
Abstract
Cytosolic glyceraldehyde-3-phosphate dehydrogenase (GAPC) catalyzes a key reaction in glycolysis and encoded by a multi-gene family which showed instability expression under abiotic stress. DNA methylation is an epigenetic modification that plays an important role in gene regulation in response to abiotic stress. The comprehension of DNA methylation at promoter region of TaGAPC1 can provide insights into the transcription regulation mechanisms of plant genes under abiotic stress. In this study, we cloned TaGAPC1 genes and its promoters from two wheat genomes, then investigated the expression patterns of TaGAPC1 under osmotic and salinity stress, and analyzed the promoter sequences. Moreover, the methylation patterns of promoters under stress were confirmed. Expression analysis indicated that TaGAPC1 was induced inordinately by stresses in two wheat genotypes with contrasting drought tolerance. Several stress-related cis-acting elements (MBS, DRE, GT1 and LTR et al.) were located in its promoters. Furthermore, the osmotic and salinity stress induced the demethylation of CG and CHG nucleotide in the promoter region of Changwu134. The methylation level of CHG and CHH in promoter of Zhengyin1 was always increased under stresses, and the CG contexts remained unchanged. The cytosine loci of stress-related cis-acting elements also showed different methylation changes in this process. These results provide insights into the relationship between promoter methylation and gene expression, promoting the function investigation of GAPC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
DNA methylation is a ubiquitous modification of genomes in eukaryotes (Zilberman et al. 2007). The most common DNA methylation is the modification of cytosine C5, and it could reach up to 25 % in the plant genome (Steward et al. 2002). There are three types of cytosine methylation in plants: CG, CHG and CHH (H means any nucleotide except guanine), while in animals methylation occurs almost only in CG sequence context (Henderson et al. 2010). As a crucial epigenetic mark in plant, DNA methylation can regulate the genomic activity by three ways: de novo methylation, maintaining and removing methylation (Liu et al. 2015). Recently, it is revealed that genome methylation was always involved in response to external factors and contributed to the evolution in plant genomes. At least three genes had been proved to be affected by DNA methylation under heat stress in Arabidopsis (Boyko et al. 2010; Naydenov et al. 2015). The level of DNA methylation that existed in genome fragment of maize hybrids was lower than that of its corresponding mid-parents, and researchers conjectured that there is an increasing trend of demethylation in the hybrids relative to their parents (Sun et al. 2015).
Wheat (Triticum aestivum L) is one of the world’s most important food crops that provide dietary carbohydrates for numerous people of the world. Water-deficit and salinity are the major abiotic factors that affect wheat production worldwide. Growth and development of plants are regulated by several proteins and genes under abiotic stresses (Li et al. 2015). Some functional genes changed in methylation patterns under abiotic stress in wheat (Sun et al. 2014; Xu et al. 2013). DNA methylation exists not only in transposable elements and repeat sequences but also in transcriptional regions of promoter (Jeon et al. 2015). And it had been proved that methylation interfered in the combination between cis-acting elements and transcription factors directly (Inamdar et al. 1991). So study on the cytosine methylation of promoter region is significant for understanding the mechanism of stress response gene in plant.
GAPC (glyceraldehyde-3-phosphate dehydrogenase, cytosolic) was once considered as a simple “housekeeping” protein and had been used as a reference in gene expression analysis (Nicholls et al. 2012). In recent years, many researches had revealed that GAPC played a positive role in responding to stresses in plants (Arumugam Pillai et al. 2002; McLoughlin et al. 2013). There are multiple genes encoding GAPC isozymes in Arabidopsis, rice and maize (Manjunath and Sachs 1997; Marri et al. 2005; Zhang et al. 2011). The Arabidopsis thaliana genome contains four phosphorylating glycolytic GAPC genes (GAPC1, GAPC2, GAPCp1, and GAPCp2) (Guo et al. 2014). 6 GAPC genes were identified in wheat cv. Chinese Spring previously, and TaGAPC1 (Traes_7DL_961822B36.2) was strongly induced by PEG (polyethylene glycol) treatment (Zeng et al. 2016). Here, TaGAPC1 and its promoter were cloned, and the expression patterns under osmotic and salinity stress of TaGAPC1 were investigated. Meanwhile, the stress-related cis-acting elements were identified from the promoters. Moreover, the methylation changes on the promoter under osmotic and salinity were analyzed with bisulfite sequence. Those studies expected to provide further insight into the molecular mechanisms of wheat tolerance and a new evidence for the correlation between promoter methylation and gene expression under abiotic stress.
