
Abstract
Methylation of DNA promoter sequences at the CpG islands has become a molecular tool for gene regulation. NAC6D gene is induced by different biotic and abiotic stimuli. The proximal promoter sequence of NAC6D was investigated for the impact of CpG methylation on its expression in response to high temperature in wheat. Gene expression was estimated by real time PCR and methylation of NAC6D promoter sequence was investigated by bisulfite sequencing. Results showed that NAC6D was highly induced by high temperature, whereas DNA methylatransferase 3 (Met3) was highly reduced by high temperature. Close investigation of NAC6D promoter methylation revealed that high temperature caused hypomehtylation of the proximal promoter sequence. Twelve CpGsites showed low difference in methylation compared to the control (normal temperature, 25°C), while 3 CpGs (–59, –169, –204) were extremely hypomethylated in response to high temperature compared to their methylation status under the normal condition. The induction of NAC6D was negatively correlated with Met3 suppression and methylation level at the CpG sites in the promoter region. Results prove that methylation greatly contribute to the regulation of NAC6D in response to high temperature. This will improve our current understanding of how plants respond to abiotic stresses at the molecular level and the integration of DNA methylation and epigenetics in the next generation plant breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The indispinsible role of epegenetics regulation of gene expression, development, shaping phenotype, and adaptation of higher organisms to their environments has been extensively investigated (Bräutigam et al., 2013; Rodríguez López and Wilkinson, 2015). DNA methylation is the most detailed studied mechanism of epigenetics that has emerged as the next generation tool for crop breeding and development (Rodríguez López and Wilkinson, 2015). In plants, DNA methylation has been reported to occur in one or more of the three nucleotide contexts CpG, CpNpG, and at CpHpH sequence context with H indicating A, T, or C (Akimoto et al., 2007). DNA methylation at these sites is recruited by different types of DNA methyltransferases (Mets) where methylation at the CpGis achieved and sustained by DNA methyltransferase I (Met1) (Law and Jacobsen, 2010; Zilberman, 2017), whereas methylation at the CpNpG is sustained by Chromomethyltransferase 3 (CMT3) (Han and Wagner, 2014). It is notewoarthy that methylation at the CpG and CpHpG sites is inherited during meiosis (Jones et al., 2001), while methylation at the CpNpN is not inherited and sustained every generation by DRM2 (Domains Rearranged Methyltransferase 2) by de novo methylation (Chan et al., 2005). In plants, DNA methylation play indispensible role in wide range of biological and metabolic processes including regulation of metabolism (Yong-Villalobos et al., 2015), differentiation and development (Ikeuchi et al., 2015), and senescence (Demeulemeester et al., 1999). It also was pointed out that DNA methylation is involved in the plant adjustment to environmental changes by the epigenetic memory (Yaish, 2017), plant growth regulation via modulation of gene expression by chromatin remodeling (Han and Wagner, 2014), plant response to abiotic stresses (Yaish et al., 2018), and controlling the function and expansion of repetitive elements (Fedoroff, 2012).
DNA methylation occurs at promoters and coding sequences and both types of methylation contribute to the regulation of gene expression (Takatsuka and Umeda, 2015). Methylation of promoter sequences as well as hypomethylation or hypermethylation of gene body sequences suppress gene expression (Suzuki and Bird, 2008; Takuno and Gaut, 2012), whereas moderate methylation of gene body sequences upregulate gene expression (Takuno and Gaut, 2012). Several line of evidence documented that hypermethylation of promoter sequences led to gene silencing (Bird, 2002; Saze et al., 2012).
Promoter hypermethylation at the CpG islands caused the repression of gene expression of several genes including MAPK12, GSTU10, and BXL1 in Arabidopsis cell culture. Mutant analysis of Mets proved that promoter hypermethylation was achieved by MET1 and DRM2 (Berdasco et al., 2008). Transcriptional gene silencing (TGS) by hypermethylation has been reported by several authors (Deng et al., 2014; Matzke et al., 1989). In the same line, hypermethylation of granule bound starch synthase I (GBSSI) promoter at –213 upstream of the transcription start site in both CpG as well as CpHpH contexts caused the suppression of GBSSI (Heilersig et al., 2006).
Recently, several reports investigated the role of promoter methylation association to gene expression level and the role of Mets in the regulation of DNA methylation (El-Shehawi et al., 2013; Elseehy, 2020; Elseehy and El-Shehawi, 2020). The T0 plants carrying the soybean isoflavone synthase (IFS) transgene controlled by either 35S promoter or Oleocin (OL) promoter showed great variations at the RNA as well as the genistein (pathway final product) levels (El-Shehawi et al., 2013). Close investigation showed that these variations were regulated by the methylation of the 35S and OL exogenous promoters (Takatsuka and Umeda, 2015). Interestingly, methylation level at the –56 and –88 CpG sites was highly correlated with the IFS expression and highly involved in the regulation of IFS transgene in T0 wheat plants (Takatsuka and Umeda, 2015). T1 plants of these T0 generation inherited the methylation level of 35S and was correlated with the expression pattern of IFS gene at the RNA level as well as the genistein level over one generation, whereas the T1 plants were able to modify methylation pattern and reconstitute the expression IFS driven by the OL exogenous promoter to normal (Suzuki and Bird, 2008).
