
Abstract
This study aimed at describing a simple and facile synthesis of pyrazolyl/isoxazolyl 1,3,4-oxadiazole derivatives from the synthetic intermediates pyrazolyl/isoxazolyl carboxylates adopting conventional and ultrasound irradiation methods. In fact ultrasound-promoted synthesis led to the formation of title compounds in higher yields and in shorter reaction times when compared with conventional methods. All the compounds were characterized by IR, 1H NMR, 13C NMR, and mass spectra. The synthesized compounds were evaluated for their antioxidant and anti-inflammatory activities. The bioassay results indicated that some title compounds were found to be more potent than standard drugs. Amongst all the tested compounds furanyl/pyridinyl linked pyrazolyl/isoxazolyl methoxyphenylsulfonylmethyloxadiazoles were identified as potential antioxidant and anti-inflammatory agents.
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The five-membered heterocyclic compounds with one or two nitrogen atoms particularly 1,3,4-oxadiazoles, pyrazoles, and isoxazoles represent an indispensable class of heteroaromatic compounds, as they are part of many biologically active pharmaceuticals vital for increasing the quality of life (Fig. 1).

Drugs containing pyrazole, isoxazole, and oxadiazole motifs
Literature searches reveal that there are several methods for synthesis of 2,5-disubstituted 1,3,4-oxadiazoles either thermal/acid catalyzed cyclization of 1,2-diacylhydrazines [1] or by oxidative cyclization of semicarbazone/hydrazone in the presence of an oxidant [2] or by microwave irradiation of hydrazide and carboxylic acid mixture [3] or cyclization of acylthiosemicarbazides. 1,3,4-Oxadiazoles and its derivatives have been attracting great attention due to their wide range of biological properties including antioxidant [4], analgesic [5], anti-inflammatory [6], anticancer [7], antidiabetic [8], anticonvulsant [9], anti-HIV [10], antihypertensive [11], antiobesity [12], antibacterial [13], antifungal [14], antitubercular [15], and cytotoxic [16] activities. Besides, many commercial drugs such as zibotentan [17], ataluren [18], raltegravir [19], tiodazosin [20], nesapidil [21], and furamizole [22] possess oxadiazole scaffold as major structural unit. They have also attracted interest in medicinal chemistry as surrogates for carboxylic acids, esters, and carboxamides [23]. On the other hand, a number of bioactive natural products and therapeutics are based on pyrazole and isoxazole motifs viz., celecoxib, tepoxalin, rimonabant, valdecoxib, leflunomide, and cloxacillin. In addition, pyrazoles and isoxazoles exhibit antibacterial [24], antioxidant [25], antidepressant [26], anticancer [27], antifungal [28], and anti-inflammatory [29] activities.
With this background heteroaromatic compounds have attracted researchers to synthesize these compounds by employing new innovative methods and techniques and to study their biological properties. Ultrasonic assisted organic synthesis, a green synthetic approach is a powerful technique to accelerate organic reactions [30, 31]. This can be considered as a processing aid in terms of energy conservation, waste minimization, more convenient and easily controlled compared to conventional heating. A large number of organic reactions can be carried out in high yields, shorter reaction time, and milder conditions under ultrasound irradiation. The characteristic feature of ultrasonicator is that the mechanical energy in the form of high-intensity, high-frequency sound waves is transferred to the reaction mixture. This results in the generation and the collapse of large number of minute bubbles throughout the mixture. This effect, which is known as cavitation, is responsible for the quick and thorough stirring of reaction mixture [32,33,34,35]. It can enhance chemical reaction, mass transfer. and offer potential for shorter reaction cycles which are inaccessible by conventional methods. Inspired by the remarkable biopotency of above mentioned heterocycles and in continuation of our interest in the synthesis of a variety of biologically active heterocycles [36,37,38], it is proposed to synthesize the molecules having two different pharmacophore units and to study their biological activity (Fig. 2).

Design strategy of the target compounds
Results and discussion
Chemistry
The synthetic route to prepare a new class of pyrazolyl/isoxazolyl 1,3,4-oxadiazoles was synthesized from the synthetic intermediates pyrazolyl/isoxazolyl carboxylates by exploiting ester functionality adopting conventional and ultrasonication methodologies as illustrated in Schemes 1, 2 and Table 1. The reaction of ethyl 5-(furan-2-yl)-1H-pyrazole-3-carboxylate (1), ethyl 5-(5-bromothiophen-2-yl)-1H-pyrazole-3-carboxylate (2), and ethyl 5-(pyridin-4-yl)-1H-pyrazole-3-carboxylate (3) with hydrazine hydrate in methanol afforded 5-(furan-2-yl)-1H-pyrazole-3-carbohydrazide (4), 5-(5-bromo-thiophen-2-yl)-1H-pyrazole-3-carbohydrazide (5), 5-(pyridin-4-yl)-1H-pyrazole-3-carbohydrazide (6), respectively. In the 1H NMR spectra of 4, 5, and 6 the absence of triplet and quartet due to ethoxy moiety and presence of two broad singlets at δ = 9.69, 9.79, 9.71 ppm (NH) and at 4.42, 4.48, 4.37 ppm (NH2) indicated the formation of acid hydrazide. The cyclocondensation of acid hydrazide 4, 5, and 6 with arylsulfonylacetic acid in the presence of phosphorus oxychloride produced 2-[5-(furan-2-yl)-1H-pyrazol-3-yl]-5-arylsulfonylmethyl-1,3,4-oxadiazole (7), 2-[5-(5-bromothiophen-2-yl)-1H-pyrazol-3-yl]-5-arylsulfonyl-methyl-1,3,4-oxadiazole (8), and 2-[5-(pyridin-4-yl)-1H-pyrazol-3-yl]-5-arylsulfonyl-methyl-1,3,4-oxadiazole (9), respectively. The 1H NMR spectra of 7a, 8a, and 9a displayed a sharp singlet at 4.15, 4.23, 4.12 ppm (CH2) and a broad singlet at 11.74, 11.93, 11.82 ppm (NH), respectively. Besides, a singlet due to C4–H of pyrazole ring appeared at downfield region and merged with aromatic protons. The signals due to NH disappeared on deuteration.


On the other hand, the treatment of compounds 4, 5, and 6 with ethyl 2-chloro-2-oxoacetate in tetrahydrofuran led to the formation of ethyl 2-[2-[5-(furan-2-yl)-1H-pyrazole-3-carbonyl]hydrazinyl]-2-oxoacetate (10), ethyl 2-[2-[5-(5-bromothiophen-2-yl)-1H-pyrazole-3-carbonyl]hydrazinyl]-2-oxoacetate (11), and ethyl 2-[2-[5-(pyridin-4-yl)-1H-pyrazole-3-carbonyl]hydrazinyl]-2-oxoacetate (12), respectively. The 1H NMR spectra of 10, 11, and 12 exhibited a triplet at 1.21, 1.24, 1.26 ppm and a quartet at 4.22, 4.26, 4.18 ppm due to methyl and methylene protons of ester group and a singlet due to C4–H of pyrazole appeared at much downfield region and merged with aromatic protons. Moreover, three broad singlets observed at 11.86, 11.82, 11.94; 9.80, 9.91, 9.78, and 9.98, 10.08, 9.84 ppm accounted for NH of pyrazole, CONH, and NHCOCOOEt, respectively. The signals of highly acidic protons disappeared when D2O was added. Furthermore, the cyclocondensation of compounds 10, 11, and 12 with POCl3 resulted in ethyl 5-[5-(furan-2-yl)-1H-pyrazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (13), ethyl 5-[5-(5-bromothiophen-2-yl)-1H-pyrazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (14), and ethyl 5-[5-(pyridin-4-yl)-1H-pyrazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (15) (Scheme 3). The 1H NMR spectra of 13, 14, and 15 showed a triplet at 1.34, 1.28, 1.33 ppm (CH3) and a quartet at 4.27, 4.31, 4.34 ppm (OCH2) in addition to the signals of aromatic protons. A broad singlet was also observed at 11.98, 11.80, 11.91 ppm (NH), which disappeared on deuteration.