Materials and methods
Primer design
The sequence of Traes_7DL_961822B36.2 was used as a query to search against the Triticum aestivum databases by Ensembl Plants (http://plants.ensembl.org/index.html). The specific primers used for gene and promoter cloning, qRT-PCR were designed by Primer Premier 5.0 and DNAMAN 6.0 based on cv. Chinese Spring sequence that differs from other TaGAPCs (Zeng et al. 2016). The wheat β-actin gene (AB181991) was used as reference for qRT-PCR. Methyl Primer Express v1.0 was used to detect the CpG islands in promoters and design the Nested amplification primers of bisulfite sequencing. Specific primer sequences were given in Table 1. Primers TaGAPC1-1FR-mF and TaGAPC1-1FR-mR were used for the first round of Nested PCR of region I, and TaGAPC1-1SR-mF and TaGAPC1-1SR-mR were employed in the second round. TaGAPC1-2FR-mF and TaGAPC1-2FR-mR were used for the first round of Nested PCR of region II, and TaGAPC1-2SR-mF and TaGAPC1-2SR-mR were employed in the second round.
Plant materials and growing conditions
Wheat cv. Changwu134 (drought-tolerant) (Weng et al. 2015) and Zhengyin1 (drought-susceptible) (Ma et al. 2015) were used in this study. The seeds were provided by the State Key Laboratory of Soil Erosion and Dryland Farming on the Loess Plateau of the Institute of Soil and Water Conservation at the Chinese Academy of Sciences. Seeds were disinfected with 0.1 % HgCl2 for 15 min and germinated on wet filter paper for 8 h at 22 °C dark, then put the germinated seeds on a petri dish (45 cm in diameter), containing half-strength Hoagland. The wheat grains were kept under a 12 h photoperiod (light/dark temperature 22 °C) for 14 days. At the two-leave stage, the solution were replaced by 20 % PEG8000 and 250 mM NaCl. The leaves were harvested and rapidly frozen in liquid nitrogen and stored at −80 °C for extraction of total RNA and genomic DNA. All experiments were repeated three times.
Homologous cloning of TaGAPC1s and sequence analysis
TaGAPC1s were amplified from wheat cDNA with PCR. The PCR reaction were performed in a final volume of 20 μL using the following PCR program: an initial denaturation step (94 °C, 5 min) was followed by 32 cycles (94 °C, 30 s; 56 °C, 45 s; 72 °C, 1 min 10 s) and a final elongation step (72 °C, 10 min). The PCR products were checked using 2 % agarose gel electrophoresis and purified, then cloned into a pMD19T (simple) (TaKaRa, Japan) vector. Several clones of each reaction were sequenced (Invitrogen, USA).
The exon-intron structures of gene were determined by aligning their coding sequences with their corresponding genomic sequences and a diagram was generated through the online Gene Structure Display Serve (http://gsds.cbi.pku.edu.cn). The sequence of genes and promoters was compared by DNAMAN6.0. The putative proteins were confirmed using the Pfam database (http://pfam.sanger.ac.uk/) and the putative localization of protein was identified using the PSORT prediction server (http://www.genscript.com/psort.html). Domains of the proteins were analyzed by the InterProScan (http://www.ebi.ac.uk/cgi-bin/iprscan/). GAPCs from Arabidopsis thaliana were searched from NCBI and phylogenetic tree was constructed by MEGA5.0 using the neighbor-joining (NJ) method.
Quantitative real-time PCR
Frozen tissues were ground in liquid nitrogen and total RNA was isolated using the RNAiso plus reagent (TaKaRa, Japan) as per the manufacturer’s specifications and treated with RNase-free DNase I (Invitrogen, USA) for 15 min to degrade any residual genomic DNA. For real-time PCR analysis, first-strand cDNAs were synthesized from DNaseI-treated total RNA using PrimeScriptTM RT-PCR Kit (TaKaRa, Japan) according to the manufacturer’s instructions. Quantitative real-time PCR (qRT-PCR) was carried out by BioEasy Master Mix (SYBR Green, BioFlux) according to the manufacturer’s instructions on the Bio-Rad CFX96 system (Bio-Rad Laboratories, USA). The PCR thermal cycle was set up as follows: 95 °C for 1 min; 40 cycles of 94 °C for 15 s, 56 °C for 15 s and 72 °C for 30s; And melting curve analysis. The quantitative analysis was accomplished with the 2−△△CT method by Bio Rad CFX Manager software and the relative expression graphs of TaGAPC1 were drawn by Origin 9.0. The wheat β-actin gene was used as an internal control. Three biological replicates were set up and all experiments were repeated three times.
