detecting gene and protein expression levels of the ERS markers GRP78, CHOP and ATF-4. Gene expression of GRP-78, CHOP and ATF-4 as well as the protein expression of GRP-78 and ATF-4 were enhanced with increasing concentrations of Hcy (Fig. 6a–c), indicating that ERS is involved in Hcy-induced apoptosis in osteoblasts. To determine whether FA alters ERS in osteoblasts exposed to Hcy, MC3T3-E1 cells were treated with different concentrations of FA after a 24-h incubation with 10–3 mol/L Hcy. Quantitative analysis by western blot and PCR showed that expression levels of GRP78, CHOP and ATF-4 were significantly decreased in response to FA treatment (Fig. 6d–f). These findings suggest that FA treatment effectively rescues the Hcy-induced ERS response in MC3T3-E1 cells.

The gene and protein expression levels of ERS related molecules. a–c Western blots and quantitative analysis were conducted to assess the expression levels of GRP78, CHOP and ATF-4 in MC3T3-E1 cells treated with Hcy. d–f The expression levels of GRP78, CHOP and ATF-4 in Hcy-induced MC3T3-E1 cells after FA intervention. Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
The present study demonstrated that Hcy inhibits the proliferation, differentiation and mineralization of osteoblasts in a concentration-dependent manner and induces osteoblast apoptosis by activating the ERS signaling pathway. However, FA reverses these Hcy-mediated effects. After intervention with FA, osteoblast morphology, cell density, cell activity and cell proliferation ability were significantly improved, and the mineralization capacity of osteoblasts was enhanced, as demonstrated by the increase in ALP levels, mineralized nodule number and calcium content. In addition, FA protected against apoptosis of osteoblasts induced by Hcy by inhibiting the PERK pathway of ERS, further confirming our hypothesis.
Osteoporosis is a skeletal disease characterized by low bone mass and microarchitectural deterioration of bone tissue, leading to increased bone fragility and fracture susceptibility, which seriously affects patient quality of life. Imbalanced osteoblastic bone formation and osteoclastic bone resorption from various causes is the primary mechanism underlying osteoporosis and osteoporotic fracture [10]. Osteoblasts are major bone-forming cells that undergo proliferation, mineralization, renewal, and repair during the bone formation process, and each stage is affected and regulated by a variety of factors, including genetic backgrounds, hormone levels and cellular regulatory factors [11]. In recent years, HHcy has been identified as a new risk factor for osteoporotic fractures. A population-based prospective study found that increased plasma Hcy is associated with low bone mineral density and a high risk of fracture, while folic acid and vitamin B12 supplementation reduce the risk of hip fracture [12]. In vivo, bone fragility at the femoral neck in Hcy-fed rats was increased by nearly 36%, the content of osteocalcin, a bone formation marker, decreased by 34%, and bone quality was consistently decreased in the presence of increased circulating Hcy levels [13]. Previous studies have shown that Hcy not only weakens the function of osteoblasts, promotes the activity of osteoclasts and increases bone resorption, but also results in a detrimental effect on bone by decreasing bone blood flow, increasing the levels of reactive oxygen species and activating the expression of matrix metalloproteinases that degrade extracellular bone matrix [6]. Furthermore, Hcy influences the formation of a stable bone matrix by inhibiting the collagen cross-linking enzyme lysyl oxidase in osteoblasts [14]. Similarly, our study found that Hcy not only reduces the density of MC3T3-E1 cells, hinders the proliferative capacity of osteoblasts but also inhibits the activity of ALP and decreases the number and calcium content of calcified nodules, suggesting a deleterious effect of Hcy on osteoblasts.