Adopting similar methodology the condensation of ethyl 5-(furan-2-yl)isoxazole-3-carboxylate (16), ethyl 5-(5-bromothiophen-2-yl)isoxazole-3-carboxylate (17), and ethyl 5-(pyridin-4-yl)isoxazole-3-carboxylate (18) with hydrazine hydrate in methanol provided 5-(furan-2-yl)isoxazole-3-carbohydrazide (19), 5-(5-bromothiophen-2-yl)isoxazole-3-carbohydrazide (20), and 5-(pyridin-4-yl)isoxazole-3-carbohydrazide (21), respectively. The absence of signals due to ester group in 1H NMR spectra of 19, 20, and 21 confirmed the formation of acid hydazide. Two broad singlets were also observed at 9.98, 10.02, 9.91 ppm, and 4.54, 4.62, 4.58 ppm due to NH and NH2, respectively, which disappeared on deuteration.
Moreover, 2-[5-(furan-2-yl)-isoxazol-3-yl]-5-arylsulfonylmethyl-1,3,4-oxadiazole (22), 2-[5-(5-bromothiophen-2-yl)-isoxazol-3-yl]-5-arylsulfonylmethyl-1,3,4-oxadiazole (23), and 2-[5-(pyridin-4-yl)-isoxazol-3-yl]-5-arylsulfonylmethyl-1,3,4-oxadiazole (24) were obtained by cyclocondensation of acid hydrazide 19, 20, and 21 with arylsulfonyl acetic acid in the presence of POCl3. The 1H NMR spectra of 22a, 23a, 24a displayed a singlet at 4.18, 4.21, and 4.22 ppm (CH2), respectively. In addition, a singlet due to C4–H of isoxazole appeared at downfield region and merged with aromatic protons. However, the resultant acid hydrazide 19, 20, and 21 was treated with ethyl 2-chloro-2-oxoacetate followed by THF gave ethyl 2-[2-[5-(furan-2-yl)isoxazole-3-carbonyl]hydrazinyl]-2-oxoacetate (25), ethyl 2-[2-[5-(5-bromothiophen-2-yl)isoxazole-3-carbonyl]hydrazinyl]-2-oxoacetate (26), and ethyl 2-[2-[5-(pyridin-4-yl)isoxazole-3-carbonyl]hydrazinyl]-2-oxoacetate (27). The 1H NMR spectra of 25, 26, and 27 presented a triplet at 1.24, 1.26, 1.20 ppm (CH3), a quartet at 4.19, 4.24, 4.17 ppm (OCH2), and two broad singlets at 9.96, 9.94, 9.89 ppm (CONH) and 10.29, 10.05, 10.07 ppm (NHCOCOOEt). The signals due to NH disappeared on deuteration. The cyclized products ethyl 5-[5-(furan-2-yl)isoxazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (28), ethyl 5-[5-(5-bromothiophen-2-yl)isoxazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (29), and ethyl 5-[5-(pyridin-4-yl)isoxazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (30) were synthesized by the cyclocondensation of diacyl hydrazide 25, 26, and 27 in the presence of POCl3. The 1H NMR spectra of 28, 29, and 30 showed a triplet and a quartet at 1.39, 1.32, 1.35 ppm and 4.35, 4.25, 4.39 ppm due to CH3 and OCH2 groups apart from signals due to C4–H of isoxazole and other aromatic protons. It was observed that target compounds were obtained in shorter reaction times and in higher yields under ultrasonication than in conventional method. The structures of all the target compounds were further ascertained by IR, 13C NMR, mass, and microanalyses.
Antioxidant activity
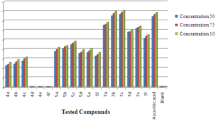
The observed data on the antioxidant activity of the test compounds and the standard drug are shown in Tables 2, 3 and 4 and Fig. 3. The results revealed that compounds 7b, 9b, 22b, and 24b showed greater radical scavenging activity in all the three methods when compared with the standard drug ascorbic acid. The compounds 7a, 8a, 8b, 9a, 22a, 23a, 23b, 24a exhibited good activity, whereas 10, 11, 12, 25, 26, 27, 28, 30 displayed moderate activity. On the other hand, the compounds 7c, 13, 14, 15, 22c, 29 exhibited low activity. However, the other compounds showed no activity. The structure–activity relationship indicated that isoxazole derivatives exhibited higher radical scavenging activity than pyrazole derivatives. Moreover, it was observed that electron donating substituents on the phenyl ring increases the activity. This may be due to mesomeric effect of methoxy group and inductive effect of methyl substituent. It was also noticed that with increasing electron donating effect the antioxidant activity increases. In fact, methoxy-substituted compounds displayed higher activity than methyl-substituted ones. Besides the compounds having chloro and bromo substituents exhibited least activity which indicated that with increasing electron withdrawing effect the activity decreases. On the other hand, the compounds 10, 11, 12, 25, 26, and 27 exhibited higher activity than compounds 13, 14, 15, 28, 29, and 30. This may be due to the increasing number of chromophoric groups. In general, pyrazole in combination with furan/pyridine and isoxazole in combination with furan/pyridine displayed excellent activity than the thiophene-linked compounds. The free radical scavenging activity of the compounds 7b, 9b, 22b, and 24b was measured at different concentrations and monitored the change in absorbance at 10, 20, and 30 min in DPPH method (Table 5). It was perceived that at these 10-min intervals the values are very close and the results exemplified that the antioxidant activity is independent of time. The IC50 value of the standard drug ascorbic acid in DPPH method was found to be 17.03 μg/cm,3 whereas IC50 values of compounds 7b, 9b, 22b, and 24b were found to be 16.69, 16.88, 16.16, and 16.41 μg/cm3, respectively. It was also observed that radical scavenging activity increases with increase in concentration in all the three methods.

The in vitro antioxidant activity of 7b, 9b, 22b, and 24b in all the three methods
Statistical analysis
All experiments are performed in triplicate (n = 3), and an ANOVA test (Microsoft Excel) is used to compare the mean values across compounds and concentrations. The results represented mean ± standard deviation (SD) of three replicated determinations. The descriptive analysis is supported by the statistical analysis. The following inferences are supported through ANOVA analysis. Since the p value of rows (compounds) is less than 0.05 (α) cannot reject the null hypothesis, and so we conclude (with 95% confidence) that there is significant difference in radical scavenging activity exhibited by the compounds for different concentrations. Since the p value of columns (concentrations) is less than 0.05 (α) cannot reject the null hypothesis, and so we conclude (with 95% confidence) that there is significant difference in radical scavenging activity displayed across the compounds. Besides, the p value (interactions) is less than 0.05, indicating that there are significant differences in the interaction between compounds and concentrations.
Anti-inflammatory activity
The results (Table 6) indicated that the compounds 7b, 9b, 22b, and 24b showed significant anti-inflammatory activity. However, 22b displayed anti-inflammatory activity equal to the standard drug phenylbutazone. The structure–activity relationship of the synthesized compounds revealed that isoxazolyl oxadiazoles exhibited greater activity than pyrazolyl oxadiazoles. In general, it was observed that electron donating methyl and methoxy substituents on the phenyl ring enhance the anti-inflammatory activity.