Inheritance of hypomethylation was experimentally documented using the methylation inhibitors 5-azacytidine and 5-azadeoxycytidine in several plant species including tobacco, rice, and brassica. The inheritance of DNA methylation methylation was proved by its association to phenotype changes across generations (Akimoto et al., 2007).
PEG-induced dehydration invoked the expression of wheat DNA Mets including Met1, Met2b, CMT, and Met3 in various wheat lines. Variations in Mets expression level was positively correlated with the wheat line resistance to biotic and abiotic stresses (Elseehy and El‑Shehawi, 2018). HSP17 promoter showed differential methylation under normal conditions compared to heat stress condition in seven wheat varieties. This was associated with methylation difference in 4 CpGs between normal and heat stress condition by 56.7–60%. The change of methylation was associated with low expression of HSP17 and high expression of Met3, whereas under heat stress high expression of HSP17 and low expression of Met3 were associated with low level of methylation of the HSP17 promoter in the 4 CpG sites (Alotaibi et al., 2020).
Genome wide methylation (methylome) has been used to estimate the impact of plant growth condition including growth, development, biotic and abiotic stresses on gene expression via whole genome sequencing (WGS) (Yaish et al., 2018; Yong-Villalobos et al., 2015). However, methylome investigation detects the collective whole genome methylation and does not give details about site-specific methylation (methylation of specific gene promoters), whereas site-specific methylation provide specific data about the role of certain CpGs in a promoter in the regulation of these genes and in most cases link methylation data (epialles) to changes in plant phenotypic characters (Elseehy and El-Shehawi, 2020).
NAC proteins are among the largest transcription families in plant kingdom (Guérin et al., 2019). NAC genes have been detected in various plant species as large gene families including rice (151 gene) (Nuruzzaman et al., 2010), soybean (101 genes) (Pinheiro et al., 2009), maize (157 genes) (Lu et al., 2015), and wheat (Triticumaestivum) (361 genes) (Saidi et al., 2017). NAC gene family plays indispensible roles in various plant biology processes including plant development, and flowering (Moyano et al., 2018; Uauy et al., 2006). They also are involved in tolerance to various abiotic stresses including drought and high temperature (Guérin et al., 2019; Guo et al., 2015; Nakashima et al., 2012; Qin et al., 2008).
Recently, NAC6D as well as other stress responsive genes including heat shock protein 17, heat shock protein 90, DREB2 were induced under heat stress in wheat (El-Shehawi, 2020). The focus of this study is to investigate the NAC6D promoter methylation status and its role in the regulation of NAC gene under heat stress.
MATERIALS AND METHODS
Plant Materials
Wheat (Triticum aestivum, Chinese spring) seeds were surface sterilized and were germinated on 1% agar water in 6 plastic vessels, 100 seeds per vessel. All vessels were kept at normal temperature for one week. Three vessels served as control (normal temperature), whereas the other three vessel were treated with 40°C for 1 h (Qin et al., 2008). Leaf samples were directly collected from both sets of plantlets and directly lyophilized for 36 h at –58°C. Dried samples were ground in TissueLyzer II (Qiagen, Germany) and used for RNA isolation.
Primers
Primers for wheat NAC6D gene, Met3, α-Tubulin, and NAC6D promoter were synthesized at Macrogen Company (Macrogen, Korea). Primers for methylation study of NAC6D promoter had Y (T+C) or R (A+G) to replace C on the forward strand or G on the reverse strand so that primers can match the original or the sequence after bisulfite treatment. Table 1 summarizes the nucleotide sequence and main information of primers used in this study.
Isolation of Genomic DNA
DNA was isolated using modified CTAB protocol (Elseehy and El-Shehawi, 2020) using lyophilized ground tissues.
Total RNA Isolation
RNA was isolated from 5 mg of the lyophilized ground plant tissue using QiaZol (Qiagen, Germany). Procedures of RNA isolation was according to the manifacturer’s guidelines.