Cloning of promoters and sequence analysis
Genomic DNA isolated by the CTAB (Hexadecyltrimethy Ammonium Bromide) method (Attitalla 2011) and the PCR reaction of promoter cloning were performed in a final volume of 20 μL using the following PCR program: an initial denaturation step (94 °C, 5 min) was followed by 32 cycles (94 °C, 30 s; 58 °C, 45 s; 72 °C, 1 min) and a final elongation step (72 °C, 10 min). The PCR products were checked using 2 % agarose gel electrophoresis and purified, then cloned into a pMD19T (simple) (TaKaRa, Japan) vector. Several clones of each reaction were sequenced (Invitrogen, USA).
PlantCARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) (Lescot et al. 2002) and PLACE (http://www.dna.affrc.Go.jp/PLACE/) were used to detect cis-elements of promoters.
Bisulfite sequencing and nested PCR
Genomic DNA was extracted with CTAB method. 1 μg DNA was modified via the EZ DNA Methylation-GoldTM kit (ZYMO RESEARCH, http://zymoresearch.com). The first round of Nested PCR reactions were performed in a final volume of 20 μL containing 2 μL bisulfite-treated DNA, 2 μL 10× buffer (Mg+2 plus), 0.8 μL dNTP, 1 μL primers, and 2.5 Units of hot-start Taq DNA polymerase (TaKaRa, Japan) with the following PCR (Touchdown PCR) program: an initial denaturation step (94 °C, 5 min) was followed by 16 touch-down cycles (cycles of 94 °C, 30 s; 60 °C to 47 °C (Region I) or 62 °C to 47 °C (Region II), 45 s, 1 °C decrease after each cycle; 72 °C, 1 min 20 s followed by 24 additional cycles of: 94 °C, 30 s; 55 °C, 45 s, 72 °C, 1 min 20 s), with a final elongation step (72 °C, 10 min). The second round of Nested PCR reactions were performed in a final volume of 50 μL containing 1 μL product of the first wheel DNA, 5 μL 10× buffer (Mg+2 plus), 4 μL dNTP, 1 μL primers, and 2.5 Units of hot-start Taq DNA polymerase with another PCR (Touchdown PCR) program: an initial denaturation step (94 °C, 5 min) was followed by 11 touch-down cycles (cycles of 94 °C, 30 s; 60 °C to 50 °C, 45 s, 1 °C decrease after each cycle; 72 °C, 1 min followed by 29 additional cycles of: 94 °C, 30 s; 55 °C, 45 s, 72 °C, 1 min), with a final elongation step (72 °C, 10 min). Size, quality and quantity of the PCR products were checked using 2 % agarose gel electrophoresis.
The PCR products were extracted using the Biospin Gel Extraction Kit (BioFlux) and ligated with the pMD™19–T Vector (TAKARA). Transferred to E.coli (DH5α) cells, at least 15 clones per insert were processed for sequencing (Invitrogen, USA). Sequences of which the cytosine transformation efficiency <97 % were removed by BiQ Analyzer software (Bock et al. 2005). The ratio of C methylation at each CG dinucleotide, and CHG and CHH trinucleotide was calculated and transformed into a percentage using KISMETH software (http://katahdin.mssm.edu/kismeth/revpage.pl) (Gruntman et al. 2008).
Statistical analysis
All of the data obtained in the study were subjected to statistical analysis by one-way ANOVA using SPSS v20. Data were considered statistically significant at P ≤ 0.05.
Results
Sequence analysis of TaGAPC1 genes and GAPC proteins
The length of TaGAPC1 cDNA were 1014 bp (Fig. 1a), and 337 amino acids were encoded by this sequence. The sequence was submitted to GeneBank and obtained an accession number of KU933269. TaGAPC1 was comprised of 11 exons and 10 introns (Fig. 2b). Alignment of GAPCs were given in Fig. 2a. The results revealed that the similarity of TaGAPC1 with AtGAPCs were over 90 %. NAD (P) binding domain (InterPro: IPR020828) and Glyceraldehyde 3-phosphate dehydrogenase, catalytic domain (InterPro: IPR020829) were detected in GAPCs (Fig. 2a). The prediction of protein subcellular locations showed that TaGAPC1 were cytoplasmic with 69.6 % possibility.

