
Abstract
Leucine (Leu) is a nutritionally essential branched-chain amino acid (BCAA) in animal nutrition. It is usually one of the most abundant amino acids in high-quality protein foods. Leu increases protein synthesis through activation of the mammalian target of rapamycin (mTOR) signaling pathway in skeletal muscle, adipose tissue and placental cells. Leu promotes energy metabolism (glucose uptake, mitochondrial biogenesis, and fatty acid oxidation) to provide energy for protein synthesis, while inhibiting protein degradation. Approximately 80 % of Leu is normally used for protein synthesis, while the remainder is converted to α-ketoisocaproate (α-KIC) and β-hydroxy-β-methylbutyrate (HMB) in skeletal muscle. Therefore, it has been hypothesized that some of the functions of Leu are modulated by its metabolites. Both α-KIC and HMB have recently received considerable attention as nutritional supplements used to increase protein synthesis, inhibit protein degradation, and regulate energy homeostasis in a variety of in vitro and in vivo models. Leu and its metabolites hold great promise to enhance the growth and health of animals (including humans, birds and fish).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Leucine (Leu) is present in all proteins (Wu 2013a) and is one of the most abundant amino acids in high-quality protein foods (Li et al. 2011b). Leucine plays a unique signaling role in skeletal muscle and adipose tissue (Li et al. 2011a), as well as other cell types, including placental cells (Kim 2007; Kim et al. 2011a, b; Li et al. 2007), mammary epithelial cells (Lei et al. 2012a), and enterocytes (Rhoads and Wu 2009). In those tissues, Leu promotes protein synthesis via activating the mammalian target of rapamycin (mTOR) signaling pathway, and also enhances energy homeostasis through augmenting mitochondrial biogenesis and fatty acid oxidation (Filhiol 2012). Moreover, Leu provides skeletal muscles with an increased flux of lipids, supplying energy substrates to support protein synthesis (Sun and Zemel 2009). However, when skeletal muscle is exposed to physiological concentrations of fatty acids in vivo, oxidation of leucine as a significant source of ATP is likely limited (Jobgen et al. 2006; Wu et al. 2014) primarily because the activity of branched-chain α-keto acid dehydrogenase activity (BCKD) is relatively low in this tissue (Wu and Thompson 1987).
The activation of AMP-activated protein kinase (AMPK) and silent information regulator transcript 1 (SIRT1) by Leu is the major event that mediates fatty acid oxidation and mitochondrial biogenesis in skeletal muscle (Liang et al. 2014). Protein degradation and synthesis are equally important processes, but little is known about the mechanisms of Leu-regulated proteolysis (Nakashima et al. 2005). The key metabolites of Leu are α-ketoisocaproate (α-KIC) and β-hydroxy-β-methylbutyrate (HMB). Both are nutritional supplements that are nitrogen-free and beneficial to the environment. It remains elusive whether the varied physiological roles in protein metabolism, insulin secretion, glucose homeostasis, fatty acid oxidation, and mitochondrial function are mediated by Leu alone, or in concert with one of Leu’s unique metabolites. Therefore, this review aims to discuss the metabolic impact of Leu and its metabolites α-KIC and HMB on protein and energy metabolism.
Overview of Leu metabolism
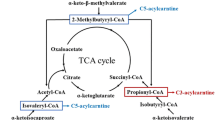
Leu metabolism in the body can be divided into two steps. The first step is the reversible transamination of Leu to form α-KIC, with the concomitant production of glutamate from α-ketoglutarate (α-KG) (Fig. 1). This reaction is catalyzed by branched-chain amino-acid aminotransferase (BCAT) (Wilkinson et al. 2013), which is highly expressed in muscle tissue (Su et al. 2012). There are two mammalian BCAT isoenzymes: a cytosolic (BCATc) and mitochondrial (BCATm) form. BCATc is present in the placenta, ovary, and brain (Suryawan et al. 1998) as well as mammary tissue and small intestine (Lei et al. 2012b, 2013; Li et al. 2009), whereas BCATm is expressed in most peripheral tissues except the liver (Zhou et al. 2010). Glutamate, a product of branched-chain amino acid (BCAA) transamination, is either amidinated with ammonia to form glutamine or transaminated with pyruvate to generate alanine in multiple tissues, including skeletal muscle (Wu et al. 1989), placenta (Self et al. 2004) and mammary tissue (Lei et al. 2012a). Interestingly, there is no BCAT isoenzyme in the liver (Hutson et al. 1992; Hall et al. 1993). The tissue supply of Leu depends on either exogenous (dietary) or endogenous (protein degradation) provision (Nissen and Abumrad 1997). Leu from dietary protein can bypass first metabolism in the liver, which results in a sharp rise of plasma Leu levels and the activation of Leu signaling in peripheral tissues in response to a meal (Lynch et al. 2006). The subsequently produced α-KIC is released into the bloodstream and taken up by different tissues where it can be oxidized or used for re-synthesis of Leu. The Leu released from the liver to the bloodstream may be used in skeletal muscle to synthesize protein or alanine and glutamine (Holecek 2002). Collectively, in extrahepatic tissues, Leu is converted to α-KIC, and then α-KIC can be re-synthesized into Leu or be further oxidized.

Pathways of leucine metabolism in animals. Leucine metabolism in the body can be divided into two steps. The first step is reversible transamination to form α-KIC with concomitant production of Glu from α-KG. This step occurs mainly in extrahepatic tissues. The second step is irreversible oxidative decarboxylation of α-KIC catalyzed by the mitochondrial BCKD complex and KICD. In the liver mitochondria, approximately 90 % of produced α-KIC is oxidized to IVA-CoA, leading to the formation of HMG-CoA and final metabolites (acetoacetate and acetyl-CoA). Within the liver cytosol, the remaining α-KIC (~5 %) is oxidized to HMB by the enzyme KICD
The second step in Leu catabolism is the irreversible oxidative decarboxylation of α-KIC, which is catalyzed by BCKD complex located within the mitochondrion (Fig. 1). As noted previously, the BCKD complex has a low activity in skeletal muscle, but is highly active in the liver, and shows intermediate activity in the heart and kidney (Suryawan et al. 1998). Therefore, the majority of α-KIC oxidation occurs in the liver. Within the liver mitochondria, approximately 90 % of all produced α-KIC is oxidized to isovaleryl CoA (IVA-CoA), leading to the formation of β-hydroxy-β-methylglutaryl-CoA (HMG-CoA) and the final metabolites acetoacetate and acetyl-CoA. Within the liver cytosol, the remaining α-KIC is oxidized to HMB by the enzyme α-KIC dioxygenase (KICD) (Nissen and Abumrad 1997). Subsequently, HMB, generated exclusively from Leu, is either eliminated by the kidneys or metabolized to HMG-CoA (Van Koevering and Nissen 1992; Panton 2000; Kovarik et al. 2010), which is the precursor for cholesterol synthesis (Rudney 1957; Holecek et al. 2009). Therefore, the second catabolic step commits the Leu carbon skeleton to the degradation pathway (She et al. 2013).
However, the amount of endogenously generated HMB is quantitatively small. About 80 % of Leu is normally used for protein synthesis while the remainder is converted to α-KIC, and only a small proportion of Leu (about 5 %) is eventually converted into HMB (Van Koevering and Nissen 1992; Molfino et al. 2013). The recommended dose of HMB is 3 g/day for adult humans. A 70-kg individual generates about 0.2–0.4 g HMB/day, depending on the Leu content in the diet (Nissen 1997). Although HMB can also be found in nature, the quantities required to increase protein synthesis and to decrease proteolysis cannot be consumed within a normal diet (Gerlinger-Romero et al. 2011). To achieve HMB’s beneficial effects, 60 g of Leu is required, but it is beyond the amount of a balanced diet (Zanchi et al. 2011). Thus, supplementation of HMB has been employed as an effective alternative nutrition strategy, as the majority of supplemental HMB is retained by the body (Fitschen et al. 2013). We will not only discuss the positive effects of Leu on protein metabolism but also those of the major Leu metabolites.