Conclusion
In summary, a series of new heteroaryl pyrazolyl/isoxazolyl 1,3,4-oxadiazole derivatives were designed and synthesized in good yields and in shorter reaction times adopting a green approach—ultrasound irradiation. The entire series of the compounds were characterized by IR, NMR, and mass spectral parameters. The title compounds were evaluated for their antioxidant and anti-inflammatory activities. The activity results showed that the methoxy substituted 1,3,4-oxadiazoles is a promising template for antioxidant and anti-inflammatory activities. It was also observed that the furanyl/pyridinyl pyrazolyl/isoxazolyl 1,3,4-oxadiazole displayed promising antioxidant and anti-inflammatory activities than the thiophenyl/pyrazolyl/isoxazolyl 1,3,4-oxadiazoles. Moreover, compounds having electron donating groups on the phenyl ring enhances the activity, when compared with the electron withdrawing groups. As a result of this study, compounds 7b, 9b, 22b, and 24b were identified as lead compounds for both activities and may be used for further studies.
Experimental
All the chemicals were purchased from commercial sources and used without further purification. Ultrasonication was performed in a Bandelin Sonorex RK 102H ultrasonic bath operating at frequency of 35 kHz. Melting points were determined in open capillaries on a Mel-Temp apparatus. The IR spectra were recorded on a Thermo Nicolet IR 200 FT-IR spectrometer as KBr pellets and the wave numbers were given in cm−1. The 1H NMR spectra were recorded in DMSO-d6 on a Bruker-400 spectrometer (400 MHz). The 13C NMR spectra were recorded in DMSO-d6 on a Bruker spectrometer operating at 100 MHz. All chemical shifts are reported in δ (ppm) using TMS as an internal standard. The high-resolution mass spectra were recorded on micromass Q-TOF micromass spectrometer using electrospray ionization. The microanalyses were performed on a Perkin–Elmer 240C elemental analyzer. The progress of the reaction was monitored by TLC using silica gel plates (silica gel 60 F254 0.25 mm) and components were visualized by observation under UV light (254 and 365 nm). The antioxidant activity was carried out using Shimadzu UV-2450 spectrophotometer. The starting compounds ethyl 5-(heteroaryl)-1H-pyrazole-3-carboxylates 1–3, 5-(heteroaryl)-1H-pyrazole-3-carbohydrazides 4–6, ethyl 5-(heteroaryl)isoxazole-3-carboxylates 16–18, and 5-(heteroaryl)-1H-isoxazole-3-carbohydrazides 19–21 were prepared as per the literature precedents [39].
General procedures for the synthesis of 5-(furan-2-yl)-1H-pyrazole-3-carbohydrazide (4), 5-(5-bromothiophen-2-yl)-1H-pyrazole-3-carbohydrazide (5), and 5-(pyridin-4-yl)-1H-pyrazole-3-carbohydrazide (6)
Conventional method
A solution of ethyl 5-(furan-2-yl)-1H-pyrazole-3-carboxylate (1), ethyl 5-(5-bromothiophen-2-yl)-1H-pyrazole-3-carboxylate (2), or ethyl 5-(pyridin-4-yl)-1H-pyrazole-3-carboxylate (3) (1.0 mmol) and hydrazine hydrate (2.0 mmol) in 3 cm3 methanol were taken in a 100-cm3 round bottomed flask fitted with reflux condenser and refluxed on a water bath for 4–6 h. It was cooled and the solid separated was filtered, dried, and recrystallized from methanol.
Ultrasonication method
Compound 1, 2, or 3 (1.0 mmol), hydrazine hydrate (2.0 mmol), and 4 cm3 methanol were subjected to ultrasound irradiation for 42–55 min. The progress of the reaction was monitored by TLC. After completion of the reaction, the contents were cooled and poured onto crushed ice. The solid separated was filtered on a Buchner funnel, washed with cold water, dried, and recrystallized from methanol to get 4, 5, 6.
General procedures for the synthesis of compounds 7a–7c, 8a–8c, and 9a–9c
Conventional method
A solution of 4, 5, or 6 (1.0 mmol), arylsulfonylacetic acid (1.0 mmol), and 3 cm3 POCl3 was taken in a 100-cm3 round-bottom flask fitted with reflux condenser carrying a calcium chloride guard-tube and refluxed for 6–9 h. The excess POCl3 was removed under reduced pressure and the residue was poured onto crushed ice. The resulting precipitate was filtered, washed with saturated sodium bicarbonate solution, and then with water, dried, and purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate-hexane (1:3) as eluent.
Ultrasonication method
A mixture of 4, 5, or 6 (1.0 mmol), arylsulfonylacetic acid (1.0 mmol), and 3 cm3 phosphorus oxychloride was subjected to ultrasound irradiation at a frequency of 35 kHz for 30–50 min at 45 °C. After completion of the reaction, the excess phosphorus oxychloride was removed under reduced pressure and the residue was poured onto crushed ice. The separated solid was collected by filtration. It was washed with saturated sodium bicarbonate solution followed by water, dried, and purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate-hexane (1:3) as eluent.
2-[5-(Furan-2-yl)-1H-pyrazol-3-yl]-5-(tosylmethyl)-1,3,4-oxadiazole (7a, C17H14N4O4S)
White solid (0.33 g, 91%); m.p.: 176–178 °C; 1H NMR (DMSO-d6): δ = 2.55 (s, 3H, Ar–CH3), 4.15 (s, 2H, CH2), 6.69–8.07 (m, 8H, Ar–H), 11.74 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 21.4 (Ar–CH3), 59.1 (CH2), 104.2, 107.8, 114.3, 126.8, 131.2, 132.3, 134.3, 138.4, 141.5, 145.7, 156.8, 160.6, 162.5 ppm; IR (KBr): \(\bar{\nu }\) = 1145, 1334 (SO2), 1565 (C=N), 1600 (C=C), 3242 (NH) cm−1; HRMS: m/z = 393.3736 ([M+Na]+).