The endoplasmic reticulum (ER) is primarily recognized as a protein-folding factory that is responsible for the biosynthesis, folding and processing of proteins. Under physiological conditions, abnormally folded or unfolded proteins can be degraded by the proteasome through ER-related degradation processes. However, in the case of pathology, the accumulation and aggregation of unfolded or misfolded proteins in the ER lumen causes ER dysfunction and triggers an adaptive signaling cascade called ERS [15]. GRP-78, an ER resident chaperone, regulates the activation of each of the three unfolded protein response pathways: inositol-requiring enzyme 1α(IRE-1α), activating transcription factor 6 (ATF-6) and PERK [16]. ATF-4, a dominant protein selectively translated in the PERK/eIF-2α, is the primary regulator of transcription of key genes for adaptive response and has many biological functions, including participation in amino acid metabolism, bone metabolism and induction of apoptosis and autophagy [17,18,19,20]. ATF-4 cooperates with many regulators of osteogenic differentiation to regulate the expression of osteogenic-related genes, thereby regulating osteogenic differentiation [21]. However, under long-term sustained or excessive ERS conditions, the PERK/eIF-2α pathway is activated, which stimulates increased biosynthesis of ATF-4 and further upregulates the expression of CHOP/GADD153, while high expression of CHOP induces osteoblast apoptosis by upregulating the expression of the pro-apoptotic protein Bax, downregulating the anti-apoptotic protein Bcl-2 and promoting the formation of apoptotic bodies and activation of caspases [22, 23]. Moreover, it has been shown that eIF-2α activates a cytoprotective intracellular degradation system, autophagy, as an alternative pathway to degrade misfolded proteins accumulated in cells to reduce bone loss [24]. Of course, it has also been suggested that the effects of autophagy induced by ERS on cell survival are different and have dual roles, including both pro-survival and pro-death, which may be dependent on the extent of ERS [25].
Studies have shown that high levels of Hcy upregulate the mRNA expression of GRP-78, ATF-4 and CHOP/GADD153 in MC3T3-E1 cells, suggesting that Hcy is an important factor mediating PERK activation-induced apoptosis in ER stress [26]. A previous study found that Hcy reduced osteoblasts viability in a dose- and time-dependent manner; at the same time, the expression of ERS-associated molecules, such as IRE-1α, GRP-78 and ATF-4 was significantly increased in Hcy-treated osteoblasts, indicating that Hcy-mediated ERS activation promotes osteoblast damage [27]. In vivo, it has been shown that overexpression of CHOP, which appears as a result of ERS induced by Hcy, contributes to osteoblast apoptosis by upregulating the expression of Bax and downregulating Bcl-2 [28, 29]. Our results are consistent with previous reports showing that along with the increase in Hcy exposure intensity, an increase in the gene expression of GRP-78, ATF-4, CHOP/GADD153 and Bax and a decrease in Bcl-2 mRNA expression were observed, further confirming that Hcy induces osteoblast apoptosis by activating the ERS pathway. As a cell survival pathway, autophagy also plays a crucial role in maintaining bone homeostasis [30]. The relevance of Hcy to autophagy has been demonstrated in other cells, including hepatocytes, cardiomyocytes, and nerve cells [31,32,33], but whether Hcy regulates osteoblast differentiation by affecting autophagy is poorly understood. Studies have suggested that Hcy induces autophagy by activating oxidative stress, ERS and mitophagy. During the early stage, Hcy-induced autophagy has a protective effect on cell survival, whereas when the body is in a long-term stress state, the levels of autophagy-related proteins and calcium ions increase, resulting in mitochondrial autophagy and injury, ultimately leading to apoptosis [34]. This study did not assess the expression of autophagy-related proteins, and the role of autophagy in these processes is still unclear. Therefore, it is necessary to explore the mechanisms of autophagy in Hcy-induced osteoblast injury in the future.
FA is involved in multiple metabolic processes, such as carbon unit transfer, DNA methylation, methionine synthesis and purine synthesis in vivo and plays an important modulatory effect on cell division, homeostasis and gene expression. Deficiency of FA can result in a variety of pathological mechanisms, such as hepatic steatosis, insulin resistance, endothelial cell dysfunction and osteoblast apoptosis. Among them, ERS has been proposed to explain observed pathological changes [35,36,37,38]. A previous study indicated that the mRNA and protein expression of the ERS-associated proteins Bip, PERK, EIF2α and CHOP in Hcy-induced hepatocytes was upregulated both in vivo and in vitro; moreover, expression of the apoptosis-associated proteins caspase-3 and caspase-12 was also increased. Most importantly, the ERS-associated apoptosis induced by Hcy was reversed after folate supplementation [35]. Another study demonstrated that FA deficiency may lead to ERS by interfering with the formation of disulfide bonds and activating the unfolded protein response, thereby activating the transcription of genes related to fat formation [36]. Hsu et al. found that folic acid deficiency is a risk factor for diabetes, which can induce ERS by upregulating CHOP expression and promoting apoptosis of insulin-producing pancreatic RINm5F islet β cells [37]. Clinical studies have reported that patients with insufficient folic acid intake have an increased risk of cardiovascular disease. The possible mechanism whereby this occurs is through to involve accumulation of Hcy causing folic acid deficiency, which triggers ERS and induces vascular endothelial cell dysfunction, promoting vascular smooth muscle cell proliferation, and further accelerating atherosclerosis formation [38].