Synthesis of cDNA and Quantitative PCR
Complementary DNA, cDNA, was generated using ImProm-II reverse transcription system (Promega, Madison, Wisconsin, United States) in 20 μL total volume reaction containing 1 μg of total RNA, 0.5 μg of random hexmer, 1X ImProm-II reaction buffer, 8 mM MgCl2, 0.5 mM of dNTPs, 1 μL ImProm-II reverse transcriptase. Real time PCR was conducted using GoTaq qPCR master mix system (Promega, Madison, Wisconsin, United States) in 20 μL reaction containing 1X GoTaq qPCR master mix, 250 nM of forward and reverse primer, 1 μL of cDNA in C1000 Thermal Cycler (BioRad, California, United States). The targeted DNA sequence was amplified using the following conditions; 95°C for 2 min, 40 cycles of 95°C for 15 s, 60°C for 1 min. The expression level of NAC6D and Met3 genes was normalized to α-tubulin mRNA level and was estimated by the 2–ΔΔCt method (Livak and Schmittgen, 2001).
DNA Treatment with Bisulfite
Bisulfite treatment of DNA was carried out in 18 μL reaction volume according to a previously published method (Li and Tollefsbol, 2011).
Amplification of Bisulfite Treated DNA
Bisulfite-treated DNA was amplified using PCR under the same IFS transgene detection PCR using primers for bisulfite PCR (Table 1). Specific primers for NAC6D promoter were designed to amplify 446 nucleotides of promoter proximal region.
Cloning and Sequencing
Bisulfite-treated DNA was purified and cloned in the pGMT-Easy vector (Promega, United States) following the manufacturer’s instructions. For each treatment, a number of 30 clones were sequenced at Macrogen (Macrogen, Korea) to disclose methylation status in a single molecule. The obtained sequences were aligned with Clustal Omega (EBI, https://www.ebi.ac.uk). Methylation status of CpGs was disclosed using CpG Viewer software (Carr et al., 2007). Cytosines (C) in the CpG contexts indicate methylated cytosine, whereas the presence of thymines (T) indicates unmethylated cytosine.
RESULTS
Gene Expression of NAC6D and Met3
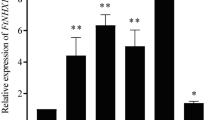
The impact of methylation at the NAC6D promoter gene on NAC6D gene expression was investigated under normal condition compared to heat stress condition in the current study. Under normal condition (control), NAC6D was low expressed (Fig. 1a). In response to high temperature, NAC6D was highly expressed up to 4.2 folds compared to the control (normal temperature) (Fig. 1a). The expression of Met3 gene was estimated to determine the role of Met3 in the methylation of NAC6D promoter sequence. Met3 showed opposite gene expression profile of NAC6D gene. Under normal condition, Met3 gene was highly expressed more than 4 folds compared to the control, whereas it was extremely downregulated up to –5 folds compared to the control group (Fig. 1b).

Quantitative PCR analysis of NAC6D (a) and Met3 (b) expression in wheat under normal (C) and high temperature (HT) conditions. Different superscripts (a, b) indicate significant changes in gene expression at p < 0.05. Errors represent standard deviation.
NAC6D Promoter Proximal Region Methylation Level
A number of 15 CpG islands were detected in the NAC6D promoter proximal region. Under normal condition, all of the detected CpG sites were methylated at different levels except the –245 site which was unmethylated. Sites –59, –62, –169, –173, –204, –220, and –224 were highly methylated with methylation level range from 60 to 70% (Figs. 2, 3). Sites –62, –169 were heavily hypermethylated with 70% methylation. Sites –59, –204, and –220 were also hypermethylated (66.7%), while sites –173, –206 as well as site –224 were also hypermethylated at 60% (Figs. 2, 3). The other CpG sites have lower methylation level at 23.3% (–46), –26.7% (–76), 10% (–178), 20% (–197, –211), 16.6 (–238) (Figs. 2, 3b). In response to high temperature, sites –173, –178, –197, –211, –224, and –245 did not have any changes in their methylation level (Figs. 2, 3b). Sites –62, –76, and –238 showed reduced methylation levels by less than 4%. Sites –59, –169, and –204 had extremely reduced methylation level by up to 50% (–169) (Figs. 2, 3b). Sites –206 and –220 had elevated methylation level by up to 10% (‒206) (Figs. 2, 3b). Overall methylation at normal temperature was 42.44% which was reduced to 33.3% in response to high temperature. These changes in methylation upon exposure to high temperature were associated with drastic change in NAC6D and Met3 expression levels (Fig. 1).

Methylation percentages in the 15 CpG islands in the promoter proximal region of NAC6D in wheat plants.

Lollipop diagram of proximal promoter region methylation status of NAC6D in wheat plants under normal conditions (a), and under high temperature (b). White circles indicate unmethylated ctosine and black circles indicate methylated cytosine.