The role of Leu and its metabolites in protein metabolism
Although the role of Leu in protein synthesis has been known for decades (Buse and Reid 1975; Anthony et al. 2000a, b), the functions of Leu metabolites in protein metabolism remain elusive. Numerous studies demonstrate that these metabolites may be partly responsible for the positive effects of Leu (Slater and Jenkins 2000; Wilson et al. 2008; Pinheiro et al. 2012). Other BCAAs, including isoleucine and valine, cannot trigger the same metabolic effects, reinforcing the supposition that Leu metabolites are critical for the anti-catabolic function of Leu (Holecek et al. 2009).
Protein synthesis
Amino acids, especially Leu, function not only as a substrate for newly synthesized protein, but also as a “trigger” (signaling molecule) for the initiation of protein synthesis (Atherton et al. 2010; Columbus et al. 2014). The modulation of protein synthesis by Leu in skeletal muscle has been reviewed extensively (Kim et al. 2007; Yin et al. 2010; Li et al. 2011a; Yao et al. 2012; Duan et al. 2015a, b). During protein synthesis in skeletal muscle, the efflux of glutamine is accompanied with the uptake of Leu via amino acid transporters, more specifically solute carrier family 1 member 5 and solute carrier family 7 member 5/SLC3A2 (Nicklin et al. 2009). The intracellular Leu concentration is detected by amino acid sensors (Han et al. 2012), which activate mediators such as Ras-related guanosine triphosphatases (Suzuki and Inoki 2011), human vacuolar sorting protein-34 (Nobukuni et al. 2005), and mitogen-activated protein kinase kinase kinase kinase-3 (Suzuki and Inoki 2011). These mediators trigger the translocation of the mTOR complex 1 (mTORC1) to the surface of late endosomes and lysosomes where it is directly activated by Ras homolog enriched in brain, which resides on the surface of the lysosome. Once mTORC1 is activated, it regulates the phosphorylation and activation of downstream proteins that modulate the initiation and/or elongation steps in translation. The main targets include 4E binding protein 1 (4E-BP1), ribosomal S6 kinases (S6K1), and eukaryotic initiation factor 2 (eIF2α) (Talvas et al. 2006). Leu can also induce protein synthesis through directly activating the eukaryotic initiation factor 4G (eIF4G) in an mTOR-independent manner (Bolster et al. 2004b).
More than 30 years ago, it was reported that α-KIC did not influence the rate of protein synthesis in skeletal muscle (Tischler 1982), and that Leu can still stimulate protein synthesis when transamination is suppressed (Mitch and Clark 1984). Based on these findings, Leu, rather than its metabolites, has been suggested as a direct nutrient signal for protein synthesis (Lynch et al. 2002, 2003; Garlick 2005). However, this idea has been challenged by an increasing amount of studies. Modification of the α-amino group (i.e., methylation) partly eliminates the ability of Leu to activate mTOR signaling in H4-EII hepatoma cells (Shigemitsu et al. 1999). In BCATm−/− mice, Leu-induced activation of mTOR is almost inhibited in perfused liver and heart, isolated adipocytes as well as in cultured primary fibroblasts (Zhou et al. 2010). Based on these observations, it is speculated that Leu metabolites (mainly α-KIC and HMB), rather than Leu itself, possess direct anabolic properties (Wilkinson et al. 2013). However, direct evidence in support of this view is still lacking.
As a dietary substitute for Leu, α-KIC’s ability to stimulate protein synthesis in a catabolic state has been examined for more than 30 years (Kang et al. 1986). At the same concentration (initiated with 148 μM/kg for 10 min and followed by 400 μM/kg/h), Leu and α-KIC enhance 4E-BP1 phosphorylation and formation of the active eIF4E·eIF4G complex. Moreover, both Leu and α-KIC stimulate muscle protein synthesis to the same extent in neonatal pigs (Escobar et al. 2010). However, whether transamination of α-KIC to form Leu is involved in α-KIC-induced protein synthesis remains controversial. An early study showed that α-KIC increases protein synthesis and reduces proteolysis in rat heart muscle without enhancing intracellular concentrations of Leu (Chua et al. 1979). In the presence or absence of an inhibitor of Leu transaminase, incubation of muscle tissue with Leu leads to the same increase in protein synthesis (Tischler 1982). A study showed that α-KIC is as effective as Leu in stimulating 4E-BP1 and S6K1 hyper-phosphorylation, although the rapid reversible transamination of α-KIC to Leu could also regulate this effect (Yoshizawa 2004). Indeed, infusion of α-KIC causes an increase in plasma Leu concentration, indicating that α-KIC is markedly converted to Leu and might not directly account for the increase in protein synthesis (Escobar et al. 2010). It is possible that transamination of Leu to α-KIC is not required for the regulatory ability of Leu, whereas the effects of α-KIC might be regulated by its conversion to Leu (Yoshizawa 2004). However, using α-KIC to increase protein synthesis is still an alternative strategy in those situations in which decreased dietary protein intake is desirable since no additional nitrogen is provided with α-KIC (Columbus et al. 2014). Overall, the role of α-KIC in stimulating protein synthesis might be an mTOR-regulated action, and reversible transamination of α-KIC to Leu might be required for the effect.
Intriguingly, HMB as a dietary substitute for Leu is better tolerated by oral administration and has no adverse effects (Baxter et al. 2005). The dosage required to stimulate protein synthesis is much lower for HMB than for Leu (Eley et al. 2007; Fuller et al. 2011). Unlike α-KIC, the metabolism of Leu to HMB is irreversible and thus the production of Leu cannot be responsible for any observed responses to HMB (Columbus et al. 2014). As with α-KIC, HMB has the potential to be used as a viable alternative to promote protein anabolism when reduced dietary protein is required. Intravenous infusion of HMB with increasing doses in neonatal piglets for 1 h leads to mTOR activation and an increase in muscle protein synthesis (Wheatley et al. 2014). The greatest response is observed for a plasma HMB concentration of 90 μM, whereas higher doses either reduce the anabolic effect or fail to induce protein synthesis above the fasting baseline level. Furthermore, HMB-stimulated protein synthesis through the mTOR pathway occurs to a similar degree as for Leu (Eley et al. 2007; Wilkinson et al. 2013). These observations suggest that mTOR signaling is also involved in HMB-stimulated protein synthesis. Additionally, HMB can enhance muscle protein synthesis under both normal and catabolic conditions (such as elevated lipopolysaccharide or cytokines levels) through multiple mechanisms (Fig. 2): (1) by increasing the activity of the growth hormone/insulin-like growth factor I (IGF-I) axis in skeletal muscle (the expression of IGF-I is induced by HMB in a dose-dependent manner) (Kornasio et al. 2009; Gerlinger-Romero et al. 2011; Zanchi et al. 2011); (2) by down-regulation of eIF2α and up-regulation of the mTOR/S6K1 pathway (Eley et al. 2007, 2008; Pimentel et al. 2011), or (3) by promoting myogenesis via the mitogen-activated protein kinase/extracellular signal-regulated protein kinase (MAPK/ERK) and phosphoinositide-3-OH kinase (PI3K)/Akt pathway, which is boosted by IGF-I. Although HMB has an immediate influence on Akt phosphorylation, it does not affect insulin receptor substrate-1 (Kornasio et al. 2009). Many of these mechanisms are also shared with Leu. Thus, HMB can be viewed as an alternative to Leu to increase muscle protein synthesis (May 2002; Smith 2005; Holecek et al. 2009; Liu et al. 2014).