Determination of electrophoresis result for homologous cloning and Nested PCR for bisulfite sequencing. cw is Changwu134 and zy is Zhengyin1; M: DNA Marker DL2000. a. Amplification of TaGAPC1. b Amplification of promoter region for TaGAPC1. c, d Nested PCR results of region I and region II, respectively, the first two lane is the first round result and last two lane is the second round result

a Multiple sequence alignment of TaGAPC1 and GAPC proteins from Arabidopsis thaliana. (Consensus: represents the same deoxynucleotide among these sequences. The identical, 75 and 50 % deoxynucleotide similarity is indicated by mazarine, pink and green color. Red underlined a, Glyceraldehyde 3-phosphate dehydrogenase, NAD (P) binding domain (IPR020828); Red underlined b, Glyceraldehyde 3-phosphate dehydrogenase, catalytic domain (IPR020829)). b Schematic diagram for exons/introns and upstream/downstream structures of TaGAPC1 (Exons, introns, upstream/downstream are indicated by yellow boxes, black horizontal lines, and blue boxes, respectively. The position of two regions about bisulfite sequencing given in arrows)
To further confirm the subfamily of this gene and discover the potential functions, the evolutionary relationship between the putative protein and other GAPDHs from Arabidopsis thaliana were analyzed by MEG 5.0 software. Result revealed that the TaGAPC1 belonged to clade of plant GAPCs, according to a previously described classification method used for Arabidopsis GAPDH family (Fig. 3) (Martin and Cerff 1986).

Phylogenetic analysis of TaGAPC1. Numbers above or below branches of the tree indicate bootstrap values. The accession number of the GAPC proteins is as follows: AtGAPC1 (P25858.2), AtGAPC2 (Q9FX54.1), AtGAPA1 (P25856.3), AtGAPA2 (Q9LPW0.1), AtGAPB (P25857.2), AtGAPCp1 (Q9SAJ6.1), AtGAPCp2 (Q5E924.1)
Expression profiles of TaGAPC1 in wheat
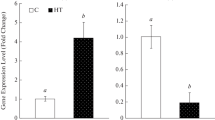
To reveal the expression of TaGAPC1 in response to osmotic and salinity stress, qRT-PCR of TaGAPC1 was performed after treated with 20 % PEG8000 and 250 mM NaCl for 24 h in two wheat cultivars, respectively. The expression of TaGAPC1 was upregulated significantly under PEG and NaCl treatment in Changwu134, peaking at 24 h (25-fold) and 6 h (13-fold), respectively (Fig. 4a). However in Zhengyin1, PEG and NaCl treatment resulted in upregulation of TaGAPC1 slightly, only reaching 3-fold and 6-fold, respectively, peaking at hours 6 and 12, respectively (Fig. 4b).

a, b The transcript abundance of TaGAPC1 after exposed to 20 % PEG8000 and 250 mM NaCl for 1, 2, 6, 12, and 24 h in Changwu134 and Zhengyin1. Standard deviation (SD) is displayed on the bar chart, significant differences were detected by the Student’s t test (**Significant at P ≤ 0.01 level; *significant at P ≤ 0.05 level). c CG, CHG and CHH context methylation change (in percentage) under stresses. d Total methylation changes of promoter in Changwu134 and Zhengyin1. Data are shown as the mean ± SD. Significant differences were detected by the Student’ s t test, ** P ≤ 0.01 were shown in the graph
Isolation the upstream region of TaGAPC1s genes from two wheat cultivars
To investigate the function of promoter in gene expression and ensure the accuracy of upstream sequences in two cultivars, the sequences were cloned by specific primers. The length of these two promoter sequences were 972 bp (Fig. 1b), which registered in the GeneBank (Accession number: KX196174). Moreover, the sequence alignment revealed that these two sequences from Changwu134 and Zhengyin1 shared 97.74 % identity (Supplementary fig. 1). Two sequences were analyzed in detail by PLACE and PlantCARE database, and the crucial elements that respond to stress and hormone were focused and shown in Table 2. The cis-acting elements that respond to drought and salinity (G-box, MBS, DRE, CGTCA-box, CBF, GT1) (Li et al. 2013a; Sun et al. 2010; Tao et al. 2015), ABA (CE1, ERE) (Luo et al. 2013; Niu et al. 2002), MeJA (CGTCA-box, TGACG-motif) (Rouster et al. 1997), low temperature (TATA-box, LTRECOREATCOR15, DRE and MYC) (Baker et al. 1994; Li et al. 2013b) and light (CATT-motif, Box I, GT1-motif and G-box) (Luo et al. 2013; Roy et al. 2012) were determined in both two wheat cultivars.