DISCUSSION
Wheat NAC6D was previously reported to be induced by high temperature (El-Shehawi, 2020). The expression of wheat NAC6D and Met3 genes was estimated under normal condition as well as high temperature condition. Wide variations were revealed in the expression of both genes; NAC6D was upregulated and Met3 was downregulated in response to elveated temperature. The methylation level of the NAC6D promoter proximal region was negatively correlated with the expression level of NAC6D, however, on the other hand methylation level of NAC6D promoter was positively correlated with the expression level of Met3 gene. This provide explanation that high temperature induced NAC6D gene through the low level of methylation at the proximal promoter sequence. This is supported by the low expression level of Met3 gene.
Mets maintain DNA methylation after meiosis and carry out the de novo methylation in response to environmental changes. Different wheat Mets presented differential expression in various tissues including embryos, sedlings, and leaves as well as different developmentla stages (Dai et al., 2005). MET1 is responsible for methylation of the hemimythelated DNA after replication across generations (Han and Wagner, 2014; Zilberman, 2017). CMT is plant specific MET which methylates the CNG sites (Han and Wagner, 2014). Met3 is responsible for the de novo methylation in response to changes in environmental or growth conditions (Dai et al., 2005), therefore its expression could be related to changes in methylation in T1 transgenic wheat plants.
Results obtained in the current study are in line with reports of previous studies. Low expression of Met3 was reported in T0 transgenic wheat plants in association with hypomethylaiton of 35S and OL exogenous promoter driving the isoflavone synthase transgene (IFS) (Elseehy and El-Shehawi, 2020). Also, the low expression profile of Met3 was inherited from high IFS exaressers driven by the 35S promoter and this was associated with high IFS expression as well as hypomethylation of the 35S promoter (Elseehy and El-Shehawi, 2018). In addition, T1 transgeneic wheat plants of T0 plants carrying the IFS drivied by 35S promoter inherited the same expression profile of IFS, methylation profile of 35S promoter, and the Met3 expression (Elseehy and El-Shehawi, 2020). On the contrary, low expressers of T1 plants carring IFS driven by OL promoter were able to change their expression profile of IFS to high level. This was correlated with hypomethylation of OL exogenous promoter and downregulation of Met3 expression (Elseehy, 2020). In the current study, change in methylation level at 3 CpG sites (–59, –169, –204) was associated with change in the NAC6D expression, methylation level in the NAC6D promoter proximal region, and the expression level in Met3. Under normal condition these 3 sites were hypermethylated, whereas upon exposure to high temperature, they were extremely hypomethylated (>50%). This was correlated with low expression of NAC6D and low Met3 expression level. This relationship was not established with other CpG sites in the NAC6D promoter, which supports the role of specific CpG sites in regulating the NAC6D promoter in response to high temperature. This was also observed in other studies (El-Shehawi, 2020; Elseehy, 2020; Elseehy and El-Shehawi, 2020). The role of certain CpG sites in the regulation of specific genes was documented in other studies. Methylation at two CpG sites (–56, –88) of the exogenous 35S promoter was strongly linked to IFS transgene expression in T0 plants which was also associated with the level of Met3 promoter (Elseehy and El-Shehawi, 2020). Also, the –106 and –151 CpG sites in the OL promoter showed high change in methylation level in T0 (Elseehy and El-Shehawi, 2020). In addition, in T1 plants the same CpG sites –56 and –88 played similar role in the regulation of IFS transgene driven by the 35S promoter. In the same line the CpG sites at –106 and –151 had the major role in regulating the IFS transgene driven by OL promoter in T1 plants (Elseehy, 2020).
In a similar fashion, the HSP17 gene promoter expression in response to high temperature was dependent on the methylation status of 4 CpG sites (–28, –45, –81, –180) in the proximal promoter region that showed up to 60% in methylation level between normal and high temperature condition (El-Shehawi, 2020). High methylation of HSP17 promoter was assocaited with low level of HSP17 expression, and high level of Met3 gene, whereas hypomethylation of HSP17 promoter was correlated with high level of HSP17 gene expression and low level of Met3 gene (El-Shehawi, 2020).
The obtained results in the current study are in parallel with results obtained by other groups that reported the association of promoter methylation level with the level of gene expression and presented promoter methylation as the molecular tool for the regulation of gene expression in plants (Bucherna et al., 2001; Tolley et al., 2012). In the maize PEPC gene promoter, methylation at 4 CpGs greatly affected the expression level of PEPC gene in different tissues including roots and leaves (Tolley et al., 2012). Several authors reported that methylation of certain promoter sequence caused the repression of gene expression levels (Akimoto et al., 2007; Berdasco et al., 2008; Bird, 2002; Lira-Medeiros et al., 2010; Saze et al., 2012; Suzuki and Bird, 2008; Takatsuka and Umeda, 2015; Wang et al., 2011). For example, in Arabidopsis, polyethylene glycol-induced drought stress was correlated with the hypermethylation of drought induced genes upstream of their transcriptional start site (TSS) (Colaneri and Jones, 2013).