2-[5-(Furan-2-yl)-1H-pyrazol-3-yl]-5-[(4-methoxyphenyl)sulfonylmethyl]-1,3,4-oxadiazole (7b, C17H14N4O5S)
White solid (0.36 g, 94%); m.p.: 187–189 °C; 1H NMR (DMSO-d6): δ = 3.95 (s, 3H, Ar–OCH3), 4.21 (s, 2H, CH2), 6.71–7.86 (m, 8H, Ar–H), 11.79 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 55.2 (Ar–OCH3), 59.6 (CH2), 104.9, 108.0, 114.7, 118.5, 130.1, 132.9, 133.6, 141.1, 145.2, 156.2, 156.4, 162.9, 164.2 ppm; IR (KBr): \(\bar{\nu }\) = 1128, 1329 (SO2), 1556 (C=N), 1610 (C=C), 3256 (NH) cm−1; HRMS: m/z = 409.3710 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)-1H-pyrazol-3-yl]-5-[(4-chlorophenyl)sulfonylmethyl]-1,3,4-oxadiazole (7c, C16H11ClN4O4S)
White solid (0.35 g, 92%); m.p.: 193–195 °C; 1H NMR (DMSO-d6): δ = 4.04 (s, 2H, CH2), 6.91–8.01 (m, 8H, Ar–H), 11.96 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 60.2 (CH2), 105.3, 108.2, 114.1, 128.2, 128.8, 133.1, 133.4, 138.1, 141.7, 146.5, 156.5, 160.2, 162.3 ppm; IR (KBr): \(\bar{\nu }\) = 1122, 1325 (SO2), 1560 (C=N), 1612 (C=C), 3227 (NH) cm−1; HRMS: m/z = 413.7886 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)-1H-pyrazol-3-yl]-5-(tosylmethyl)-1,3,4-oxadiazole (8a, C17H13BrN4O3S2)
White solid (0.41 g, 89%); m.p.: 234–236 °C; 1H NMR (DMSO-d6): δ = 2.51 (s, 3H, Ar–CH3), 4.23 (s, 2H, CH2), 6.52–7.95 (m, 7H, Ar–H), 11.93 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 21.6 (Ar–CH3), 59.5 (CH2), 103.8, 113.5, 126.3, 128.7, 131.8, 132.5, 132.9, 135.7, 138.1, 143.7, 145.9, 160.9, 161.8 ppm; IR (KBr): \(\bar{\nu }\) = 1134, 1337 (SO2), 1587 (C=N), 1619 (C=C), 3263 (NH) cm−1; HRMS: m/z = 488.3289 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)-1H-pyrazol-3-yl]-5-[(4-methoxyphenyl)sulfonylmethyl]-1,3,4-oxadiazole (8b, C17H13BrN4O4S2)
White solid (0.40 g, 85%); m.p.: 261–263 °C; 1H NMR (DMSO-d6): δ = 3.89 (s, 3H, Ar–OCH3), 4.18 (s, 2H, CH2), 6.47–7.93 (m, 7H, Ar–H), 11.87 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 55.8 (Ar–OCH3), 60.9 (CH2), 105.5, 113.8, 118.1, 128.0, 130.4, 132.3, 133.1, 133.8, 143.2, 145.3, 159.7, 161.4, 164.7 ppm; IR (KBr): \(\bar{\nu }\) = 1136, 1331 (SO2), 1584 (C=N), 1626 (C=C), 3210 (NH) cm−1; HRMS: m/z = 504.3296 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)-1H-pyrazol-3-yl]-5-[(4-chlorophenyl)sulfonylmethyl]-1,3,4-oxadiazole (8c, C16H10BrClN4O3S2)
Pale yellow solid (0.43 g, 90%); m.p.: 268–270 °C; 1H NMR (DMSO-d6): δ = 4.09 (s, 2H, CH2), 6.58–8.02 (m, 7H, Ar–H), 11.92 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 60.4 (CH2), 105.8, 113.3, 127.7, 128.4, 129.5, 132.2, 132.8, 133.5, 138.6, 143.5, 145.8, 159.5, 162.6 ppm; IR (KBr): \(\bar{\nu }\) = 1129, 1332 (SO2), 1578 (C=N), 1621 (C=C), 3254 (NH) cm−1; HRMS: m/z = 508.7459 ([M+Na]+).
2-[5-(Pyridin-4-yl)-1H-pyrazol-3-yl]-5-(tosylmethyl)-1,3,4-oxadiazole (9a, C18H15N5O3S)
White solid (0.32 g, 86%); m.p.: 181–183 °C; 1H NMR (DMSO-d6): δ = 2.58 (s, 3H, Ar–CH3), 4.12 (s, 2H, CH2), 6.61–8.20 (m, 9H, Ar–H), 11.82 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 21.1 (Ar–CH3), 60.1 (CH2), 104.3, 125.7, 126.6, 131.5, 132.4, 134.8, 136.1, 138.3, 145.6, 147.5, 160.3, 161.2 ppm; IR (KBr): \(\bar{\nu }\) = 1140, 1320 (SO2), 1572 (C=N), 1617 (C=C), 3236 (NH) cm−1; HRMS: m/z = 404.3991 ([M+Na]+).
2-[5-(Pyridin-4-yl)-1H-pyrazol-3-yl]-5-[(4-methoxyphenyl)sulfonylmethyl]-1,3,4-oxadiazole (9b, C18H15N5O4S)
White solid (0.32 g, 82%); m.p.: 197–199 °C; 1H NMR (DMSO-d6): δ = 3.91 (s, 3H, Ar–OCH3), 4.26 (s, 2H, CH2), 6.49–8.09 (m, 9H, Ar–H), 11.92 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 54.6 (Ar–OCH3), 59.3 (CH2), 104.7, 118.8, 125.9, 126.5, 131.9, 132.8, 136.8, 145.4, 147.3, 160.8, 161.5, 164.5 ppm; IR (KBr): \(\bar{\nu }\) = 1138, 1324 (SO2), 1553 (C=N), 1630 (C=C), 3250 (NH) cm−1; HRMS: m/z = 420.3995 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)-1H-pyrazol-3-yl]-5-[(4-chlorophenyl)sulfonylmethyl]-1,3,4-oxadiazole (9c, C17H12ClN5O3S)
Pale yellow solid (0.34 g, 87%); m.p.: 203–205 °C; 1H NMR (DMSO-d6): δ = 4.17 (s, 2H, CH2), 6.73–8.24 (m, 9H, Ar–H), 11.89 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 59.8 (CH2), 105.4, 125.2, 128.7, 129.5, 133.6, 134.7, 136.4, 138.5, 146.1, 147.8, 159.1, 162.4 ppm; IR (KBr): \(\bar{\nu }\) = 1125, 1326 (SO2), 1564 (C=N), 1629 (C=C), 3223 (NH) cm−1; HRMS: m/z = 424.8139 ([M+Na]+).
General procedures for the synthesis of compounds 10–12
Conventional method
The compound 4, 5, or 6 (1.5 mmol) was dissolved in 20 cm3 dry tetrahydrofuran and ethyl 2-chloro-2-oxoacetate (1.8 mmol) was added dropwise. The mixture was stirred at room temperature for 13–16 h (monitored by TLC). Then, the reaction mixture was poured onto crushed ice and extracted with dichloromethane. The solvent was removed under reduced pressure and the resultant residue was purified by recrystallization from acetone/methanol.
Ultrasonication method
To a solution of 4, 5, or 6 (1.5 mmol) in 20 cm3 THF, ethyl 2-chloro-2-oxoacetate (1.8 mmol) was added and kept under ultrasonication for 28–42 min. The progress of the reaction was monitored by TLC. After completion of the reaction, the contents of the flask were poured onto crushed ice and extracted with dichloromethane. Removal of the solvent under vacuum gave a residue which was purified by recrystallization from acetone/methanol.
Ethyl 2-[2-[5-(furan-2-yl)-1H-pyrazole-3-carbonyl]hydrazinyl]-2-oxoacetate (10, C12H12N4O5)
White solid (0.39 g, 89%); m.p.: 152–154 °C; 1H NMR (DMSO-d6): δ = 1.21 (t, 3H, CH3, J = 7.2 Hz), 4.22 (q, 2H, OCH2, J = 7.2 Hz), 7.04–7.68 (m, 4H, Ar–H), 9.80 (bs, 1H, NH), 9.98 (bs, 1H, NH), 11.86 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 14.3 (CH3), 61.1 (CH2), 106.4, 109.1, 115.3, 136.8, 143.5, 148.2, 155.7, 159.7 (NHCO), 164.3 (CO), 164.9 (CONH) ppm; IR (KBr): \(\bar{\nu }\) = 1583 (C=N), 1618 (C=C), 1665 (CO–NH), 1732 (CO–O), 3244 (NH) cm−1; HRMS: m/z = 315.2415 ([M+Na]+).