Bisulfite sequence result of promoter in two regions
The bisulfite sequencing PCR (BSP) was used to examine the cytosine methylation status of two regions (I and II) at the promoter region. As showed in Fig. 2b, region I covered from 208 to 596 bp, and region II covered from 714 to 1119 bp. The BSP result of two regions in normal growth of Changwu134 and Zhengyin1 displayed a cytosine methylated map in Fig. 5a, b. The results showed that the region I were heavily (>60 %) CG cytosine methylated and moderately (>20 %) CHG cytosine methylated, but slightly (<20 %) CHH cytosine methylated in studied two cultivars. It was obvious that the methylation rate ranking of the three cytosine methylation types in promoter region I was CG > CHG > CHH, which follow the same pattern of average methylation levels (Cokus et al. 2008). But in region II, it was irregular that all of the cytosine were slightly (<20 %) methylated in region II, even approaching to zero approximately. It is revealed that the cytosine methylation mainly occurred in the upstream of promoter.

Bisulfite sequencing for cytosine methylation of two regions (I and II) within the promoter of TaGAPC1 in plants under normal conditions of two wheat cultivars. All three types of cytosine residues, CG (red circles), CHG (blue circles), and CHH (green circles) were shown in the map. Filled and empty circles denote methylated and unmethylated cytosines, respectively. A row represents a sequencing result of a positive clone. a Cytosine methylation maps for region I in Changwu134 and Zhengyin1 orderly. b Cytosine methylation maps for region II in Changwu134 and Zhengyin1 orderly
The methylation rate of promoters under 20 % PEG8000 and 250 mM NaCl conditions were also detected. The results showed that the CG and CHG context was demethylated under PEG and NaCl stress in Changwu134 (Fig. 4c). CG methylation rate of PEG group were lower than CK group (0.85-fold), and CHG methylation rate of NaCl group were also lower than CK (0.67-fold). But the cytosine methylation level was increased in Zhengyin1 (Fig. 4c), the CG context methylation level was constant and CHG and CHH was increased slightly under PEG and NaCl treatment. The C methylation change of cis-elements which involved in stress response was given in Table 3. The cytosines of G-box, MBS, ABRERATCAL, MYBCORE and MYCCONSENSUSAT were demethylated by stresses in Changwu134. The cytosines of CE1 were demethylated by all the stresses in two wheat genotypes.
Discussion
Water deficit and salinity stress have adverse effects on the growth and production of crops. Many efforts have been made to elucidate the mechanisms of stress response in crops. The fine tuning of gene expression in response to abiotic stress was a key strategy for crops to cope with the changing environment (Petrillo et al., 2014). Recent researches have investigated variety functions of GAPC proteins in plants, including the stress-response (Guo et al. 2012; Hancock et al. 2005). In poaceae plants, OsGAPC2 had been proven that involved in responding to drought, salt, heat and ABA stresses (Zhang et al. 2011), and ZmGAPC1 was response to anoxia (Manjunath and Sachs 1997). In this research, phylogenic analysis investigate that TaGAPC1 belonged to one clade of AtGAPC1 and AtGAPC2, which was involved in responding to heat, anaerobic, salt and drought (Guo et al. 2012; Yang et al. 1993; Zhang et al. 2011). So study on the expression of GAPC gene under abiotic stress was significant for understanding the functions of stress response in wheat. Further result of qRT-PCR confirmed that the expression of TaGAPC1 was strongly induced by stresses in drought-tolerant wheat genotype and slightly induced in drought-sensitive wheat genotype. It was indicated that TaGAPC1 may play some roles of stress response via unknown mechanism in wheat.
Epigenetic modification contributes to the expression divergence of functional gene expression, including histone acetylation and DNA methylation (Hu et al., 2013; Xu et al., 2013). Growing evidences from recent studies have suggested that demethylation of promoter region could enhance the expression of genes when plants exposed to stresses (Paszkowski and Whitham, 2001; Choi and Sano, 2007). It has been found that hypomethylation and hypermethylation are more frequent by drought stress in drought-tolerant and drought-sensitive rice genotypes, respectively (Gayacharan and Joel, 2013). In recent researches, numerous diversely methylated regions associated with differential expression of genes were found important for abiotic stress response in some rice cultivars with different resistance (Garg et al., 2015). Moreover, in plants, the three patterns of DNA methylation (CG, CHG, and CHH) were accomplished, maintained, and demethylated by different enzymes or protein families, also being affected by respective regulatory pathways (Law and Jacobsen 2010; Stroud et al. 2014). So study on the methylation of gene’s upstream region is significant for understanding the mechanism of stress response gene in plants. In present study, CG and CHG context of promoter region were demethylated by stresses in drought-tolerant wheat. However in drought-susceptible wheat, CG context was maintained methylation and CHG (CHH) methylation was established by their pathways. This difference may is a reason why gene transcribe significantly in drought-tolerant wheat.