Possible mechanisms for HMB to stimulate protein synthesis in skeletal muscle. HMB increases protein synthesis in the muscle through MAPK/ERK signaling, IGF-1 axis, PI3K/Akt pathway, mTOR/S6K1 signaling
Circulating insulin has been suggested to promote protein synthesis (Anthony et al. 2002), so it is speculated that Leu-induced protein synthesis is attributed in part to insulin. However, supplementation with physiological levels of Leu promotes the small-intestinal development and whole-body growth in sow-reared piglets without affecting the plasma level of insulin, growth hormone or IGF-I (Sun et al. 2015). Leu and α-KIC are important nutrient signals that increase insulin secretion in islet cells (Zhou et al. 2010), while HMB consumption does not lead to a change in plasma insulin concentration (Wilkinson et al. 2013) (discussed below). Inhibiting insulin signaling via PI3K activity does not suppress stimulation of the mTORC1 pathway to induce protein synthesis by Leu or other essential amino acids (O’Connor et al. 2003; Bolster et al. 2004a). Neither Leu administration nor rapamycin treatment influences the phosphorylation of insulin regulators such as Akt, tuberous sclerosis complex 2, or AMPK (Suryawan et al. 2008). Taken together, insulin might facilitate, but is not required for, protein synthesis induced by Leu or its metabolites.
Protein degradation
Muscle protein degradation requires the proteasome and the enhanced expression of two ubiquitin ligases (E3): muscle ring finger 1 (MuRF1) (Bodine et al. 2001) and muscle atrophy F-box (MAFbx), also named atrogin-1 (Gomes et al. 2001). Expression of the proteasome subunits and MuRF1 is regulated via activation of the transcription factor nuclear factor-κB (NF-κB), while forkhead box O transcription factors are required for the expression of MAFbx (Russell and Tisdale 2009). In response to amino acid or energy deficiency, cells simultaneously reduce their rates of protein synthesis and switch to protein degradation (Talvas et al. 2006). The suppression of protein degradation by Leu requires its transamination, and depends on the stimulation of protein degradation by host- or tumor-derived regulators (Tischler 1982; Mitch and Clark 1984; Baracos and Mackenziey 2006). For example, incubation of muscle cells from healthy rats in medium containing Leu decreases the rate of protein degradation, whereas muscle cells from tumor-bearing rats do not respond to Leu (Busquets et al. 2002). However, the exact mechanism by which Leu inhibits protein degradation remains unclear. Leu and its metabolites might prevent skeletal muscle proteolysis via mTOR signaling. Unlike for insulin, the upstream kinase PI3K might not be required (Mitchell et al. 2004). Other researchers disagree with this assumption, and show that inhibition of proteolysis by Leu is not through mTOR signaling in mouse C2C12 myotubes and isolated rat hepatocytes (Mordier et al. 2000; Kanazawa et al. 2004), but through PI3K and protein kinase C (PKC) signaling, which inhibits myofibrillar proteolysis (Nakashima et al. 2005). Additionally, Leu (10 mM) can inhibit proteolysis in skeletal muscle by serving as a negative-feedback mediator of the lysosomal proteolytic system and reducing gene expression of ATP-ubiquitin-dependent proteolytic players (Busquets et al. 2000). However, α-KIC is much more effective in suppressing proteolytic-related genes resulting in a decrease in myofibrillar proteolysis of skeletal muscle tissue (Nakashima et al. 2007). It should be borne in mind that the use of 10 mM Leu in the study, which is about 50 times its plasma concentration, is highly unphysiological and the nutritional relevance of the results is questionable.
Similarly, HMB supplementation might also positively affect the inhibition of protein degradation (Smith 2004; Van Someren et al. 2005; Holecek et al. 2009; Russell and Tisdale 2009; Gerlinger-Romero et al. 2011; Noh et al. 2014). HMB (50 μM) completely attenuates total protein degradation induced by angiotensin II and tumor necrosis factor-α/interferon-γ in murine myotubes. It also attenuates the increase of reactive oxygen species (ROS) formation via activation of p38 MAPK and the activity of both caspase-3 and -8. These results suggest that HMB plays a critical role in protein degradation and muscle damage (Eley et al. 2008). Thus, HMB as a nutritional supplement has been used to effectively reduce muscle loss (Caperuto et al. 2007; Hao 2011; Alway et al. 2013; Fitschen et al. 2013; Hasselgren 2014). Contrary to previous supposition, one recent study provides evidence that the effect of Leu on protein degradation is not regulated by HMB (Baptista et al. 2013), which might be more potent than Leu in inhibiting muscle protein degradation, especially in tumor-bearing animals (Baracos and Mackenziey 2006). Unlike Leu, which acts via the class III PI3K pathway, HMB transduces its signal through the class I PI3K pathway (Drummond and Rasmussen 2008). Overall, HMB attenuates protein degradation through multiple mechanisms (Fig. 3). First, HMB suppresses PKC signaling, which in turn attenuates the ubiquitin–proteosome proteolytic pathway (Smith 2004, 2005); second, HMB reduces the activity of mitochondrial-associated caspases and the subsequent production of ROS. These actions lead to suppression of myonuclear apoptosis (Hao 2011). Third, HMB increases cell membrane integrity through cholesterol synthesis (circulating cholesterol does not increase), which is assumed, but not confirmed, to decrease tissue damage-induced proteolysis (Nissen and Abumrad 1997; Stancliffe 2012). Finally, it is possible that HMB serves as a structural component within the cell membrane (Nissen et al. 1996), but how this may impact protein turnover is not clear.

Possible mechanisms for HMB to inhibit protein degradation in skeletal muscle. HMB inhibits protein degradation through multiple mechanisms, including suppressing ubiquitin–proteasome system and enhancing cell membrane integrity
The role of Leu and its metabolites in energy metabolism
Protein and energy metabolism are tightly coupled, and protein synthesis is inhibited when there are insufficient energy substrates. mTORC1, AMPK, and myokines are involved in the regulation of Leu and the cross-talk between protein and energy metabolism (Duan et al. 2015a). Leu plays a key role in energy metabolism apart from its pivotal function in protein turnover (Liang et al. 2014). It can promote energy partition from adipocytes to myocytes, causing reduced lipid storage in adipocytes and enhanced fatty acids utilization in muscle cells (Sun and Zemel 2007). Activation of AMPK and SIRT1 by Leu is the major event that mediates fatty acid oxidation and mitochondrial biogenesis leading to increased energy expenditure in skeletal muscle cells (Liang et al. 2014). However, it remains unclear whether energy metabolism is mediated directly by Leu itself, or if its metabolites α-KIC and/or HMB contribute to the observed effects. Another possible mechanism for Leu to regulate energy metabolism is through the modulation of synthesis of nitric oxide (Dai et al. 2013; Yang et al. 2015). The following section will focus on the regulatory role of Leu and its metabolites in insulin secretion, glucose homeostasis, fatty acid oxidation, and mitochondria biosynthesis.
Insulin secretion
As mentioned above, Leu and α-KIC are insulin secretagogues, whereas HMB is not (Zhou et al. 2010; Wilkinson et al. 2013). The key questions are (1) whether Leu and α-KIC catabolism are required for the stimulation of insulin secretion, and (2) what underlying mechanisms are responsible for Leu- and α-KIC-stimulated insulin secretion (LSIS and KICSIS, respectively).
Short chain acyl-CoAs (generated by Leu catabolism) and α-KG (produced from Leu-activated glutamate oxidation) are required for the promotion of insulin secretion (MacDonald et al. 2005; MacDonald 2007). Mevalonate derived from Leu catabolism might be a signal for insulin secretion by interacting with succinate (Fahien and MacDonald 2002). These observations and previous studies all indicate that Leu and α-KIC metabolism is essential for these nutrients to stimulate insulin secretion (Hutton et al. 1979, 1980; MacDonald et al. 2005; MacDonald 2007). However, there are conflicting observations that noted no impact of Leu on insulin secretion in BCATm−/− islets (Zhou et al. 2010). Leu-induced glutamate dehydrogenase (GDH) activation contributes to LSIS. GDH can oxidize glutamate to α-KG, which is accompanied by the production of NADPH, an intracellular signal for insulin secretion (Sener and Malaisse 1980; Rabaglia et al. 2005). Interestingly, Leu serves as the allosteric activator of GDH. However, insulin secretion stimulated by Leu and α-KIC involves distinct mechanisms. The metabolites of Leu and/or α-KIC (such as acetoacetate) might stimulate the insulinotropic effect of α-KIC, and α-KIC oxidation increased the ratios of ATP/ADP, NADH/NAD+, and NADPH/NADP+ to promote insulin secretion. Amino-oxyacetic acid (an aminotransferase inhibitor) prevents KICSIS but promotes LSIS, implicating a possibly important role for α-KG, while loss of α-KIC/glutamate transamination to Leu/α-KG inhibits KICSIS (Zhou et al. 2010). Thus, transamination might play a crucial role in KICSIS, whereas activation of GDH is more important for LSIS.