To date, few studies have reported on the correlation between FA and bone metabolism. Clinical studies have found that the risk of osteoporosis and fracture in patients with folate deficiency is significantly increased, but the specific molecular mechanism remains unclear [39]. Adequate FA intake can prevent DNA damage, reduce oxidative stress and prevent apoptosis, which may be a potential mechanism for the protective effect of FA on bone cells [40]. Studies have shown that FA deficiency directly activates autophagy and induces cell death [41], but the effect of FA deficiency-induced autophagy on osteoblasts has not been reported. In addition, FA deficiency interferes with bone collagen connections and fiber formation by affecting Hcy metabolism, subsequently affecting bone mineralization capacity [42]. However, FA intervention reduces Hcy concentration in ovariectomized rats, increases levels of ALP in bone tissue homogenate, increases the bone mineral density of the lumbar spine and femur, improves the internal quality of the lumbar spine, and enhances the hardness and flexibility of the lumbar spine [8]. In our study, FA was used to interfere with Hcy-induced MC3T3-E1 cells to explore whether FA affected the proliferation, differentiation and calcification of osteoblasts through the ERS pathway. It was concluded that FA significantly attenuates the deleterious effects of Hcy on osteoblasts, as demonstrated by increasing cell viability and number, enhancing ALP activity and the mineralization ability of osteoblasts. Moreover, with increasing FA intervention concentration, the mRNA expression of GRP-78, ATF-4, CHOP/GADD153 and BAX in MC3T3-E1 cells was downregulated, while the mRNA expression of Bcl-2 gradually increased. Similar results were observed at the protein level, which further confirmed that FA has a beneficial effect on bone metabolism and partially attenuates the damage to osteoblasts induced by Hcy through the PERK-mediated ERS pathway.
In conclusion, this study clearly demonstrates that the adverse effects of Hcy on osteoblast proliferation and differentiation are dose-dependent. FA not only significantly enhances cell viability and protects against osteoblasts apoptosis but also has a direct osteogenic effect on osteoblasts; furthermore, these actions could be partly mediated by inhibition of the PERK-mediated ERS pathway. However, whether FA achieves these bone-protective effects by inhibiting two other ERS pathways, IRE-1α and ATF-6, remains unclear. Therefore, additional basic and clinical studies are still needed to further confirm the effects of FA on bone metabolism.
References
Ganguly P, Alam SF (2015) Role of homocysteine in the development of cardiovascular disease. Nutr J 14:6
Weekman EM, Woolums AE, Sudduth TL, Wilcock DM (2017) Hyperhomocysteinemia-induced gene expression changes in the cell types of the brain. ASN Neuro 9:1759091417742296
Huang P, Wang F, Sah BK, Jiang J, Ni Z, Wang J, Sun X (2015) Homocysteine and the risk of age-related macular degeneration: a systematic review and meta-analysis. Sci Rep 5:10585
Zhang D, Wen X, Wu W, Guo Y, Cui W (2015) Elevated homocysteine level and folate deficiencyassociated with increased overall risk of carcinogenesis: meta-analysis of 83 case-control studies involving 35,758 individuals. PLoS ONE 10:e0123423
Zhai Y, Behera J, Tyagi SC, Tyagi N (2019) Hydrogen sulfide attenuates homocysteine-induced osteoblast dysfunction by inhibiting mitochondrial toxicity. J Cell Physiol 234:18602–18614
Vacek TP, Kalani A, Voor MJ, Tyagi SC, Tyagi N (2013) The role of homocysteine in bone remodeling. Clin Chem Lab Med 51:579–590
Moshal KS, Singh M, Sen U, Rosenberger DS, Henderson B, Tyagi N, Zhang H, Tyagi SC (2006) Homocysteine-mediated activation and mitochondrial translocation of calpain regulates MMP-9 in MVEC. Am J Physiol Heart Circ Physiol 291:H2825-2835