Ethyl 2-[2-[5-(5-bromothiophen-2-yl)-1H-pyrazole-3-carbonyl]hydrazinyl]-2-oxoacetate (11, C12H11BrN4O4S)
White solid (0.54 g, 93%); m.p.: 189–191 °C; 1H NMR (DMSO-d6): δ = 1.24 (t, 3H, CH3, J = 7.1 Hz), 4.26 (q, 2H, OCH2, J = 7.1 Hz), 6.94–7.65 (m, 3H, Ar–H), 9.91 (bs, 1H, NH), 10.08 (bs, 1H, NH), 11.82 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 14.5 (CH3), 61.5 (CH2), 106.8, 115.1, 129.4, 131.8, 136.3, 143.9, 148.6, 158.9 (NHCO), 164.1 (CO), 164.5 (CONH) ppm; IR (KBr): \(\bar{\nu }\) = 1580 (C=N), 1610 (C=C), 1649 (CO–NH), 1738 (CO–O), 3271 (NH) cm−1; HRMS: m/z = 410.1989 ([M+Na]+).
Ethyl 2-[2-[5-(pyridin-4-yl)-1H-pyrazole-3-carbonyl]hydrazinyl]-2-oxoacetate (12, C13H13N5O4)
White solid (0.38 g, 84%); m.p.: 158–160 °C; 1H NMR (DMSO-d6): δ = 1.26 (t, 3H, CH3, J = 7.2 Hz), 4.18 (q, 2H, OCH2, J = 7.2 Hz), 7.10–8.16 (m, 5H, Ar–H), 9.78 (bs, 1H, NH), 9.84 (bs, 1H, NH), 11.94 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 13.9 (CH3), 60.9 (CH2), 106.3, 129.5, 135.7, 136.1, 148.3, 148.7, 158.5 (NHCO), 164.5 (CO), 164.9 (CONH) ppm; IR (KBr): \(\bar{\nu }\) = 1585 (C=N), 1615 (C=C), 1658 (CO–NH), 1716 (CO–O), 3210 (NH) cm−1; HRMS: m/z = 326.2670 ([M+Na]+).
General procedures for the synthesis of compounds 13–15
Conventional method
A mixture of 10, 11, or 12 (1.5 mmol) and 5 cm3 POCl3 was taken in a 100-cm3 round-bottomed flask fitted with reflux condenser carrying a calcium chloride guard-tube and refluxed for 22–24 h (monitored by TLC). After completion of the reaction, the contents of the flask were cooled to room temperature and poured onto crushed ice. The separated solid was filtered, washed with saturated sodium bicarbonate solution followed by water, and dried. It was purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate-hexane (1:3) as eluent.
Ultrasonication method
The compound 10, 11, or 12 (1.5 mmol) and 5 cm3 POCl3 were subjected to ultrasound irradiation at a frequency of 35 kHz for 80–95 min at 40 °C. The progress of the reaction was monitored by TLC. After completion of the reaction, the contents of the flask were cooled to room temperature and poured onto crushed ice. The separated solid was filtered, washed with saturated sodium bicarbonate solution followed by water, and dried. It was purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate-hexane (1:3) as eluent.
Ethyl 5-[5-(furan-2-yl)-1H-pyrazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (13, C12H10N4O4)
White solid (0.33 g, 82%); m.p.: 141–142 °C; 1H NMR (DMSO-d6): δ = 1.34 (t, 3H, CH3, J = 6.8 Hz), 4.27 (q, 2H, OCH2, J = 6.8 Hz), 7.11–8.01 (m, 4H, Ar–H), 11.98 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 14.8 (CH3), 61.2 (CH2), 103.3, 108.8, 114.5, 131.7, 140.4, 144.9, 153.2, 159.0, 159.4 (CO), 160.7 ppm; IR (KBr): \(\bar{\nu }\) = 1579 (C=N), 1623 (C=C), 1725 (CO–O), 3238 (NH) cm−1; HRMS: m/z = 297.2269 ([M+Na]+).
Ethyl 5-[5-(5-bromothiophen-2-yl)-1H-pyrazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (14, C12H9BrN4O3S)
White solid (0.49 g, 90%); m.p.: 173–175 °C; 1H NMR (DMSO-d6): δ = 1.28 (q, 3H, CH3, J = 7.0 Hz), 4.31 (q, 2H, OCH2, J = 7.0 Hz), 6.86–8.08 (m, 3H, Ar–H), 11.80 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 14.6 (CH3), 60.9 (CH2), 103.8, 114.2, 128.4, 131.3, 132.9, 142.6, 144.1, 158.7, 159.1 (CO), 160.5 ppm; IR (KBr): \(\bar{\nu }\) = 1566 (C=N), 1614 (C=C), 1720 (CO–O), 3235 (NH) cm−1; HRMS: m/z = 392.1839 ([M+Na]+).
Ethyl 5-[5-(pyridin-4-yl)-1H-pyrazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (15, C13H11N5O3)
White solid (0.40 g, 94%); m.p.: 155–157 °C; 1H NMR (DMSO-d6): δ = 1.33 (t, 3H, CH3, J = 6.9 Hz), 4.34 (q, 2H, OCH2, J = 6.9 Hz), 6.90–8.27 (m, 5H, Ar–H), 11.91 (bs, 1H, pyrazole-NH) ppm; 13C NMR (DMSO-d6): δ = 13.8 (CH3), 61.5 (CH2), 103.5, 123.5, 131.5, 135.9, 144.7, 146.9, 159.1, 159.7 (CO), 160.3 ppm; IR (KBr): \(\bar{\nu }\) = 1561 (C=N), 1628 (C=C), 1718 (CO–O), 3249 (NH) cm−1; HRMS: m/z = 308.2539 [M+Na]+.
General procedures for the synthesis of 5-(furan-2-yl)isoxazole-3-carbohydrazide (19), 5-(5-bromothiophen-2-yl)isoxazole-3-carbohydrazide (20), and 5-(pyridin-4-yl)isoxazole-3-carbohydrazide (21)
Conventional method
The compound ethyl 5-(furan-2-yl)isoxazole-3-carboxylate (16), ethyl 5-(5-bromothiophen-2-yl)isoxazole-3-carboxylate (17), or ethyl 5-(pyridin-4-yl)isoxazole-3-carboxylate (18) (1.0 mmol) and hydrazine hydrate (2.0 mmol) in 3 cm3 methanol was taken in a 100 cm3 round-bottomed flask fitted with reflux condenser and refluxed on a water bath for 3–4 h. The contents were cooled and poured onto crushed ice. The solid separated was filtered, dried, and recrystallized from methanol.
Ultrasonication method
A mixture of 16, 17, or 18 (1.0 mmol), hydrazine hydrate (2.0 mmol), and 4 cm3 methanol was taken and heated at reflux conditions under ultrasound irradiation at a frequency of 35 kHz for 38–50 min. The progress of the reaction was monitored by TLC. After completion of the reaction, the contents were cooled and poured onto crushed ice. The solid separated was filtered on a Buchner funnel, washed with cold water, dried, and recrystallized from methanol.
General procedures for the synthesis of compounds 22a–22c, 23a–23c, and 24a–24c
Conventional method
A mixture of 19, 20, or 21 (1.0 mmol) arylsulfonylacetic acid (1.0 mmol), and 3 cm3 POCl3 was taken in a 100-cm3 round-bottomed flask fitted with reflux condenser carrying a calcium chloride guard-tube and refluxed for 4.5–7 h. The excess phosphorus oxychloride was removed under reduced pressure and the residue was poured onto crushed ice. The resulting precipitate was filtered, neutralized with saturated sodium bicarbonate solution and then with water, dried, and further purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate-hexane (1:3) as eluent.