Glucose homeostasis
Circulating insulin binds and activates its specific receptors, which in turn initiates a series of intracellular signaling cascades (mainly mTOR and PI3K signaling), resulting in protein synthesis and translocation of the glucose transporter 4 from the cytosol to the plasma membrane. This process promotes glucose uptake into skeletal muscle cells or adipocytes (Kleinert et al. 2011). Leu mediates satiety and modulates glucose metabolism through peripheral and central mechanisms (Lynch et al. 2002, 2006; Nishitani et al. 2002; Zhou et al. 2010; Su et al. 2012). It acts via PKC rather than protein kinase B, which is activated by insulin (Nishitani et al. 2002). However, one recent study provides new mechanistic insights into the facilitation of insulin-induced glucose uptake and insulin signaling by Leu in a dose-dependent manner via both mTORC1 and mTORC2, and demonstrated that Leu alone cannot affect glucose uptake (Liu et al. 2014). Leu (2 mM) stimulates glucose uptake in the muscle for 15–45 min, whereas longer (>60 min) stimulation leads to loss of stimulatory action. Lower concentrations have little or no impact (Nishitani et al. 2002). One potential mechanism may be that Leu feedback suppresses insulin signaling and reduces glucose utilization in skeletal muscle by activation of mTOR signaling (Zhang et al. 2007). It raises the possibility that appropriate mTOR activity is required for the full activation of insulin signaling, while too much activation results in insulin resistance (Liu et al. 2014). However, Leu has no impact on the stability of blood glucose concentrations and net glucose transport in skeletal muscle (Baum et al. 2005). The decrease in ATP levels caused by reduced glucose availability is only about twofold, thus glucose is unlikely the major energy donor for protein synthesis (Dennis et al. 2001; Hay and Sonenberg 2004). To provide energy for protein synthesis, Leu may function to promote fat loss (Zhang et al. 2007). Furthermore, the ability of α-KIC to stimulate glucose uptake is weaker than Leu at the same concentration (2 mM), and transamination of α-KIC to Leu cannot be ruled out (Nishitani et al. 2002).
Fatty acid oxidation
Skeletal muscle plays a crucial role in energy homeostasis by clearing serum free fatty acid (FFA), whole-body FA oxidation, and lipid utilization. Numerous studies have shown that Leu can mediate adipocyte lipid metabolism to provide skeletal muscle with an increased flux of FFA, supplying energy substrates to support protein synthesis (Sun and Zemel 2007, 2009). However, it is hypothesized that Leu metabolites such as HMB may contribute to the effect. HMB might also regulate lipid metabolism apart from protein metabolism (Hasselgren 2014). HMB caused a greater decrease in body fat (−1.1 vs. −0.5 %), and a greater increase in lean body mass (1.4 vs. 0.9 kg) compared with a placebo (Panton et al. 2000), and increased oxidation of the FA palmitate by 30 % (Ransone et al. 2003). It also increases lipolysis and decreases the content of adipose tissue with no change in body mass, leading to an increase of lipid availability and plasma FFA concentration (Gerlinger-Romero et al. 2011; Pinheiro et al. 2012). In addition, increased FFA oxidation decreases glucose utilization (Turner et al. 2007; Pinheiro et al. 2012), and increased muscle mass can effectively enhance fat oxidation (Wilson et al. 2008). The success of increasing fat loss is related with increased mitochondrial content and size (Toledo et al. 2006). Therefore, we will discuss the role of Leu and its metabolites in mitochondria biosynthesis in more detail.
Mitochondrial biogenesis
When mitochondrial function is impaired in obese or diabetic animals, the concentrations of BCAA in the plasma are markedly elevated (Fu et al. 2005; Jobgen et al. 2006, Tekwe et al. 2013; Wu et al. 2007a, b). In contrast, improvements in mitochondrial function prevents such as increase in BCAA in obese or diabetic subjects (Tekwe et al. 2013, Yang et al. 2015). Furthermore, mitochondria play a key role in mediating adipocyte lipid metabolism and cellular energy metabolism (Vankoningsloo et al. 2005). Thus, mitochondrial dysfunction leads to a decreased mitochondrial number and oxidative capacity, resulting in enhanced free radical production and subsequent oxidative stress (Stancliffe et al. 2011; Stancliffe and Zemel 2012). As a functional amino acid (Wu et al. 2013b), Leu is a key element that can increase mitochondrial biogenesis and fatty acid oxidation in muscle cells. Leu (0.5 mM) can promote mitochondrial biogenesis in both C2C12 myocytes and 3T3-L1 adipocytes, and modulates skeletal muscle energy metabolism by regulating peroxisome proliferator-activated receptor gamma coactivator 1α and SIRT-1 expression levels (Sun and Zemel 2009). C2C12 myotubes were treated with physiologically relevant concentrations of Leu, α-KIC, or HMB to investigate the direct role of Leu versus its metabolites in mitochondrial biogenesis and fatty acid oxidation (Stancliffe 2012). The results show that both α-KIC and HMB increase mitochondrial biogenesis and fatty acid oxidation to a similar extent as Leu. Leu and HMB also promoted mitochondrial biogenesis of the myotubes by about 50 %, and induced expression of mitochondrial regulatory and component genes. Given the vast functional overlap between Leu and HMB, it is speculated that HMB has a similar function to Leu (Wilson et al. 2008). To confirm this, BCAT, BCKD, or KICD siRNA were transfected to murine myoblasts to examine the role of intact Leu versus HMB on myoblast mitochondrial abundance (Stancliffe 2012). Leu-stimulated mitochondrial mass and expression of mitochondrial genes were inhibited upon BCAT and KICD knockdown. These observations indicate that the effects of Leu on muscle mitochondrial biogenesis and fatty acid oxidation are actually regulated by the metabolite HMB.
Summary and perspectives
Our basic knowledge of the regulation of protein and energy metabolism by Leu and its metabolites has been greatly expanded over the past years. Understanding the important relationship of Leu and its metabolites in protein synthesis and energy balance may provide new strategies to enhance body composition in animals and humans. Importantly, both α-KIC and HMB can be used as efficient nitrogen-free stimulators of protein and energy metabolism in skeletal muscle and adipose tissues, and both may act as a viable alternative to Leu. Nonetheless, some key questions remain unanswered. First, how do α-KIC and HMB enter the cell? Are they taken up through the same amino acid transporters as Leu or transporters for ketoacids? Second, are there sensors in the cytosol unique to α-KIC and HMB? Or how can they activate mTOR signaling? Third, among Leu, α-KIC, and HMB, which is most effective at increasing protein synthesis under normal and catabolic conditions? Finally, although recent studies focus on the needs of BCAA and their nitrogenous metabolites by animals (Wu et al. 2009, 2013a, b, 2014), what are optimal requirements of dietary Leu and related amino acids to enhance growth, development, and survival of mammals, birds and fish? Further studies are essential to clearly address these questions.