The cis-elements, which presented in the promoter region of stress-responsive genes, play a crucial role in interacting with transcription factors to regulate the gene expression under stress (Bjornson et al. 2016). The cis-elements were the key factors in regulating of promoter activity, so the methylation of their sequences worth been further explored. In this study, the prediction results of promoters revealed that there were regulatory elements that response to abiotic stress, such as low temperature, ABA, salinity, and drought (Fig. 4). The statistics result summarized the methylation frequency of cis-elements which related to stress tolerance (Table 3) (Wang et al. 2014). Some of the stress-induced alterations in C methylation occurred at sites that located within known cis-elements, such as G-box and MBS (drought and salinity response), were demethylated in Changwu134, but established in Zhengyin1; CE1 (ABA response) was reduced by osmotic stress and lower by salinity stress. It was conclusion that TaGAPC1 was responding to both osmotic and salinity stress in drought-tolerant wheat genotypes, indicating that there may some unknown pathways for combining transcriptional regulatory proteins, which can recognize methylation of cytosine in the cis-elements of promoter regions.
In conclusion, the TaGAPC1 gene was expressed differentially under stress condition in wheat genotypes with contrasting drought tolerance. Further results revealed that the methylation patterns of promoter region, which induced by stress differentially in wheat genotypes with contrasting drought tolerance, were an important regulatory mechanism for sensing and responding to the stress conditions. Our investigation also suggested the potential role of some specific loci methylation change on cis-elements for gene responding to stresses. These findings allow us to further known the function of TaGAPC1 genes, and understand of epigenetic regulation of gene expression in abiotic stress responses.
References
Arumugam Pillai M, Lihuang Z, Akiyama T (2002) Molecular cloning, characterization, expression and chromosomal location of OsGAPDH, a submergence responsive gene in rice (Oryza sativa L.). Theor Appl Gen 105:34–42. doi:10.1007/s00122-001-0833-9
Attitalla IH (2011) Modified CTAB method for high quality genomic DNA extraction from medicinal plants. Pak J Biol Sci 14:998–999. doi:10.3923/pjbs.2011.998.999
Baker SS, Wilhelm KS, Thomashow MF (1994) The 5′-region of Arabidopsis thaliana cor15a has cis-acting elements that confer cold-, drought- and ABA-regulated gene expression. Plant Mol Biol 24:701–713. doi:10.1007/BF00029852
Bjornson M, Dandekar A, Dehesh K (2016) Determinants of timing and amplitude in the plant general stress response. J Integr Plant Biol 58:119–126. doi:10.1111/jipb.12373
Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T (2005) BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 21:4067–4068. doi:10.1093/bioinformatics/bti652
Boyko A et al (2010) Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS One 5:e9514. doi:10.1371/journal.pone.0009514
Choi CS, Sano H (2007) Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants Mol Genet Genomics 277:589–600 doi:10.1007/s00438-007-0209-1
Cokus SJ et al (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452:215–219. doi:10.1038/nature06745
Gayacharan, Joel AJ (2013) Epigenetic responses to drought stress in rice (Oryza sativa L.) Physiol Mol Biol Plants 19:379–387 doi:10.1007/s12298-013-0176-4
Garg R, Chevala VN, Shankar R, Jain M (2015) Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response Scientific Reports 5:14922 doi:10.1038/srep14922
Gruntman E, Qi Y, Slotkin RK, Roeder T, Martienssen RA, Sachidanandam R (2008) Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics 9:371. doi:10.1186/1471-2105-9-371
Guo L, Devaiah SP, Narasimhan R, Pan X, Zhang Y, Zhang W, Wang X (2012) Cytosolic glyceraldehyde-3-phosphate dehydrogenases interact with phospholipase Ddelta to transduce hydrogen peroxide signals in the Arabidopsis response to stress. Plant Cell 24:2200–2212. doi:10.1105/tpc.111.094946