Ultrasonication method
To an equimolar (1.0 mmol) solution of 19, 20, or 21 and arylsulfonylacetic acid, 4 cm3 POCl3 was added and subjected to ultrasound irradiation for 21–35 min at 45 °C. After completion of the reaction, the excess phosphorus oxychloride was removed under reduced pressure and the residue was poured onto crushed ice. The separated solid was collected by filtration. It was washed with saturated sodium bicarbonate solution followed by water, dried, and purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate-hexane (1:3) as eluent.
2-[5-(Furan-2-yl)isoxazol-3-yl]-5-(tosylmethyl)-1,3,4-oxadiazole (22a, C17H13N3O5S)
White solid (0.30 g, 83%); m.p.: 180–182 °C; 1H NMR (DMSO-d6): δ = 2.59 (s, 3H, Ar–CH3), 4.18 (s, 2H, CH2), 6.92–8.06 (m, 8H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 21.8 (Ar–CH3), 60.3 (CH2), 107.3, 109.6, 115.4, 127.2, 131.7, 134.1, 138.6, 144.0, 149.3, 153.2, 154.6, 160.7, 162.0 ppm; IR (KBr): \(\bar{\nu }\) = 1138, 1336 (SO2), 1576 (C=N), 1617 (C=C) cm−1; HRMS: m/z = 394.3557 ([M+Na]+).
2-[5-(Furan-2-yl)isoxazol-3-yl]-5-[(4-methoxyphenyl)sulfonylmethyl]-1,3,4-oxadiazole (22b, C17H13N3O6S)
White solid (0.36 g, 95%); m.p.: 188–190 °C; 1H NMR (DMSO-d6): δ = 3.98 (s, 3H, Ar–OCH3), 4.26 (s, 2H, CH2), 6.79–7.94 (m, 8H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 56.1 (Ar–OCH3), 61.8 (CH2), 107.7, 109.3, 115.0, 117.8, 128.1, 131.3, 144.9, 149.3, 153.6, 154.2, 160.9, 162.3, 165.1 ppm; IR (KBr): \(\bar{\nu }\) = 1125, 1324 (SO2), 1572 (C=N), 1623 (C=C) cm−1; HRMS: m/z = 410.3563 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)isoxazol-3-yl]-5-[(4-chlorophenyl)sulfonylmethyl]-1,3,4-oxadiazole (22c, C16H10ClN3O5S)
White solid (0.33 g, 86%); m.p.: 192–194 °C; 1H NMR (DMSO-d6): δ = 4.12 (s, 2H, CH2), 6.70–8.13 (m, 8H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 60.5 (CH2), 107.1, 109.1, 115.7, 126.3, 131.2, 134.8, 138.4, 144.3, 149.7, 153.8, 154.1, 160.1, 162.9 ppm; IR (KBr): \(\bar{\nu }\) = 1162, 1320 (SO2), 1567 (C=N), 1614 (C=C) cm−1; HRMS: m/z = 414.7710 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)isoxazol-3-yl]-5-(tosylmethyl)-1,3,4-oxadiazole (23a, C17H12BrN3O4S2)
White solid (0.37 g, 80%); m.p.: 243–245 °C; 1H NMR (DMSO-d6): δ = 2.53 (s, 3H, Ar–CH3), 4.21 (s, 2H, CH2), 6.58–8.08 (m, 7H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 21.3 (Ar–CH3), 60.6 (CH2), 113.2, 126.9, 127.3, 130.8, 131.3, 134.5, 134.9, 138.8, 150.4, 153.1, 154.8, 161.3, 163.1 ppm; IR (KBr): \(\bar{\nu }\) = 1136, 1325 (SO2), 1573 (C=N), 1619 (C=C) cm−1; HRMS: m/z = 489.3146 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)isoxazol-3-yl]-5-[(4-methoxyphenyl)sulfonylmethyl]-1,3,4-oxadiazole (23b, C17H12BrN3O5S2)
White solid (0.45 g, 94%); m.p.: 265–267 °C; 1H NMR (DMSO-d6): δ = 3.92 (s, 3H, Ar-OCH3), 4.15 (s, 2H, CH2), 6.43–8.00 (m, 7H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 55.7 (Ar–OCH3), 59.2 (CH2), 113.6, 118.3, 127.0, 128.6, 130.1, 131.2, 134.4, 149.9, 153.4, 154.3, 160.4, 162.7, 164.8 ppm; IR (KBr): \(\bar{\nu }\) = 1143, 1319 (SO2), 1575 (C=N), 1612 (C=C) cm−1; HRMS: m/z = 505.3137 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)isoxazol-3-yl]-5-[(4-chlorophenyl)sulfonylmethyl]-1,3,4-oxadiazole (23c, C16H9BrClN3O4S2)
White solid (0.43 g, 89%); m.p.: 270–272 °C; 1H NMR (DMSO-d6): δ = 4.17 (s, 2H, CH2), 6.51–7.14 (m, 7H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 60.9 (CH2), 113.1, 126.5, 127.5, 130.7, 131.7, 134.1, 134.4, 138.9, 149.6, 153.7, 154.0, 160.8, 163.4 ppm; IR (KBr): \(\bar{\nu }\) = 1162, 1320 (SO2), 1597 (C=N), 1614 (C=C) cm−1; HRMS: m/z = 509.7294 ([M+Na]+).
2-[5-(Pyridin-4-yl)isoxazol-3-yl]-5-(tosylmethyl)-1,3,4-oxadiazole (24a, C18H14N4O4S)
White solid (0.32 g, 86%); m.p.: 196–198 °C; 1H NMR (DMSO-d6): δ = 2.57 (s, 3H, Ar–CH3), 4.22 (s, 2H, CH2), 6.74–8.16 (m, 9H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 20.7 (Ar–CH3), 59.7 (CH2), 125.1, 126.5, 131.0, 134.7, 136.2, 138.3, 147.9, 149.5, 152.8, 154.9, 161.6, 163.6 ppm; IR (KBr): \(\bar{\nu }\) = 1132, 1328 (SO2), 1580 (C=N), 1622 (C=C) cm−1; HRMS: m/z = 405.3827 ([M+Na]+).
2-[5-(Pyridin-4-yl)isoxazol-3-yl]-5-[(4-methoxyphenyl)sulfonylmethyl]-1,3,4-oxadiazole (24b, C18H14N4O5S)
White solid (0.35 g, 88%); m.p.: 203–205 °C; 1H NMR (DMSO-d6): δ = 3.95 (s, 3H, Ar–OCH3), 4.24 (s, 2H, CH2), 7.56–8.21 (m, 9H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 55.5 (Ar–OCH3), 60.1 (CH2), 118.1, 125.6, 128.4, 131.7, 136.5, 147.2, 150.0, 153.9, 154.7, 159.8, 162.8, 164.3 ppm; IR (KBr): \(\bar{\nu }\) = 1151, 1336 (SO2), 1572 (C=N), 1617 (C=C) cm−1; HRMS: m/z = 421.3816 ([M+Na]+).