Wang HM, Tang XL, Tian DZ (2011) Protective effect of folic acid on osteoporosis in ovariectomized rats. Chin J Endocrinol Metab 27:594–598
Ratisoontorn C, Seto ML, Broughton KM, Cunningham ML (2005) In vitro differentiation profifile of osteoblasts derived from patients with Saethre-Chotzen syndrome. Bone 36:627–634
Seeman E (2002) Pathogenesis of bone fragility in women and men. Lancet 359:1841
Franz-Odendaal TA, Hall BK, Witten PE (2006) Buried alive: how osteoblasts become osteocytes. Dev Dyn 235:176–190
Gjesdal CG, Vollset SE, Ueland PM, Refsum H, Meyer HE, Tell GS (2007) Plasma homocysteine, folate, and vitamin B 12 and the risk of hip fracture: the hordaland homocysteine study. J Bone Miner Res 22:747–756
Herrmann M, Wildemann B, Claes L, Klohs S, Ohnmacht M, Taban-Shomal O, Hübner U, Pexa A, Umanskaya N, Herrmann W (2007) Experimental hyperhomocysteinemia reduces bone quality in rats. Clin Chem 53:1455–1461
Thaler R, Agsten M, Spitzer S, Paschalis EP, Karlic H, Klaushofer K, Varga F (2011) Homocysteine suppresses the expression of the collagen cross-linker lysyl oxidase involving IL-6, Fli1, and epigenetic DNA methylation. J Biol Chem 286:5578–5588
Zhang K, Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462
Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ (1999) Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J 339:135–141
Kilberg MS, Shan J, Su N (2009) ATF4-dependent transcriptionmediates signaling of amino acid limitation. Trends Endocrinol Metab 20:436–443
Nakamura S, Miki H, Kido S, Nakano A, Hiasa M, Oda A, Amou H, Watanabe K, Harada T, Fujii S, Takeuchi K, Kagawa K, Ozaki S, Matsumoto T, Abe M (2013) Activating transcription factor 4, an ER stress mediator, is required for, but excessive ER stress suppresses osteoblastogenesis by bortezomib. Int J Hematol 98:66–73
Oakes SA, Papa FR (2015) The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 10:173–194
Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL (2010) Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 29:4424–4435
Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, Hanauer A, Karsenty G (2004) ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 117:387–398
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389
Chen Y, Gao H, Yin Q, Chen L, Dong P, Zhang X, Kang J (2013) ER stress activating ATF4/CHOP-TNF-α signaling pathway contributes to alcohol-induced disruption of osteogenic lineage of multipotential mesenchymal stem cell. Cell Physiol Biochem 32:743–754
Li J, Li X, Liu D, Hamamura K, Wan Q, Na S, Yokota H, Zhang P (2019) eIF2α signaling regulates autophagy of osteoblasts and the development of osteoclasts in OVX mice. Cell Death Dis 10:921
Song S, Tan J, Miao Y, Li M, Zhang Q (2017) Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol 232:2977–2984
Kanazawa I, Tomita T, Miyazaki S, Ozawa E, Yamamoto LA, Sugimoto T (2017) Bazedoxifene ameliorates homocysteine-induced apoptosis and accumulation of advanced glycation end products by reducing oxidative stress in MC3T3-E1 cells. Calcif Tissue Int 100:286–297
Park SJ, Kim KJ, Kim WU, Oh IH, Cho CS (2012) Involvement of endoplasmic reticulum stress in homocysteine-induced apoptosis of osteoblastic cells. J Bone Miner Metab 30:474–484
Shirakawa K, Maeda S, Gotoh T, Hayashi M, Shinomiya K, Ehata S, Nishimura R, Mori M, Onozaki K, Hayashi H, Uematsu S, Akira S, Ogata E, Miyazono K, Imamura T (2006) CCAAT/enhancer-binding protein homologous protein (CHOP) regulates osteoblast differentiation. Mol Cell Biol 26:6105–6116
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12:982–995
Li W, Zhang S, Liu J, Liu Y, Liang Q (2019) Vitamin K2 stimulates MC3T3-E1 osteoblast differentiation and mineralization through autophagy induction. Mol Med Rep 19:3676–3684
Yang A, Jiao Y, Yang S, Deng M, Yang X, Mao C, Sun Y, Ding N, Li N, Zhang M, Jin S, Zhang H, Jiang Y (2018) Homocysteine activates autophagy by inhibition of CFTR expression via interaction between DNA methylation and H3K27me3 in mouse liver. Cell Death Dis 9:169