DNA methylation is involved in eppiallele and their contribution to next generation breeding. Several studies have discussed the role of eppialles in crop improvement (Song et al., 2017; Wei et al., 2017). The potential of apiallele application has been substantiated by the inheritance of them across generations which was established by several lines of evidence (Akimoto et al., 2007). Priming provided a supporting evidence for transgeneration inheritance of epialleles. In priming, plants can acquire the capability to tolerate biotic or abiotic stresses by the ability to recognize specific environmental stimuli which gives the plants protection via provoking faster response for the same signal upond second exposure (Conrath, 2011). Several studies supported the transgeneration of priming methylation marks (Becker and Weigel, 2012; Hauben et al., 2009; Kathiria et al., 2010; Tricker et al., 2013). The obtained results of the current study are in line with previous studies that promoter methylation is one major factor of gene regulation in response to abiotic stresses. This is translated into a linkage between methylation pattern and specific phenotype. All together can contribute toward next generation of plant breeding using epiallele strategies and epifingerprinting.
REFERENCES
Akimoto, K., Katakami, H., Kim, H.J., Ogawa, E., Sano, C.M., Wada, Y., and Sano, H., Epigenetic inheritance in rice plants, Ann. Bot., 2007, vol. 100, no. 2, pp. 205–217. https://doi.org/10.1093/aob/mcm110
Alotaibi, S.S., El-Shehawi, A.M., and Elseehy, M., Heat shock proteins expression is regulated by promoter cpg methylation/demethylation under heat stress in wheat varieties, Pak. J. Biol. Sci., 2020, vol. 23, no. 10, pp. 1310–1320. https://doi.org/10.3923/pjbs.2020.1310.1320
Becker, C. and Weigel, D., Epigenetic variation: origin and transgenerational inheritance, Curr. Opin. Plant Biol., 2012, vol. 15, no. 5, pp. 562–567. https://doi.org/10.1016/j.pbi.2012.08.004
Berdasco, M., Alcázar, R., Garcia-Ortiz, M.V., Ballestar, E., Fernández, A.F., Roldán-Arjona, T., Tiburcio, A.F., Altabella, T., Buisine, N., Quesneville, H., Baudry, A., Lepiniec, L., Alaminos, M., Rodríguez, R., Lloyd, A., Colot, V., Bender, J., Canal, M.J., Esteller, M., and Fraga, M.F., Promoter DNA hypermethylation and gene repression in undifferentiated Arabidopsis cells, PLoS One, 2008, vol. 3, no. 10, art. ID e3306. https://doi.org/10.1371/journal.pone.0003306
Bird, A., DNA methylation patterns and epigenetic memory, Genes Dev., 2002, vol. 16, no. 1, pp. 6–21. https://doi.org/10.1101/gad.947102
Bräutigam, K., Vining, K.J., Lafon-Placette, C., Fossdal, C.G., Mirouze, M., Marcos, J.G., Fluch, S., Fraga, M.F., Guevara, M., Abarca, D., Johnsen, O., Maury, S., Strauss, S.H., Campbell, M.M., Rohde, A., Díaz-Sala, C., and Cervera, M.T., Epigenetic regulation of adaptive responses of forest tree species to the environment, Ecol. Evol., 2013, vol. 3, no. 2, pp. 399–415. https://doi.org/10.1002/ece3.461
Bucherna, N., Szabó, E., Heszky, L.E., and Nagy, I., DNA methylation and gene expression differences during alternative in vitro morphogenetic processes in eggplant (Solanum melongena L.), In Vitro Cell. Dev. Biol.-Plant, 2001, vol. 37, no. 5, pp. 672–677.
Carr, I.M., Valleley, E.M., Cordery, S.F., Markham, A.F., and Bonthron, D.T., Sequence analysis and editing for bisulphite genomic sequencing projects, Nucleic Acids Res., 2007, vol. 35, no. 10, art. ID e79. https://doi.org/10.1093/nar/gkm330
Chan, S.W., Henderson, I.R., and Jacobsen, S.E., Gardening the genome: DNA methylation in Arabidopsis thaliana, Nat. Rev. Genet., 2005, vol. 6, no. 5, pp. 351–360. https://doi.org/10.1038/nrg1601
Colaneri, A.C. and Jones, A.M., Genome-wide quantitative identification of DNA differentially methylated sites in Arabidopsis seedlings growing at different water potential, PLoS One, 2013, vol. 8, no. 4, art. ID e59878. https://doi.org/10.1371/journal.pone.0059878
Conrath, U., Molecular aspects of defence priming, Trends Plant Sci., 2011, vol. 16, no. 10, pp. 524–531. https://doi.org/10.1016/j.tplants.2011.06.004
Dai, Y., Ni, Z., Dai, J., Zhao, T., and Sun, Q., Isolation and expression analysis of genes encoding DNA methyltransferase in wheat (Triticum aestivum L.), Biochim. Biophys. Acta, 2005, vol. 1729, no. 2, pp. 118–125. https://doi.org/10.1016/j.bbaexp.2005.04.001
Demeulemeester, M., Van Stallen, N., and De Proft, M., Degree of DNA methylation in chicory (Cichorium intybus L.): influence of plant age and vernalization, Plant Sci., 1999, vol. 142, no. 1, pp. 101–108.