Abbreviations
- α-KG:
-
α-Ketoglutarate
- α-KIC:
-
α-Ketoisocaproate
- 4E-BP1:
-
4E-binding protein 1
- AMPK:
-
AMP-activated protein kinase
- ATP:
-
Adenosine triphosphate
- BCAA:
-
Branched-chain amino acid
- BCAT:
-
Branched-chain amino-acid aminotransferase
- BCKD:
-
Branched-chain α-keto acid dehydrogenase
- eIF2α:
-
Eukaryotic initiation factor 2α
- eIF4E:
-
Eukaryotic initiation factor 4E
- eIF4G:
-
Eukaryotic initiation factor-4G
- FFA:
-
Free fatty acid
- GDH:
-
Glutamate dehydrogenase
- HMB:
-
β-Hydroxy-β-methylbutyrate
- HMG-CoA:
-
β-Hydroxy-β-methylglutaryl-CoA
- IGF-I:
-
Insulin-like growth factor I
- IVA-CoA:
-
Isovaleryl CoA
- KICD:
-
KIC dioxygenase
- KICSIS:
-
KIC-stimulated insulin secretion
- LSIS:
-
Leucine-stimulated insulin secretion
- MAFbx:
-
Muscle atrophy F-box
- MAPK:
-
Mitogen-activated protein kinase
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
Mammalian target of rapamycin complex 1
- MuRF1:
-
Muscle ring finger 1
- NF-κB:
-
Nuclear factor-κB
- PI3K:
-
Phosphoinositide-3-OH kinase
- PKC:
-
Protein kinase C
- ROS:
-
Reactive oxygen species
- S6K1:
-
p70 ribosomal protein S6 kinase
- SIRT1:
-
Silent information regulator transcript 1
References
Alway SE, Pereira SL, Edens NK, Hao Y, Bennett BT (2013) beta-Hydroxy-beta-methylbutyrate (HMB) enhances the proliferation of satellite cells in fast muscles of aged rats during recovery from disuse atrophy. Exp Gerontol 48(9):973–984
Anthony JC, Anthony TG, Kimball SR, Vary TC, Jefferson LS (2000a) Orally administered leucine stimulates protein synthesis in skeletal muscle of post-absorptive rats in association with increased eIF4F formation. J Nutr 130:139–145
Anthony JC, Yoshizawa F, Anthony TG, Kimball SR, Jefferson LS (2000b) Leucine stimulates translation initiation in skeletal muscle of postabsorptive rats via a rapamycin-sensitive pathway. J Nutr 130:2413–2419
Anthony JC, Lang CH, Crozier SJ, Anthony TG, MacLean DA, Kimball SR, Jefferson LS (2002) Contribution of insulin to the translational control of protein synthesis in skeletal muscle by leucine. Am J Physiol Endocrinol Metab 282(5):E1092–E1101
Atherton PJ, Smith K, Etheridge T, Rankin D, Rennie MJ (2010) Distinct anabolic signalling responses to amino acids in C2C12 skeletal muscle cells. Amino Acids 38(5):1533–1539
Baptista IL, Silva WJ, Artioli GG, Guilherme JPLF, Leal ML, Aoki MS, Miyabara EH, Moriscot AS (2013) Leucine and HMB differentially modulate proteasome system in skeletal muscle under different sarcopenic conditions. PLoS ONE 8(10):e76752
Baracos VE, Mackenziey ML (2006) Investigations of branched-chain amino acids and their metabolites in animal models of cancer. J Nutr 136(1):237S–242S
Baum JI, O’Connor JC, Seyler JE, Anthony TG, Freund GG, Layman DK (2005) Leucine reduces the duration of insulin-induced PI 3-kinase activity in rat skeletal muscle. Am J Physiol Endocrinol Metab 288(1):E86–E91
Baxter JH, Carlos JL, Thurmond J, Rehani RN, Bultman J, Frost D (2005) Dietary toxicity of calcium beta-hydroxy-beta-methyl butyrate (CaHMB). Food Chem Toxicol 43(12):1731–1741
Bodine SC, Latres E, Baumhueter S, Lai VKM, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na EQ, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294(5547):1704–1708
Bolster DR, Jefferson LS, Kimball SR (2004a) Regulation of protein synthesis associated with skeletal muscle hypertrophy by insulin-, amino acid- and exercise-induced signalling. Proc Nutr Soc 63(2):351–356
Bolster DR, Vary TC, Kimball SR, Jefferson LS (2004b) Leucine regulates translation initiation in rat skeletal muscle via enhanced eIF4G phosphorylation. J Nutr 134(7):1704–1710
Buse MG, Reid SS (1975) Leucine. A possible regulator of protein turnover in muscle. J Clin Invest 56(5):1250–1261
Busquets S, Alvarez B, Llovera M, Agell N, Lopez-Soriano FJ, Argiles JM (2000) Branched-chain amino acids inhibit proteolysis in rat skeletal muscle: mechanisms involved. J Cell Physiol 184(3):380–384
Busquets S, Alvarez B, Lopez-Soriano FJ, Argiles JM (2002) Branched-chain amino acids: a role in skeletal muscle proteolysis in catabolic states? J Cell Physiol 191:283–289
Caperuto EC, Tomatieli RV, Colquhoun A, Seelaender MCL, Rosa LFBPC (2007) beta-hydoxy-beta-methylbutyrate supplementation affects Walker 256 tumor-bearing rats in a time-dependent manner. Clin Nutr 26(1):117–122
Chua BD, Siehl L, Morgan HE (1979) Effect of leucine and metabolites of branched chain amino acids on protein turnover in heart. J Biol Chem 254:8358–8362
Columbus DA, Fiorotto ML, Davis TA (2014) Leucine is a major regulator of muscle protein synthesis in neonates. Amino Acids 47:259–270
Dai ZL, Wu ZL, Yang Y, Wang JJ, Satterfield MC, Meininger CJ, Bazer FW, Wu G (2013) Nitric oxide and energy metabolism in mammals. BioFactors 39:383–391
Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G (2001) Mammalian TOR: a homeostatic ATP sensor. Science 294(5544):1102–1105
Drummond MJ, Rasmussen BB (2008) Leucine-enriched nutrients and the regulation of mammalian target of rapamycin signalling and human skeletal muscle protein synthesis. Curr Opin Clin Nutr Metab Care 11(3):222–226
Duan YH, Li FN, Liu HN, Li YH, Liu YY, Kong XF, Zhang YZ, Deng D, Tang YL, Feng ZM, Wu GY, Yin YL (2015a) Nutritional and regulatory roles of leucine in muscle growth and fat reduction. Front Biosci (Landmark) 20:796–813
Duan YH, Li FN, Tan KR, Liu HN, Li YH, Liu YY, Kong XF, Tang YL, Wu GY, Yin YL (2015b) Key mediators of intracellular amino acids signaling to mTORC1 activation. Amino Acids 47(5):857–867
Eley HL, Russell ST, Baxter JH, Mukerji P, Tisdale MJ (2007) Signaling pathways initiated by beta-hydroxy-beta-methylbutyrate to attenuate the depression of protein synthesis in skeletal muscle in response to cachectic stimuli. Am J Physiol Endocrinol Metab 293(4):E923–E931
Eley HL, Russell ST, Tisdale MJ (2008) Mechanism of attenuation of muscle protein degradation induced by tumor necrosis factor-alpha and angiotensin II by beta-hydroxy-beta-methylbutyrate. Am J Physiol Endocrinol Metab 295(6):E1417–E1426
Escobar J, Frank JW, Suryawan A, Nguyen HV, Van Horn CG, Hutson SM, Davis TA (2010) Leucine and alpha-ketoisocaproic acid, but not norleucine, stimulate skeletal muscle protein synthesis in neonatal pigs. J Nutr 140(8):1418–1424
Fahien LA, MacDonald MJ (2002) The succinate mechanism of insulin release. Diabetes 51(9):2669–2676
Filhiol TM (2012) The Effects of Leucine on Mitochondrial Biogenesis and Cell Cycle in A-375 Melanoma Cells. The University of Tennessee, Knoxville
Fitschen PJ, Wilson GJ, Wilson JM, Wilund KR (2013) Efficacy of beta-hydroxy-beta-methylbutyrate supplementation in elderly and clinical populations. Nutrition 29(1):29–36