2-[5-(5-Bromothiophen-2-yl)isoxazol-3-yl]-5-[(4-chlorophenyl)sulfonylmethyl]-1,3,4-oxadiazole (24c, C17H11ClN4O4S)
White solid (0.37 g, 92%); m.p.: 210–212 °C; 1H NMR (DMSO-d6): δ = 4.20 (s, 2H, CH2), 6.84–8.30 (m, 9H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 59.9 (CH2), 125.3, 126.1, 131.0, 134.5, 136.9, 138.2, 147.7, 149.8, 153.5, 154.4, 160.3, 162.5 ppm; IR (KBr): \(\bar{\nu }\) = 1129, 1334 (SO2), 1570 (C=N), 1630 (C=C) cm−1; HRMS: m/z = 425.7996 ([M+Na]+).
General procedures for the synthesis of compounds 25–27
Conventional method
To a solution of 19, 20, or 21 (1.5 mmol), 20 cm3 dry THF, 1.8 cm3 ethyl 2-chloro-2-oxoacetate were taken in a 100-cm3 round-bottomed flask carrying a calcium chloride guard-tube and the mixture was stirred at laboratory temperature for 10–14 h. After completion of the reaction, the reaction mixture was poured onto crushed ice and extracted with dichloromethane. The solvent was removed under reduced pressure and the resultant residue was purified by recrystallization from acetone/methanol.
Ultrasonication method
The compound 19, 20, or 21 (1.5 mmol) was dissolved in 25 cm3 THF. To this ethyl 2-chloro-2-oxoacetate (4.0 mmol) was added and subjected to ultrasound irradiation for 25–40 min. After completion of the reaction (monitored by TLC), the contents of the flask were poured onto crushed ice and extracted with dichloromethane. The solvent was removed in vacuo. The resultant residue was purified by recrystallization from acetone/methanol.
Ethyl 2-[2-[5-(furan-2-yl)isoxazole-3-carbonyl]hydrazinyl]-2-oxoacetate (25, C12H11N3O6)
White solid (0.36 g, 83%); m.p.: 158–160 °C; 1H NMR (DMSO-d6): δ = 1.24 (t, 3H, CH3, J = 6.4 Hz), 4.20 (q, 2H, OCH2, J = 6.4 Hz), 7.29–7.68 (m, 4H, Ar–H), 9.96 (bs, 1H, NH), 10.29 (bs, 1H, NH) ppm; 13C NMR (DMSO-d6): δ = 14.6 (CH3), 60.7 (CH2), 105.3, 109.6, 116.4, 143.2, 151.7, 155.1, 157.6, 159.9 (NHCO), 164.7 (CO), 165.1 (CONH) ppm; IR (KBr): \(\bar{\nu }\) = 1568 (C=N), 1625 (C=C), 1660 (CO–NH), 1726 (CO–O), 3235 (NH) cm−1; HRMS: m/z = 316.2256 ([M+Na]+).
Ethyl 2-[2-[5-(5-bromothiophen-2-yl)isoxazole-3-carbonyl]hydrazinyl]-2-oxoacetate (26, C12H10BrN3O5S)
White solid (0.52 g, 91%); m.p.: 206–208 °C; 1H NMR (DMSO-d6): δ = 1.26 (t, 3H, CH3, J = 6.4 Hz), 4.24 (q, 2H, OCH2, J = 6.4 Hz), 7.01–7.71 (m, 3H, Ar–H), 9.94 (bs, 1H, NH), 10.05 (bs, 1H, NH) ppm; 13C NMR (DMSO-d6): δ = 14.8 (CH3), 61.3 (CH2), 105.7, 116.0, 129.8, 131.3, 136.8, 151.2, 157.4, 159.5 (NHCO), 164.2 (CO), 164.8 (CONH) ppm; IR (KBr): \(\bar{\nu }\) = 1563 (C=N), 1620 (C=C), 1668 (CO–NH), 1720 (CO–O), 3257 (NH) cm−1; HRMS: m/z = 411.1830 ([M+Na]+).
Ethyl 2-[2-[5-(pyridin-4-yl)isoxazole-3-carbonyl]hydrazinyl]-2-oxoacetate (27, C13H12N4O5)
White solid (0.36 g, 79%); m.p.: 163–165 °C; 1H NMR (DMSO-d6): δ = 1.20 (t, 3H, CH3, J = 6.6 Hz), 4.17 (q, 2H, OCH2, J = 6.6 Hz), 7.15–8.20 (m, 5H, Ar–H), 9.89 (bs, 1H, NH), 10.07 (bs, 1H, NH) ppm; 13C NMR (DMSO-d6): δ = 14.3 (CH3), 61.5 (CH2), 105.1, 120.7, 139.6, 142.1, 151.5, 159.0 (NHCO), 160.4, 164.9 (CO), 165.5 (CONH) ppm; IR (KBr): \(\bar{\nu }\) = 1570 (C=N), 1622 (C=C), 1656 (CO–NH), 1729 (CO–O), 3266 (NH) cm−1; HRMS: m/z = 327.2532 ([M+Na]+).
General procedures for the synthesis of compounds 28–30
Conventional method
The compound 25, 26, or 27 (1.5 mmol) dissolved in 5 cm3 POCl3 was taken in a 100-cm3 round-bottom flask fitted with reflux condenser carrying a calcium chloride guard-tube and refluxed for 20–24 h. The progress of the reaction was monitored by TLC. After completion of the reaction, the contents of the flask were cooled to room temperature and poured onto crushed ice. The resulting precipitate was filtered, washed with saturated sodium bicarbonate solution followed by water, and dried. It was purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate-hexane (1:3) as eluent.
Ultrasonication method
To the compound 25, 26, or 27 (1.6 mmol) 5 cm3 POCl3 was added and kept under ultrasonication for 75–90 min at 40 °C. The progress of the reaction was monitored by TLC. After completion of the reaction, the contents of the flask were cooled to room temperature and poured onto crushed ice. The resulting precipitate was filtered, washed with saturated sodium bicarbonate solution followed by water and dried. It was purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate-hexane (1:3) as eluent.
Ethyl 5-[5-(furan-2-yl)isoxazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (28, C12H9N3O5)
White solid (0.39 g, 90%); m.p.: 144–146 °C; 1H NMR (DMSO-d6): δ = 1.39 (t, 3H, CH3, J = 6.4 Hz), 4.35 (q, 2H, OCH2, J = 6.4 Hz), 6.92–8.21 (m, 4H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 14.7 (CH3), 61.8 (CH2), 106.8, 109.7, 115.4, 144.1, 149.2, 150.2, 153.3, 159.5 (CO), 159.9, 162.5 ppm; IR (KBr): \(\bar{\nu }\) = 1575 (C=N), 1625 (C=C), 1697 (CO–O) cm−1; HRMS: m/z = 298.2091 ([M+Na]+).
Ethyl 5-[5-(5-bromothiophen-2-yl)isoxazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (29, C12H8BrN3O4S)
White solid (0.51 g, 87%); m.p.: 198–200 °C; 1H NMR (DMSO-d6): δ = 1.32 (t, 3H, CH3, J = 6.8 Hz), 4.25 (q, 2H, OCH2, J = 6.8 Hz), 6.91–8.15 (m, 3H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 14.1 (CH3), 61.3 (CH2), 105.8, 113.0, 127.2, 130.5, 134.8, 149.0, 153.9, 159.5, 160.4 (CO), 163.0 ppm; IR (KBr): \(\bar{\nu }\) = 1562 (C=N), 1618 (C=C), 1712 (CO–O) cm−1; HRMS: m/z = 393.1677 ([M+Na]+).