Deng, S., Dai, H., Arenas, C., Wang, H., Niu, Q.W., and Chua, N.H., Transcriptional silencing of Arabidopsis endogenes by single-stranded RNAs targeting the promoter region, Plant Cell Physiol., 2014, vol. 55, no. 4, pp. 823–833.
Elseehy, M.M. and El-Shehawi, A.M., Expression profile of wheat DNA methyltransferases genes in egyptian wheat (Triticum Aestivum) varieties under peg induced dehydration, Alexandria Sci. Exch. J., 2018, vol. 39, pp. 695–701.
Elseehy, M.M. and El-Shehawi, A.M., Methylation of exogenous promoters regulates soybean isoflavone synthase (GmIFS) transgene in T0 transgenic wheat (Triticum aestivum), Cytol. Genet., 2020, vol. 54, no. 3, pp. 271–282. https://doi.org/10.3103/S0095452720030032
Elseehy, M.M., Differential transgeneration methylation of exogenous promoters in T1 transgenic wheat (Triticum aestivum), Cytol. Genet., 2020, vol. 54, no. 5, pp. 493–504.
El-Shehawi, A., Heat Shock Proteins expression is regulated by promoter CpG methylation/demethylation under heat stress in wheat varieties, Pak. J. Biol. Sci., vol. 23, no. 10, pp. 1310–1320.
El-Shehawi, A.M., Fahmi, A.I., Elseehy, M.M., and Nagaty, H.A., Enhancement of nutritional quality of wheat (Triticum aestivum) by metabolic engineering of isoflavone pathway, Am. J. Biochem. Biotechnol., 2013, vol. 9, no. 4, pp. 404–414.
Fedoroff, N.V., Transposable elements, epigenetics, and genome evolution, Science, 2012, vol. 338, no. 6108, pp. 758–767.
Guérin, C., Roche, J., Allard, V., Ravel, C., Mouzeyar, S., and Bouzidi, M.F., Genome-wide analysis, expansion and expression of the NAC family under drought and heat stresses in bread wheat (T. aestivum L.), PLoS One, 2019, vol. 14, no. 3, art. ID e0213390. https://doi.org/10.1371/journal.pone.0213390
Guo, W., Zhang, J., Zhang, N., Xin, M., Peng, H., Hu, Z., Ni, Z., and Du, J., The wheat NAC transcription factor TaNAC2L is regulated at the transcriptional and post-translational levels and promotes heat stress tolerance in transgenic Arabidopsis, PLoS One, 2015, vol. 10, no. 8, art. ID e0135667. https://doi.org/10.1371/journal.pone.0135667
Han, S.K. and Wagner, D., Role of chromatin in water stress responses in plants, J. Exp. Bot., 2014, vol. 65, no. 10, pp. 2785–2799. https://doi.org/10.1093/jxb/ert403
Hauben, M., Haesendonckx, B., Standaert, E., Van Der Kelen, K., Azmi, A., Akpo, H., Van Breusegem, F., Guisez, Y., Bots, M., Lambert, B., Laga, B., and De Block, M., Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield, Proc. Natl. Acad. Sci. U. S. A., 2009, vol. 106, no. 47, pp. 20109–20114. https://doi.org/10.1073/pnas.0908755106
Heilersig, B.H., Loonen, A.E., Janssen, E.M., Wolters, A.M., and Visser, R.G., Efficiency of transcriptional gene silencing of GBSSI in potato depends on the promoter region that is used in an inverted repeat, Mol. Genet. Genomics, 2006, vol. 275, no. 5, pp. 437–449. https://doi.org/10.1007/s00438-006-0101-4
Ikeuchi, M., Iwase, A., and Sugimoto, K., Control of plant cell differentiation by histone modification and DNA methylation, Curr. Opin. Plant Biol., 2015, vol. 28, pp. 60–67. https://doi.org/10.1016/j.pbi.2015.09.004
Jones, L., Ratcliff, F., and Baulcombe, D.C., RNA-directed transcriptional gene silencing in plants can be inherited independently of the RNA trigger and requires Met1 for maintenance, Curr. Biol., 2001, vol. 11, no. 10, pp. 747–757. https://doi.org/10.1016/s0960-9822(01)00226-3
Kathiria, P., Sidler, C., Golubov, A., Kalischuk, M., Kawchuk, L.M., and Kovalchuk, I., Tobacco mosaic virus infection results in an increase in recombination frequency and resistance to viral, bacterial, and fungal pathogens in the progeny of infected tobacco plants, Plant Physiol., 2010, vol. 153, no. 4, pp. 1859–1870. https://doi.org/10.1104/pp.110.157263