Fu WJ, Haynes TE, Kohli R, Hu J, Shi W, Spencer TE, Carroll RJ, Meininger CJ, Wu G (2005) Dietary l-arginine supplementation reduces fat mass in Zucker diabetic fatty rats. J Nutr 135:714–721
Fuller JC Jr, Sharp RL, Angus HF, Baier SM, Rathmacher JA (2011) Free acid gel form of beta-hydroxy-beta-methylbutyrate (HMB) improves HMB clearance from plasma in human subjects compared with the calcium HMB salt. Br J Nutr 105(3):367–372
Garlick PJ (2005) The role of leucine in the regulation of protein metabolism. J Nutr 135(6):1553S–1556S
Gerlinger-Romero F, Guimaraes-Ferreira L, Giannocco G, Nunes MT (2011) Chronic supplementation of beta-hydroxy-beta methylbutyrate (HMbeta) increases the activity of the GH/IGF-I axis and induces hyperinsulinemia in rats. Growth Horm IGF Res 21(2):57–62
Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL (2001) Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci 98(25):14440–14445
Hall TR, Wallin R, Reinhart GD, Hutson SM (1993) Branched-chain aminotransferase isoenzymes—purification and characterization of the rat-brain isoenzyme. J Biol Chem 268(5):3092–3098
Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, Ha SH, Ryu SH, Kim S (2012) Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149(2):410–424
Hao Y, Jackson JR, Wang Y, Edens N, Pereira SL, Alway SE (2011) β-Hydroxy-β-methylbutyrate reduces myonuclear apoptosis during recovery from hind limb suspension-induced muscle fiber atrophy in aged rats. Am J Physiol Regul Integr Comp Physiol 301:R701–R715
Hasselgren PO (2014) beta-Hydroxy-beta-methylbutyrate (HMB) and prevention of muscle wasting. Metabolism 63(1):5–8
Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. GENE DEV 18(16):1926–1945
Holecek M (2002) Relation between glutamine, branched-chain amino acids, and protein metabolism. Nutrition 18(2):130–133
Holecek M, Muthny T, Kovarik M, Sispera L (2009) Effect of beta-hydroxy-beta-methylbutyrate (HMB) on protein metabolism in whole body and in selected tissues. Food Chem Toxicol 47(1):255–259
Hutson SM, Wallin R, Hall TR (1992) Identification of mitochondrial branched-chain aminotransferase and its isoforms in rat-tissues. J Biol Chem 267(22):15681–15686
Hutton JC, Sener A, Malaisse WJ (1979) Metabolism of 4-methyl-2-oxopentanoate in rat pancreatic-islets. Biochem J 184(2):291–301
Hutton JC, Sener A, Malaisse WJ (1980) Interaction of branched-chain amino-acids and keto acids upon pancreatic-islet metabolism and insulin-secretion. J Biol Chem 255(15):7340–7346
Jobgen WS, Fried SK, Fu WJ, Meininger CJ, Wu G (2006) Regulatory role for the arginine-nitric oxide pathway in metabolism of energy substrates. J Nutr Biochem 17:571–588
Kanazawa T, Taneike I, Akaishi R, Yoshizawa F, Furuya N, Fujimura S, Kadowaki M (2004) Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem 279:8452–8459
Kang CW, Tungsanga K, Walser M (1986) Effect of the level of dietary protein on the utilization of alpha-ketoisocaproate for protein synthesis. Am J Clin Nutr 43(4):504–509
Kim SW, Mateo RD, Yin YL, Wu GY (2007) Functional amino acids and fatty acids for enhancing production performance of sows and piglets. Asian Austral J Anim 20(2):295–306
Kim JY, Burghardt RC, Wu G, Johnson GA, Spencer TE, Bazer FW (2011a) Select nutrients in the ovine uterine lumen: VII. Effects of arginine, leucine, glutamine, and glucose on trophectoderm cell signaling, proliferation, and migration. Biol Reprod 84:62–69
Kim JY, Burghardt RC, Wu G, Johnson GA, Spencer TE, Bazer FW (2011b) Select nutrients in the ovine uterine lumen: IX. Differential effects of arginine, leucine, glutamine and glucose on interferon tau, orinithine decarboxylase and nitric oxide synthase in the ovine conceptus. Biol Reprod 84:1139–1147
Kleinert M, Liao YH, Nelson JL, Bernard JR, Wang WY, Ivy JL (2011) An amino acid mixture enhances insulin-stimulated glucose uptake in isolated rat epitrochlearis muscle. J Appl Physiol 111(1):163–169
Kornasio R, Riederer I, Butler-Browne G, Mouly V, Uni Z, Halevy O (2009) Beta-hydroxy-beta-methylbutyrate (HMB) stimulates myogenic cell proliferation, differentiation and survival via the MAPK/ERK and PI3K/Akt pathways. Biochim Biophys Acta 1793(5):755–763
Kovarik M, Muthny T, Sispera L, Holecek M (2010) Effects of beta-hydroxy-beta-methylbutyrate treatment in different types of skeletal muscle of intact and septic rats. J Physiol Biochem 66(4):311–319
Lei J, Feng DY, Zhang YL, Zhao FQ, Wu ZL, San Gabriel A, Fujishima Y, Uneyama H, Wu G (2012a) Nutritional and regulatory role of branched-chain amino acids in lactation. Front Biosci 17:2725–2739
Lei J, Feng DY, Zhang YL, Dahanayaka S, Li XL, Yao K, Wang JJ, Wu ZL, Dai ZL, Wu G (2012b) Regulation of leucine catabolism by metabolic fuels in mammary epithelial cells. Amino Acids 43:2179–2189
Lei J, Feng DY, Zhang YL, Dahanayaka S, Li XL, Yao K, Wang JJ, Wu ZL, Dai ZL, Wu G (2013) Hormonal regulation of leucine catabolism in mammary epithelial cells. Amino Acids 45:531–541
Li P, Yin YL, Li DF, Kim SW, Wu GY (2007) Amino acids and immune function. Br J Nutr 98:237–252
Li P, Knabe DA, Kim SW, Lynch CJ, Hutson SM, Wu G (2009) Lactating porcine mammary tissue catabolizes branched-chain amino acids for glutamine and aspartate synthesis. J Nutr 139:1502–1509
Li F, Yin Y, Tan B, Kong X, Wu G (2011a) Leucine nutrition in animals and humans: mTOR signaling and beyond. Amino Acids 41(5):1185–1193
Li XL, Rezaei R, Li P, Wu G (2011b) Composition of amino acids in feed ingredients for animal diets. Amino Acids 40:1159–1168
Liang C, Curry BJ, Brown PL, Zemel MB (2014) Leucine modulates mitochondrial biogenesis and SIRT1-AMPK signaling in C2C12 myotubes. J Nutr Metab 2014:239750
Liu H, Liu R, Xiong YF, Li X, Wang XL, Ma Y, Guo HL, Hao LP, Yao P, Liu LG, Wang D, Yang XF (2014) Leucine facilitates the insulin-stimulated glucose uptake and insulin signaling in skeletal muscle cells: involving mTORC1 and mTORC2. Amino Acids 46(8):1971–1979
Lynch CJ, Patson BJ, Anthony J, Vaval A, Jefferson LS, Vary TC (2002) Leucine is a direct-acting nutrient signal that regulates protein synthesis in adipose tissue. Am J Physiol Endocrinol Metab 283(3):E503–E513
Lynch CJ, Halle B, Fujii H, Vary TC, Wallin R, Damuni ZH, Hutson SM (2003) Potential role of leucine metabolism in the leucine-signaling pathway involving mTOR. Am J Physiol Endocrinol Metab 285(4):E854–E863
Lynch CJ, Gern B, Lloyd C, Hutson SM, Eicher R, Vary TC (2006) Leucine in food mediates some of the postprandial rise in plasma leptin concentrations. Am J Physiol Endocrinol Metab 291(3):E621–E630
MacDonald MJ (2007) Synergistic potent insulin release by combinations of weak secretagogues in pancreatic islets and INS-1 cells. J Biol Chem 282(9):6043–6052
MacDonald MJ, Fahien LA, Brown LJ, Hasan NM, Buss JD, Kendrick MA (2005) Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am J Physiol Endocrinol Metab 288(1):E1–E15