Guo L, Ma F, Wei F, Fanella B, Allen DK, Wang X (2014) Cytosolic phosphorylating glyceraldehyde-3-phosphate dehydrogenases affect Arabidopsis cellular metabolism and promote seed oil accumulation. Plant Cell 26:3023–3035. doi:10.1105/tpc.114.126946
Hancock JT et al (2005) Proteomic identification of glyceraldehyde 3-phosphate dehydrogenase as an inhibitory target of hydrogen peroxide in Arabidopsis. Plant Physiol Biochem 43:828–835. doi:10.1016/j.plaphy.2005.07.012
Hu ZR, Han ZF, Song N, Chai LL, Yao YY, Peng HR, Ni ZF, Sun QX (2013) Epigenetic modification contributes to the expression divergence of three TaEXPA1 homoeologs in hexaploid wheat (Triticum aestivum) New Phytologist 197:1344–1352 doi:10.1111/nph.12131
Henderson IR, Chan SR, Cao X, Johnson L, Jacobsen SE (2010) Accurate sodium bisulfite sequencing in plants. Epigenetics 5:47–49. doi:10.4161/epi.5.1.10560
Inamdar NM, Ehrlich KC, Ehrlich M (1991) CpG methylation inhibits binding of several sequence-specific DNA-binding proteins from pea, wheat, soybean and cauliflower. Plant Mol Biol 17:111–123. doi:10.1007/BF00036811
Jeon J, Choi J, Lee GW, Park SY, Huh A, Dean RA, Lee YH (2015) Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci Rep 5:8567. doi:10.1038/srep08567
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204–220. doi:10.1038/nrg2719
Lescot M et al (2002) PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res 30:325–327. doi:10.1093/nar/27.1.295
Li CN, Ng CKY, Fan LM (2015) MYB transcription factors, active players in abiotic stress signaling. Environ Exp Bot 114:80–91. doi:10.1016/j.envexpbot.2014.06.014
Li F, Han YY, Feng YA, Xing SC, Zhao MR, Chen YH, Wang W (2013a) Expression of wheat expansin driven by the RD29 promoter in tobacco confers water-stress tolerance without impacting growth and development. J Biotechnol 163:281-291. doi:10.1016/j.jbiotec.2012.11.008
Li M et al. (2013b) Strength comparison between cold-inducible promoters of Arabidopsis cor15a and cor15b genes in potato and tobacco. Plant Physiol Bioch 71:77-86. doi:10.1016/j.plaphy.2013.06.021
Liu J, Feng L, Li J, He Z (2015) Genetic and epigenetic control of plant heat responses. Front Plant Sci 6:267. doi:10.3389/fpls.2015.00267
Luo QL, Li YG, Gu HQ, Zhao L, Gu XP, Li WB (2013) The promoter of soybean photoreceptor GmPLP1 gene enhances gene expression under plant growth regulator and light stresses. Plant Cell Tiss Org 114:109–119. doi:10.1007/s11240-013-0310-6
Ma X et al (2015) Identification and comparative analysis of differentially expressed miRNAs in leaves of two wheat (Triticum aestivum L.) genotypes during dehydration stress. BMC Plant Biol 15:21. doi:10.1186/s12870-015-0413-9
Manjunath S, Sachs MM (1997) Molecular characterization and promoter analysis of the maize cytosolic glyceraldehyde 3-phosphate dehydrogenase gene family and its expression during anoxia. Plant Mol Biol 33:97–112. doi:10.1023/A:1005729112038
Marri L, Sparla F, Pupillo P, Trost P (2005) Co-ordinated gene expression of photosynthetic glyceraldehyde-3-phosphate dehydrogenase, phosphoribulokinase, and CP12 in Arabidopsis thaliana. J Exp Bot 56:73–80. doi:10.1093/jxb/eri020
Martin W, Cerff R (1986) Prokaryotic features of a nucleus-encoded enzyme. cDNA sequences for chloroplast and cytosolic glyceraldehyde-3-phosphate dehydrogenases from mustard (Sinapis alba). Eur J Biochem 159:323–331. doi:10.1111/j.1432-1033.1986.tb09871.x
McLoughlin F et al (2013) Identification of novel candidate phosphatidic acid-binding proteins involved in the salt-stress response of Arabidopsis thaliana roots. Biochem J 450:573–581. doi:10.1042/BJ20121639
Naydenov M, Baev V, Apostolova E, Gospodinova N, Sablok G, Gozmanova M, Yahubyan G (2015) High-temperature effect on genes engaged in DNA methylation and affected by DNA methylation in Arabidopsis. Plant Physiol Biochem 87:102–108. doi:10.1016/j.plaphy.2014.12.022
Nicholls C, Li H, Liu JP (2012) GAPDH: a common enzyme with uncommon functions. Clin Exp Pharmacol Physiol 39:674–679. doi:10.1111/j.1440-1681.2011.05599.x
Niu X, Helentjaris T, Bate NJ (2002) Maize ABI4 binds coupling element1 in abscisic acid and sugar response genes. Plant Cell 14:2565–2575. doi:10.1105/tpc.003400