Law, J.A. and Jacobsen, S.E., Establishing, maintaining and modifying DNA methylation patterns in plants and animals, Nat. Rev. Genet., 2010, vol. 11, no. 3, pp. 204–220. https://doi.org/10.1038/nrg2719
Li, Y. and Tollefsbol, T.O., DNA methylation detection: bisulfite genomic sequencing analysis, Methods Mol. Biol., 2011, vol. 791, pp. 11–21. https://doi.org/10.1007/978-1-61779-316-5_2
Lira-Medeiros, C.F., Parisod, C., Fernandes, R.A., Mata, C.S., Cardoso, M.A., and Ferreira, P.C., Epigenetic variation in mangrove plants occurring in contrasting natural environment, PLoS One, 2010, vol. 5, no. 4, art. ID e10326. https://doi.org/10.1371/journal.pone.0010326
Livak, K.J. and Schmittgen, T.D., Analysis of relative gene expression data using real-time quantitative pcr and the 2−ΔΔCT method, Methods, 2001, vol. 25, no. 4, pp. 402–408. https://doi.org/10.1006/meth.2001.1262
Lu, M., Sun, Q.P., Zhang, D.F., Wang, T.Y., and Pan, J.B., Identification of 7 stress-related NAC transcription factor members in maize (Zea mays L.) and characterization of the expression pattern of these genes, Biochem. Biophys. Res. Commun., 2015, vol. 462, no. 2, pp. 144–150. https://doi.org/10.1016/j.bbrc.2015.04.113
Matzke, M.A., Primig, M., Trnovsky, J., and Matzke, A.J., Reversible methylation and inactivation of marker genes in sequentially transformed tobacco plants, Embo J., 1989, vol. 8, no. 3, pp. 643–649.
Moyano, E., Martínez-Rivas, F.J., Blanco-Portales, R., Molina-Hidalgo, F.J., Ric-Varas, P., Matas-Arroyo, A.J., Caballero, J.L., Muñoz-Blanco, J., and Rodríguez-Franco, A., Genome-wide analysis of the NAC transcription factor family and their expression during the development and ripening of the Fragaria × ananassa fruits, PLoS One, 2018, vol. 13, no. 5, art. ID e0196953. https://doi.org/10.1371/journal.pone.0196953
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K., NAC transcription factors in plant abiotic stress responses, Biochim. Biophys. Acta, Gene Regul. Mech., 2012, vol. 1819, no. 2, pp. 97–103. https://doi.org/10.1016/j.bbagrm.2011.10.005
Nuruzzaman, M., Manimekalai, R., Sharoni, A.M., Satoh, K., Kondoh, H., Ooka, H., and Kikuchi, S., Genome-wide analysis of NAC transcription factor family in rice, Gene, 2010, vol. 465, nos. 1–2, pp. 30–44. https://doi.org/10.1016/j.gene.2010.06.008
Pinheiro, G.L., Marques, C.S., Costa, M.D., Reis, P.A., Alves, M.S., Carvalho, C.M., Fietto, L.G., and Fontes, E.P., Complete inventory of soybean NAC transcription factors: sequence conservation and expression analysis uncover their distinct roles in stress response, Gene, 2009, vol. 444, nos. 1–2, pp. 10–23. https://doi.org/10.1016/j.gene.2009.05.012
Qin, D., Wu, H., Peng, H., Yao, Y., Ni, Z., Li, Z., Zhou, C., and Sun, Q., Heat stress-responsive transcriptome analysis in heat susceptible and tolerant wheat (Triticum aestivum L.) by using Wheat Genome Array, BMC Genomics, 2008, vol. 9, art. ID 432. https://doi.org/10.1186/1471-2164-9-432
Rodríguez López, M. and Wilkinson, M.J., Epi-fingerprinting and epi-interventions for improved crop production and food quality, Front. Plant Sci., 2015, vol. 6, art. ID 397. https://doi.org/10.3389/fpls.2015.00397
Saidi, M.N., Mergby, D., and Brini, F., Identification and expression analysis of the NAC transcription factor family in durum wheat (Triticum turgidum L. ssp. durum), Plant Physiol. Biochem., 2017, vol. 112, pp. 117–128.https://doi.org/10.1016/j.plaphy.2016.12.028
Saze, H., Tsugane, K., Kanno, T., and Nishimura, T., DNA methylation in plants: relationship to small RNAs and histone modifications, and functions in transposon inactivation, Plant Cell Physiol., 2012, vol. 53, no. 5, pp. 766–784.https://doi.org/10.1093/pcp/pcs008