Ethyl 5-[5-(pyridin-4-yl)isoxazol-3-yl]-1,3,4-oxadiazole-2-carboxylate (30, C13H10N4O4)
White solid (0.38 g, 85%); m.p.: 156–158 °C; 1H NMR (DMSO-d6): δ = 1.35 (t, 3H, CH3, J = 7.0 Hz), 4.39 (q, 2H, OCH2, J = 7.0 Hz), 6.98–8.34 (m, 5H, Ar–H) ppm; 13C NMR (DMSO-d6): δ = 14.5 (CH3), 60.7 (CH2), 106.2, 125.2, 136.1, 147.5, 149.7, 154.2, 159.3 (CO), 160.1, 162.9 ppm; IR (KBr): \(\bar{\nu }\) = 1559 (C=N), 1622 (C=C), 1716 (CO–O) cm−1; HRMS: m/z = 309.2360 ([M+Na]+).
Antioxidant activity
The compounds 7–15 and 22–30 were tested for antioxidant property by 2,2-diphenyl-1-picrylhydrazyl (DPPH), hydrogen peroxide (H2O2), and nitric oxide (NO) methods at four different concentrations 25, 50, 75, and 100 µg/cm3 using ascorbic acid as the standard drug [40].
Anti-inflammatory activity
The in vivo anti-inflammatory activity of the title compounds 7–15 and 22–30 was studied using carrageenan-induced hind paw edema test in male albino rats (150–180 g) of Wistar strain at 100 mg/kg body weight using phenylbutazone as the standard [41].
References
Liras S, Allen MP, Segelstein BE (2000) Synth Commun 30:437
Gaonkar SL, Rai KML, Prabhuswamy B (2006) Eur J Med Chem 41:841
Khan KM, Ullah Z, Rani M, Perveen S, Haider SM, Choudhary MI, Rahman A, Voelter W (2004) Lett Org Chem 1:50
(a) Mihailovic N, Markovic V, Matic IZ, Stanisavljevic NS, Jovanovi ZS, Trifunovic SZ, Joksovic L (2017) RSC Adv 7:8550; (b) Reddy GM, Muralikrishna A, Padmavathi V, Padmaja A, Tilak TK, Rao CA (2013) Chem Pharm Bull 61:1291
Almasirad A, Mousavi Z, Tajik M, Assarzadeh M, Shafiee A (2014) Daru J Pharm Sci 22:1
Husain A, Ahmad A, Alam MM, Ajmal M, Ahuja P (2009) Eur J Med Chem 44:3798
El-Din MMG, El-Gamal MI, Abdel-Maksoud MS, Yoo KH, Oh CH (2015) Eur J Med Chem 90:45
Shingalapur RV, Hosamani KM, Keri RS, Hugar MH (2010) Eur J Med Chem 45:1753
Chaudhary SK, Chaudhary M, Chaudhari A, Parmar SS (1978) J Pharm Sci 67:1507
El-Emam AA, Al-Deeb OA, Al-Omar M, Lehmann J (2004) Bioorg Med Chem 12:5107
Dolman SJ, Gosselin F, O’Shea PD, Davies IW (2006) J Org Chem 71:9548
Biftu T, Feng DD, Liang GB, Kuo H, Qian X, Naylor EM, Colandrea VJ, Candelore MR, Cascieri MA, Colwell LF, Forrest MJ, Hom GJ, MacIntyre DE, Stearns RA, Strader CD, Wyvratt MJ, Fisher MH, Weber AE (2000) Bioorg Med Chem Lett 10:1431
Khalilullah H, Khan S, Nomani MDS, Ahmed B (2016) Arab J Chem 9:S1029
Salahuddin, Shaharyar M, Mazumder A, Abdullah MM (2017) Arab J Chem 10:503
Kumar GVS, Rajendraprasad Y, Mallikarjuna BP, Chandrashekar SM, Kistayya C (2010) Eur J Med Chem 45:2063
Padmavathi V, Reddy GS, Padmaja A, Kondaiah P, Shazia A (2009) Eur J Med Chem 44:2106
James ND, Growcott JW (2009) Drugs Future 34:624
Jones AM, Helm JM (2009) Drugs 69:1903
Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M (2008) J Med Chem 51:5843
Vardan S, Smulyan H, Mookherjee S, Eich R (1983) Clin Pharmacol Ther 34:290
Schlecker R, Thieme PC (1988) Tetrahedron 44:3289
Desai NC, Dodiya AM, Rajpara KM, Rupala YM (2014) J Saudi Chem Soc 18:255
Warmus JS, Flamme C, Zhang LY, Barrett S, Bridges A, Chen H, Gowan R, Kaufman M, Sebolt-Leopold J, Leopold W, Merriman R, Ohren J, Pavlovsky A, Przybranowski S, Tecle H, Valik H, Whitehead C, Zhang E (2008) Bioorg Med Chem Lett 18:6171
Bondock S, Fadaly W, Metwally MA (2009) Eur J Med Chem 44:4813
Padmaja A, Payani T, Reddy GD, Padmavathi V (2009) Eur J Med Chem 44:4557
Yu LF, Eaton JB, Fedolak A, Zhang HK, Hanania T, Brunner D, Lukas RJ, Kozikowski AP (2012) J Med Chem 55:9998
Kumbhare RM, Kosurkar UB, Ramaiah MJ, Dadmal TL, Pushpavalli SN, Pal-Bhadra M (2012) Bioorg Med Chem Lett 22:5424
Srinivas A, Nagaraj A, Reddy CS (2010) Eur J Med Chem 45:2353
Kankala S, Kankala RK, Gundepaka P, Thota N, Nerella S, Gangula MR, Guguloth H, Kagga M, Vadde R, Vasam CS (2013) Bioorg Med Chem Lett 23:1306
Rajesh N, Palakshi Reddy B, Vijayakumar V (2012) Ultrason Sonochem 19:522
Bretanha LC, Teixeira VE, Marina R, Siqueira GM, Cunico W, Pereira CMP, Freitag RA (2011) Ultrason Sonochem 18:704
Singh V, Kaur KP, Khurana A, Kad GL (1998) Resonance 3:56
Mason TJ (1997) Chem Soc Rev 26:443
Einhorn C, Einhorn J, Luche JL (1989) Synthesis 11:787
Estager J, Leveque JM, Turgis R, Draye M (2007) Tetrahedron Lett 48:755
Sravya G, Yamini G, Padmavathi V, Padmaja A (2016) Eur J Med Chem 122:647
Madhu Sekhar M, Nagarjuna U, Padmavathi V, Padmaja A, Vasudeva Reddy N, Vijaya T (2018) Eur J Med Chem 145:1
Sowmya DV, Lakshmi Teja G, Padmaja A, Kamal Prasad V, Padmavathi V (2018) Eur J Med Chem 143:891
Zih-Yang L, Hsiu-Ming K, Chung KL (2017) Tetrahedron 73:1650
Yamini G, Sravya G, Padmavathi V, Padmaja A, Appa Rao C, Rajasekhar A (2017) J Heterocycl Chem 54:3498
Sowmya DV, Basha SS, Maheswari Devi PU, Lavanyalatha Y, Padmaja A, Padmavathi V (2017) Med Chem Res 26:1010
Acknowledgements
The author G. Yamini is thankful to the University Grants Commission (UGC), New Delhi, for the sanction of UGC-BSR fellowship. One of the authors, M. Madhu Sekhar is grateful to UGC-New Delhi, India, for the sanction of SRF fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gudi, Y., Mangali, M.S., Gundala, S. et al. Synthesis, characterization, and bioassay of a new class of pyrazolyl/isoxazolyl oxadiazoles. Monatsh Chem 149, 2311–2326 (2018). https://doi.org/10.1007/s00706-018-2295-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-018-2295-7