Paszkowski J, Whitham SA (2001) Gene silencing and DNA methylation processes Current Opinion in Plant Biology 4:123-129 doi:10.1016/S1369-5266(00)00147-3
Petrillo E,Godoy Herz MA,Barta A,Kalyna M,Kornblihtt AR (2014) Let there be light: Regulation of gene expression in plants RNA Biology 11:1215-20 doi:10.4161/15476286.2014.972852
Rouster J, Leah R, Mundy J, Cameron-Mills V (1997) Identification of a methyl jasmonate-responsive region in the promoter of a lipoxygenase 1 gene expressed in barley grain. Plant J 11:513–523. doi:10.1039/cc9960000015
Roy S, Choudhury SR, Singh SK, Das KP (2012) Functional analysis of light-regulated promoter region of AtPollambda gene. Planta 235:411–432. doi:10.1007/s00425-011-1517-6
Steward N, Ito M, Yamaguchi Y, Koizumi N, Sano H (2002) Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J Biol Chem 277:37741–37746. doi:10.1074/jbc.M204050200
Stroud H et al (2014) Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol 21:64–72. doi:10.1038/nsmb.2735
Sun H et al (2014) DNA methylation pattern of Photoperiod-B1 is associated with photoperiod insensitivity in wheat (Triticum aestivum). New Phytol 204:682–692. doi:10.1111/Nph.12948
Sun LF, Liu TJ, Shan XH, Su SZ, Li SP, Yuan YP, Zhang J (2015) Analysis of DNA cytosine methylation patterns in maize hybrids and their parents. Biol Plant 59:266–272. doi:10.1007/s10535-015-0490-5
Sun Q, Gao F, Zhao L, Li K, Zhang J (2010) Identification of a new 130 bp cis-acting element in the TsVP1 promoter involved in the salt stress response from Thellungiella halophila. BMC Plant Biol 10:90. doi:10.1186/1471-2229-10-90
Tao Y et al (2015) Cloning and functional analysis of the promoter of a stress-inducible gene (ZmRXO1) in maize. Plant Mol Biol Rep 33:200–208. doi:10.1007/s11105-014-0741-1
Wang M et al (2014) Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol 55:1354–1365. doi:10.1093/pcp/pcu059
Weng MY, Cui LR, Liu FL, Zhang M, Shan L, Yang SS, Deng XP (2015) Effects of drought stress on antioxidant enzymes in seedlings of different wheat genotypes. Pak J Bot 47:49–56
Xu CH, Wang M, Zhou L, Quan TY, Xia GM (2013) Heterologous Expression of the Wheat Aquaporin Gene TaTIP2;2 Compromises the Abiotic Stress Tolerance of Arabidopsis thaliana. PloS one 8. doi:10.1371/journal.pone.0079618
Yang Y, Kwon HB, Peng HP, Shih MC (1993) Stress responses and metabolic regulation of glyceraldehyde-3-phosphate dehydrogenase genes in Arabidopsis. Plant Physiol 101:209–216. doi:10.1104/pp.101.1.209
Zeng L, Deng R, Guo Z, Yang S, Deng X (2016) Genome-wide identification and characterization of Glyceraldehyde-3-phosphate dehydrogenase genes family in wheat (Triticum aestivum). BMC Genomics 17:240. doi:10.1186/s12864-016-2527-3
Zhang XH, Rao XL, Shi HT, Li RJ, Lu YT (2011) Overexpression of a cytosolic glyceraldehyde-3-phosphate dehydrogenase gene OsGAPC3 confers salt tolerance in rice. Plant Cell Tiss Org 107:1–11. doi:10.1007/s11240-011-9950-6
Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39:61–69. doi:10.1038/ng1929
Acknowledgments
This work was supported by The National Natural Science Foundation of China (No. 31271625 and No. 51479189), The State Key Laboratory of Soil Erosion and Dryland Farming on Loess Plateau, Institute of Soil and Water Conservation, Chinese Academy of Sciences (No. 10502), and The National Basic Research Program of China (2015CB150402).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Néstor Carrillo
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary fig. 1
Comparison of promoter sequences of TaGAPC1s. Identical similarity is shaded in gray; cis-acting elements in two promoters from different wheat cultivars are in rectangles; Translational start sites are shown in italics. 5′ UTR are shown in underline. (TIF 18,721 kb)
Rights and permissions
About this article

Cite this article
Fei, Y., Xue, Y., Du, P. et al. Expression analysis and promoter methylation under osmotic and salinity stress of TaGAPC1 in wheat (Triticum aestivum L). Protoplasma 254, 987–996 (2017). https://doi.org/10.1007/s00709-016-1008-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-016-1008-5