Vacek TP, Vacek JC, Tyagi N, Tyagi SC (2012) Autophagy and heart failure: a possible role for homocysteine. Cell Biochem Biophys 62:1–11
Zhao Y, Huang G, Chen S, Gou Y, Dong Z, Zhang X (2016) Homocysteine aggravates cortical neural cell injury through neuronal autophagy overactivation following rat cerebral ischemia-reperfusion. Int J Mol 17:1196
Tripathi M, Zhang CW, Singh BK, Sinha RA, Moe KT, DeSilva DA, Yen PM (2016) Hyperhomocysteinemia causes ER stress and impaired autophagy that is reversed by Vitamin B supplementation. Cell Death Dis 7:e2513
Yang A, Sun Y, Mao C, Yang S, Huang M, Deng M, Ding N, Yang X, Zhang M, Jin S, Jiang Y, Huang Y (2017) Folate protects hepatocytes of hyperhomocysteinemia mice from apoptosis via cystic fibrosis transmembrane conductance regulator (CFTR)-activated endoplasmic reticulum stress. J Cell Biochem 118:2921–2932
Shinohara M, Ji C, Kaplowitz N (2010) Differences in betaine-homocysteine methyltransferase expression, endoplasmic reticulum stress response, and liver injury between alcohol-fed mice and rats. Hepatology 51:796–805
Hsu HC, Chiou JF, Wang YH, Chen CH, Mau SY, Ho CT, Chang PJ, Liu TZ, Chen CH (2013) Folate deficiency triggers an oxidative-nitrosative stress-mediated apoptotic cell death and impedes insulin biosynthesis in RINm5F pancreatic islet β-cells: relevant to the pathogenesis of diabetes. PLoS ONE 8:e77931
Gérard N, Chanson-Rollé A, Rock E, Brachet P (2014) Proteomic analysis identifies cytoskeleton-interacting proteins as major downstream targets of altered folate status in the aorta of adult rat. Mol Nutr Food Res 58:2307–2319
Akpolat V, Bilgin HM, Celik MY, Erdemoglu M, Isik B (2013) An evaluation of nitric oxide, folate, homocysteine levels and lipid peroxidation in postmenopausal osteoporosis. Adv Clin Exp Med 22:403–409
Luckock M (2000) Folic acid: nutritional biochemestry, molecular biology, and role in disease processes. Mol Genet Metab 71:121–138
Yin X, Gao R, Geng Y, Chen X, Liu X, Mu X, Ding Y, Wang Y, He J (2019) Autophagy regulates abnormal placentation induced by folate deficiency in mice. Mol Hum Reprod 25:305–319
Herrmann M, Peter Schmidt J, Umanskaya N, Wagner A, Taban-Shomal O, Widmann T, Colaianni G, Wildemann B, Herrmann W (2007) The role of hyperhomocysteinemia as well as folate, vitamin B (6) and B (12) deficiencies in osteoporosis: a systematic review. Clin Chem Lab Med 45:1621–1632
Acknowledgements
This work was supported by the Diabetes Clinical Research Project of Shanghai Medical and Health Development Foundation (Phase I 10 Study) [Grant number: DMRFP-I-10]; Special Research Fund of Standardized Metabolic Disease Management Center [Grant number: 2018-mmczxjj-3]; Natural Science Foundation of Gansu Province [Grant number: 21JR1RA096, 1308RJZA254, 1606RJZA347] and the Fundamental Research Funds for the Central Universities [Grant number: 2022142zrk012].
Author information
Authors and Affiliations
Contributions
SS, DZ and JJL completed the project, analyzed the data, and drafted the manuscript; XLT and HXC designed the study, interpreted the data and contributed to critically revising the manuscript; HYZ, QMW and WNR participated in experimental operations; DHZ provided financial support and supervised data collection and manuscript writing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors have no interests to disclose.
Ethical approval
All authors have read the Journal’s position on issues involved in ethical publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Su, S., Zhang, D., Liu, J. et al. Folate ameliorates homocysteine-induced osteoblast dysfunction by reducing endoplasmic reticulum stress-activated PERK/ATF-4/CHOP pathway in MC3T3-E1 cells. J Bone Miner Metab 40, 422–433 (2022). https://doi.org/10.1007/s00774-022-01313-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-022-01313-x