Song, Q., Zhang, T., Stelly, D.M., and Chen, Z.J., Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons, Genome Biol., 2017, vol. 18, no. 1, art. ID 99. https://doi.org/10.1186/s13059-017-1229-8
Suzuki, M.M. and Bird, A., DNA methylation landscapes: provocative insights from epigenomics, Nat. Rev. Genet., 2008, vol. 9, no. 6, pp. 465–476. https://doi.org/10.1038/nrg2341
Takatsuka, H. and Umeda, M., Epigenetic control of cell division and cell differentiation in the root apex, Front. Plant Sci., 2015, vol. 6, art. ID 1178. https://doi.org/10.3389/fpls.2015.01178
Takuno, S. and Gaut, B.S., Body-methylated genes in Arabidopsis thaliana are functionally important and evolve slowly, Mol. Biol. Evol., 2012, vol. 29, no. 1, pp. 219–227. https://doi.org/10.1093/molbev/msr188
Tolley, B.J., Woodfield, H., Wanchana, S., Bruskiewich, R., and Hibberd, J.M., Light-regulated and cell-specific methylation of the maize PEPC promoter, J. Exp. Bot., 2012, vol. 63, no. 3, pp. 1381–1390. https://doi.org/10.1093/jxb/err367
Tricker, P.J., López, C.M., Gibbings, G., Hadley, P., and Wilkinson, M.J., Transgenerational, dynamic methylation of stomata genes in response to low relative humidity, Int. J. Mol. Sci., 2013, vol. 14, no. 4, pp. 6674–6689. https://doi.org/10.3390/ijms14046674
Uauy, C., Distelfeld, A., Fahima, T., Blechl, A., and Dubcovsky, J., A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat, Science, 2006, vol. 314, no. 5803, pp. 1298–1301. https://doi.org/10.1126/science.1133649
Wang, W.S., Pan, Y.J., Zhao, X.Q., Dwivedi, D., Zhu, L.H., Ali, J., Fu, B.Y., and Li, Z.K., Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.), J. Exp. Bot., 2011, vol. 62, no. 6, pp. 1951–1960. https://doi.org/10.1093/jxb/erq391
Wei, X., Song, X., Wei, L., Tang, S., Sun, J., Hu, P., and Cao, X., An epiallele of rice AK1 affects photosynthetic capacity, J. Integr. Plant Biol., 2017, vol. 59, no. 3, pp. 158–163. https://doi.org/10.1111/jipb.12518
Yaish, M.W., Al-Lawati, A., Al-Harrasi, I., and Patankar, H.V., Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula), BMC Genomics, 2018, vol. 19, no. 1, art. ID 78. https://doi.org/10.1186/s12864-018-4484-5
Yaish, M.W., Editorial: epigenetic modifications associated with abiotic and biotic stresses in plants: an implication for understanding plant evolution, Front. Plant Sci., 2017, vol. 8, art. ID 1983. https://doi.org/10.3389/fpls.2017.01983
Yong-Villalobos, L., González-Morales, S.I., Wrobel, K., Gutiérrez-Alanis, D., Cervantes-Perez, S.A., Hayano-Kanashiro, C., Oropeza-Aburto, A., Cruz-Ramirez, A., Martínez, O., and Herrera-Estrella, L., Methylome analysis reveals an important role for epigenetic changes in the regulation of the Arabidopsis response to phosphate starvation, Proc. Natl. Acad. Sci. U. S. A., 2015, vol. 112, no. 52, pp. E7293–7302. https://doi.org/10.1073/pnas.1522301112
Zilberman, D., An evolutionary case for functional gene body methylation in plants and animals, Genome Biol., 2017, vol. 18, no. 1, art. ID 87. https://doi.org/10.1186/s13059-017-1230-2
Funding
The current work was funded by Taif University Researcher Supporting Project number (TURSP–2020/75), Taif University, Taif, Saudi Arabia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
About this article

Cite this article
El-Shehawi, A.M., Elseehy, M.A. & Elseehy, M.M. CpG Methylation of the Proximal Promoter Region Regulates the Expression of NAC6D Gene in Response to High Temperature in Wheat (Triticum aestivum). Cytol. Genet. 56, 449–457 (2022). https://doi.org/10.3103/S009545272205005X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S009545272205005X