May PE, Barber A, D’Olimpio JT, Hourihane A, Abumrad NN (2002) Reversal of cancer-related wasting using oral supplementation with a combination of β-hydroxy-β-methylbutyrate, arginine, and glutamine. Am J Surg 183:471–479
Mitch WE, Clark AS (1984) Specificity of the effects of leucine and its metabolites on protein degradation in skeletal muscle. Biochem J 222:579–586
Mitchell JC, Evenson AR, Tawa NE (2004) Leucine inhibits proteolysis by the mTOR kinase signaling pathway in skeletal muscle. J Surg Res 121(2):311
Molfino A, Gioia G, Rossi Fanelli F, Muscaritoli M (2013) Beta-hydroxy-beta-methylbutyrate supplementation in health and disease: a systematic review of randomized trials. Amino Acids 45(6):1273–1292
Mordier S, Deval C, Bechet D, Tassa A, Ferrara M (2000) Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin-independent signaling pathway. J Biol Chem 275(38):29900–29906
Nakashima K, Ishida A, Yamazaki M, Abe H (2005) Leucine suppresses myofibrillar proteolysis by down-regulating ubiquitin-proteasome pathway in chick skeletal muscles. Biochem Biophys Res Commun 336(2):660–666
Nakashima K, Yakabe Y, Ishida A, Yamazaki M, Abe H (2007) Suppression of myofibrillar proteolysis in chick skeletal muscles by alpha-ketoisocaproate. Amino Acids 33(3):499–503
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136(3):521–534
Nishitani S, Matsumura T, Fujitani S, Sonaka I, Miura Y, Yagasaki K (2002) Leucine promotes glucose uptake in skeletal muscles of rats. Biochem Biophys Res Commun 299(5):693–696
Nissen SL, Abumrad NN (1997) Nutritional role of the leucine metabolite β-hydroxy β-methylbutyrate (HMB). Nutr Biochem 8(300–331):301
Nissen S, Sharp R, Ray M, Rathmacher JA, Rice D, Fuller JC, Connelly AS, Abumrad N (1996) Effect of leucine metabolite beta-hydroxy-beta-methylbutyrate on muscle metabolism during resistance-exercise training. J Appl Physiol 81(5):2095–2104
Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, Zwartkruis FJ, Thomas G (2005) Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci 102(40):14238–14243
Noh KK, Chung KW, Choi YJ, Park MH, Jang EJ, Park CH, Yoon C, Kim ND, Kim MK, Chung HY (2014) beta-Hydroxy beta-methylbutyrate improves dexamethasone-induced muscle atrophy by modulating the muscle degradation pathway in SD rat. PLoS One 9(7):e102947
O’Connor PMJ, Kimball SR, Suryawan A, Bush JA, Nguyen HV, Jefferson LS, Davis TA (2003) Regulation of translation initiation by insulin and amino acids in skeletal muscle of neonatal pigs. Am J Physiol Endocrinol Metab 285(1):E40–E53
Panton LB, Rathmacher JA, Baier S, Nissen S (2000) Nutritional supplementation of the leucine metabolite β-Hydroxy-β-Methylbutyrate (HMB) during resistance training. Nutrition 16(9):734–739
Pimentel GD, Rosa JC, Lira FS, Zanchi NE, Ropelle ER, Oyama LM, Oller do Nascimento CM, de Mello MT, Tufik S, Santos RV (2011) beta-Hydroxy-beta-methylbutyrate (HMbeta) supplementation stimulates skeletal muscle hypertrophy in rats via the mTOR pathway. Nutr Metab (Lond) 8(1):11
Pinheiro CH, Gerlinger-Romero F, Guimaraes-Ferreira L, de Souza-Jr AL, Vitzel KF, Nachbar RT, Nunes MT, Curi R (2012) Metabolic and functional effects of beta-hydroxy-beta-methylbutyrate (HMB) supplementation in skeletal muscle. Eur J Appl Physiol 112(7):2531–2537
Rabaglia ME, Gray-Keller MP, Frey BL, Shortreed MR, Smith LM, Attie AD (2005) alpha-Ketoisocaproate-induced hypersecretion of insulin by islets from diabetes-susceptible mice. Am J Physiol Endocrinol Metab 289(2):E218–E224
Ransone J, Neighbors K, Lefavi R, Chromiak J (2003) The effect of β-Hydroxy β-Methylbutyrate on muscular strength and body composition in collegiate football players. The Journal of Strength and Conditioning Research 17(1):34–39
Rhoads JM, Wu G (2009) Glutamine, arginine, and leucine signaling in the intestine. Amino Acids 37:111–122
Rudney H (1957) The biosynthesis of Beta-Hydroxy-Beta-Methylglutaric acid. J Biol Chem 227(1):363–377
Russell ST, Tisdale MJ (2009) Mechanism of attenuation by beta-hydroxy-beta-methylbutyrate of muscle protein degradation induced by lipopolysaccharide. Mol Cell Biochem 330(1–2):171–179
Self JT, Spencer TE, Johnson GA, Hu J, Bazer FW, Wu G (2004) Glutamine synthesis in the developing porcine placenta. Biol Reprod 70:1444–1451
Sener A, Malaisse WJ (1980) l-Leucine and a nonmetabolized analog activate pancreatic-islet glutamate-dehydrogenase. Nature 288(5787):187–189
She PX, Olson KC, Kadota Y, Inukai A, Shimomura Y, Hoppel CL, Adams SH, Kawamata Y, Matsumoto H, Sakai R, Lang CH, Lynch CJ (2013) Leucine and protein metabolism in obese zucker rats. PLoS One 8(3):e59443
Shigemitsu K, Tsujishita Y, Miyake H, Hidayat S, Tanaka N, Hara K, Yonezawa K (1999) Structural requirement of leucine for activation of p70 S6 kinase. FEBS Lett 447(2–3):303–306
Slater GJ, Jenkins D (2000) beta-hydroxy-beta-methylbutyrate (HMB) supplementation and the promotion of muscle growth and strength. Sports Med 30(2):105–116
Smith HJ, Wyke SM, Tisdale MJ (2004) Mechanism of the attenuation of proteolysis-inducing factor stimulated protein degradation in muscle by β-hydroxy-β-methylbutyrate. Cancer Res 64:8731–8735
Smith HJ, Mukerji P, Tisdale MJ (2005) Attenuation of proteasome-induced proteolysis in skeletal muscle by β-Hydroxy-β-Methylbutyrate in cancer-induced muscle loss. Cancer Res 65(1):277–283
Stancliffe RA (2012) Role of beta-hydroxy-beta-methylbutyrate (hmb) in leucine stimulation of mitochondrial biogenesis and fatty acid oxidation. The University of Tennessee, Knoxville
Stancliffe RA, Zemel MB (2012) Role of beta-hydroxy-beta-methylbutyrate (HMB) in leucine stimulation of muscle mitochondrial biogenesis. FASEB J 26:251.6
Stancliffe RA, Eades M, Smart K, Zemel MB (2011) Role of mTOR and beta-hydroxy-beta-methylbutyrate (HMB) in leucine stimulation of muscle mitochondrial biogenesis and fatty acid oxidation. FASEB J 25:606.1
Su Y, Lam TKT, He W, Pocai A, Bryan J, Aguilar-Bryan L, Gutierrez-Juarez R (2012) Hypothalamic leucine metabolism regulates liver glucose production. Diabetes 61(1):85–93
Sun X, Zemel MB (2007) Leucine and calcium regulate fat metabolism and energy partitioning in murine adipocytes and muscle cells. Lipids 42(4):297–305
Sun X, Zemel MB (2009) Leucine modulation of mitochondrial mass and oxygen consumption in skeletal muscle cells and adipocytes. Nutr Metab (Lond) 6:26
Sun YL, Wu ZL, Li W, Zhang C, Sun KJ, Ji Y, Wang B, Jiao N, He BB, Wang WW, Dai ZL, Wu G (2015) Dietary l-leucine supplementation enhances intestinal development in suckling piglets. Amino Acids 47(8):1517–1525
Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE, Hutson SM (1998) A molecular model of human branched-chain amino acid metabolism. Am J Clin Nutr 68(1):72–81
Suryawan A, Jeyapalan AS, Orellana RA, Wilson FA, Nguyen HV, Davis TA (2008) Leucine stimulates protein synthesis in skeletal muscle of neonatal pigs by enhancing mTORC1 activation. Am J Physiol Endocrinol Metab 295(4):E868–E875
Suzuki T, Inoki K (2011) Spatial regulation of the mTORC1 system in amino acids sensing pathway. Acta Biochim Biophys Sin 43(9):671–679
Talvas J, Obled A, Fafournoux P, Mordier S (2006) Regulation of protein synthesis by leucine starvation involves distinct mechanisms in mouse C2C12 myoblasts and myotubes. J Nutr 136(6):1466–1471
Tekwe CD, Lei J, Yao K, Rezaei R, Li XL, Dahanayaka S, Carroll RJ, Meininger CJ, Bazer FW, Wu G (2013) Oral administration of interferon tau enhances oxidation of energy substrates and reduces adiposity in Zucker diabetic fatty rats. BioFactors 39:552–563
Tischler ME, Desautels M, Goldberg AL (1982) Does leucine, leucyl-tRNA, or some metabolite of leucine regulate protein synthesis and degradation in skeletal and cardiac muscle? J Biol Chem 257:1613–1621
Toledo FG, Watkins S, Kelley DE (2006) Changes induced by physical activity and weight loss in the morphology of inter-myofibrillar mitochondria in obese men and women. J Clin Endocrinol Metab 92(5):1827–1833
Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, Cooney GJ (2007) Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle—evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes 56(8):2085–2092
Van Koevering M, Nissen S (1992) Oxidation of leucine and alpha-ketoisocaproate to beta-hydroxy-beta-methylbutyrate in vivo. Am J Physiol 262(1 Pt 1):E27–E31
Van Someren KA, Edwards AJ, Howatson G (2005) Supplementation with β-hydroxy- β-methylbutyrate (HMB) and α-ketoisocaproic acid (KIC) reduces signs and symptoms of exercise-induced muscle damage in man. Int J Sport Nutr Exerc Metab 15:413–424
Vankoningsloo S, Piens M, Lecocq C, Gilson A, De Pauw A, Renard P, Demazy C, Houbion A, Raes M, Arnould T (2005) Mitochondrial dysfunction induces triglyceride accumulation in 3T3-L1 cells: role of fatty acid beta-oxidation and glucose. J Lipid Res 46(6):1133–1149
Wheatley SM, El-Kadi SW, Suryawan A, Boutry C, Orellana RA, Nguyen HV, Davis SR, Davis TA (2014) Protein synthesis in skeletal muscle of neonatal pigs is enhanced by administration of beta-hydroxy-beta-methylbutyrate. Am J Physiol Endocrinol Metab 306(1):E91–E99
Wilkinson DJ, Hossain T, Hill DS, Phillips BE, Crossland H, Williams J, Loughna P, Churchward-Venne TA, Breen L, Phillips SM, Etheridge T, Rathmacher JA, Smith K, Szewczyk NJ, Atherton PJ (2013) Effects of leucine and its metabolite beta-hydroxy-beta-methylbutyrate on human skeletal muscle protein metabolism. J Physiol 591(Pt 11):2911–2923
Wilson GJ, Wilson JM, Manninen AH (2008) Effects of beta-hydroxy-beta-methylbutyrate (HMB) on exercise performance and body composition across varying levels of age, sex, and training experience: a review. Nutr Metab (Lond) 5:1
Wu G (2013a) Amino acids: biochemistry and nutrition. CRC Press, Boca Raton
Wu G (2013b) Functional amino acids in nutrition and health. Amino Acids 45:407–411
Wu G (2014) Dietary requirements of synthesizable amino acids by animals: a paradigm shift in protein nutrition. J Anim Sci Biotechnol 5:34
Wu G, Thompson JR (1987) Ketone bodies inhibit leucine degradation in chick skeletal muscle. Int J Biochem 19:937–943
Wu G, Thompson JR, Sedgwick G, Drury M (1989) Formation of alanine and glutamine in chick (Gallus domesticus) skeletal muscle. Comp Biochem Physiol 93B:609–613
Wu G, Collins JK, Perkins-Veazie P, Siddiq M, Dolan KD, Kelly KA, Heaps CL, Meininger CJ (2007a) Dietary supplementation with watermelon pomace juice enhances arginine availability and ameliorates the metabolic syndrome in Zucker diabetic fatty rats. J Nutr 137:2680–2685
Wu GY, Bazer FW, Davis TA, Johnson GA, Kim SW, Knabe DA, Spencer TE, Yin YL (2007b) Important roles for arginine-family amino acids in swine nutrition and production. Livestock Science 122:8–22
Wu GY, Bazer FW, Davis TA, Kim SW, Li P, Rhoads JM, Satterfield MC, Smith SB, Spencer TE, Yin YL (2009) Arginine metabolism and nutrition in growth, health and disease. Amino Acids 37:169–175
Wu G, Wu ZL, Dai ZL, Yang Y, Wang WW, Liu C, Wang B, Wang JJ, Yin YL (2013a) Dietary requirements of “nutritionally nonessential amino acids” by animals and humans. Amino Acids 44:1107–1113
Wu G, Bazer FW, Satterfield MC, Li XL, Wang XQ, Johnson GA, Burghardt RC, Dai ZL, Wang JJ, Wu ZL (2013b) Impacts of arginine nutrition on embryonic and fetal development in mammals. Amino Acids 45:241–256
Wu G, Bazer FW, Dai ZL, Li DF, Wang JJ, Wu ZL (2014) Amino acid nutrition in animals: protein synthesis and beyond. Annu Rev Anim Biosci 2:387–417
Yang Y, Wu ZL, Meininger CJ, Wu G (2015) l-Leucine and NO-mediated cardiovascular function. Amino Acids 47:435–447
Yao K, Yin YL, Li XL, Xi PB, Wang JJ, Lei J, Hou YQ, Wu GY (2012) Alpha-ketoglutarate inhibits glutamine degradation and enhances protein synthesis in intestinal porcine epithelial cells. Amino Acids 42(6):2491–2500
Yin Y, Yao K, Liu Z, Gong M, Ruan Z, Deng D, Tan B, Liu Z, Wu G (2010) Supplementing L-leucine to a low-protein diet increases tissue protein synthesis in weanling pigs. Amino Acids 39(5):1477–1486
Yoshizawa F, Sekizawa H, Hirayama S, Yamazaki Y, Nagasawa T, Sugahara K (2004) Tissue specific regulation 4E-BP1 and S6K1 phosphorylation by α-ketoisocaproate. J Nutr Sci Vitaminol (Tokyo) 50:56–60
Zanchi NE, Gerlinger-Romero F, Guimaraes-Ferreira L, de Siqueira MA, Felitti V, Lira FS, Seelaender M, Lancha AH (2011) HMB supplementation: clinical and athletic performance-related effects and mechanisms of action. Amino Acids 40(4):1015–1025
Zhang YY, Guo KY, LeBlanc RE, Loh D, Schwartz GJ, Yu YH (2007) Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes 56(6):1647–1654
Zhou Y, Jetton TL, Goshorn S, Lynch CJ, She P (2010) Transamination is required for alpha-ketoisocaproate but not leucine to stimulate insulin secretion. J Biol Chem 285(44):33718–33726
Acknowledgments
This study was jointly supported by National Basic Research Program of China (2013CB127305, 2012CB124704), National Nature Science Foundation of China (31110103909, 31330075), The Chinese Academy of Science STS Project (KFJ-EW-STS-063), Key Projects in the National Science & Technology Pillar Program (2013BAD21B04), the Hubei Hundred Talent program, and Texas A&M AgriLife Research (H-8200).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Rights and permissions
About this article

Cite this article
Duan, Y., Li, F., Li, Y. et al. The role of leucine and its metabolites in protein and energy metabolism. Amino Acids 48, 41–51 (2016). https://doi.org/10.1007/s00726-015-2067-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-015-2067-1