
Abstract
A new class of bis and tris heterocycles–pyrazolyl indoles and thiazolyl pyrazolyl indoles were prepared from the Michael acceptor (E)-3-(1H-indol-3-yl)-1-arylprop-2-en-1-ones by ultrasound irradiation technique and tested for antioxidant activity. The thiazolyl pyrazolyl indoles and pyrazolyl indoles showed greater radical scavenging activity than pyrazolinyl indoles. Amongst all the tested compounds, 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-p-tolyl-1H-pyrazol-5′-yl)-1H-indole (7b) and 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-(p-methoxyphenyl)-1H-pyrazol-5′-yl)-1H-indole (7c) displayed promising antioxidant activity when compared with standard drug ascorbic acid. The compounds having electron donating groups (CH3, OCH3) on the phenyl ring exhibited greater antioxidant activity than those with electron withdrawing groups (Cl, Br, NO2).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nitrogen containing five-memberedand six-membered heterocyclic systems are scaffolds of many efficacious drugs. One such class of compounds are indoles, pyrazoles and thiazoles. Indole and their derivatives possess anticancer (Chen et al. 1996), antioxidant (Suzen and Buyukbingol 2000), antidepressant (Zhou et al. 2008), anticonvulsant (Ahuja and Siddiqui 2014), antifungal (Zhang et al. 2013), antiviral (Zhang et al. 2015), anti-inflammatory (Radwan et al. 2007), anti-rheumatoidal (Buyukbingol et al. 1994) and anti-HIV (Suzen and Buyukbingol 1998) activities. Many indole derivatives are considered as the most potent scavenger of free radicals (Chyan et al. 1999). In addition, pyrazole is endowed with diverse pharmacological activities, such as antimicrobial (Sridhar et al. 2004), anti-inflammatory (Raffa et al. 2010), anticancer (Altintop et al. 2014), antiviral (El-Sabbagh et al. 2009), anticonvulsant and antidepressant (Abdel-Aziz et al. 2009). Several pyrazole drugs for example, celecoxib demonstrates anti-inflammation effect and inhibits COX-2, rimonabant functions as cannabinoid receptor and is utilized in obesity treatment, fomepizole inhibits alcohol dehydrogenase and sildenafil inhibits phosphodiesterase. Thiazoles have also attracted a great deal of interest due to their presence in natural products and pharmaceutical agents. Thiazole derivatives exhibit antioxidant, antibacterial, antifungal, antitubercular, diuretic, anti-inflammatory and anticancer activities (Siddiqui et al. 2009). Riluzole, a novel neuroprotective drug; sulfathiazole, an antimicrobial drug; bleomycin, an antineoplastic drug; epothiloneA (Wu and Yang 2011), an anticancer drug consist of thiazole containing drugs. It is well known that the combination of two or more types of heterocycles into one molecule could yield a novel entity, with enhanced biological properties. Prompted by the above observations and in continuation of our studies towards the synthesis of a variety of bioactive heterocycles, herein we describe the synthesis and antioxidant activity of a new class of pyrazolyl indoles and thiazolyl pyrazolyl indoles.
Results and discussion
Chemistry
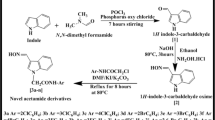
The 4′,5′-dihydro-5′-(1H-indol-3-yl)-3′-arylpyrazole-1′-carbothioamide (4), 5′-(1H-indol-3-yl)-3′-aryl-1H-pyrazole-1′-carbothioamide (5) and 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-aryl-1H-pyrazol-5′-yl)-1H-indole (7) were synthesized from the Michael acceptor (E)-3-(1H-indol-3-yl)-1-arylprop-2-en-1-one (3) (Scheme 1). In fact, the compound 3 was prepared by the Claisen–Schmidt reaction of indole-3-carboxaldehyde (1) and aryl ketones (2) in the presence of NaOH in methanol by ultrasound irradiation. The 1H NMR spectrum of 3a displayed two doublets at δ 8.06 and 7.66 ppm due to olefin protons, HA and HB. The coupling constant value J AB = 16.2 Hz indicated their trans geometry. Adopting similar methodology, the reaction of compound 3 with thiosemicarbazide in the presence of NaOH in ethanol led to the formation of 4. The 1H NMR spectrum of 4a exhibited an AMX splitting pattern for pyrazoline ring protons. The three doublets of doublets appeared at δ 4.87, 3.75, 3.02 ppm were assigned to HA, HM and HX, respectively. The coupling constant values J AM = 12.5, J MX = 10.8 and J AX = 6.7 Hz indicated that HA, HM are cis, HA, Hx are trans and HM, HX are geminal. In addition, two broad singlets were observed at δ 10.01, 5.53 ppm due to NH and NH2, respectively, which disappeared on deuteration. Oxidation of 4 with chloranil in xylene furnished the aromatized product 5. The 1H NMR spectrum of 5a showed a singlet at δ 6.72, and two broad singlets at 10.06 and 5.56 ppm due to C4′–H, NH and NH2, respectively. The signals due to highly acidic protons disappeared when D2O was added. Furthermore, compound 7 was obtained by exploiting thioamide group in 5. Thus the cyclocondensation reaction of 5 with p-chlorophenacyl bromide (6) under ultrasonication provided 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-aryl-1H-pyrazol-5′-yl)-1H-indole (7). The 1H NMR spectrum of 7a displayed a singlet at δ 6.75 due to C4′–H. Another singlet corresponding to C5′′–H was observed at downfield region and merged with aromatic protons. The structures of all the compounds were further established by infrared spectroscopy (IR), carbon-13 nuclear magnetic resonance (13C NMR), mass spectra and elemental analyses.

Synthesis of pyrazolyl indoles and thiazolyl pyrazolyl indoles
Biological evaluation
Antioxidant activity
The compounds 4, 5 and 7 were tested for antioxidant activity by 2,2-diphenylpicrylhydrazyl (DPPH), nitric oxide (NO) and hydrogen peroxide (H2O2) methods. The experimental data on the antioxidant activity of the compounds 4, 5 and 7 and control drug are presented in Tables 1–3, respectively. The mean antioxidant values are shown visually in Figs. 1–3. Amongst all the tested compounds, 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-p-tolyl-1H-pyrazol-5′-yl)-1H-indole (7b) and 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-(p-methoxyphenyl)-1H-pyrazol-5′-yl)-1H-indole) (7c) displayed promising radical scavenging activity in all the three methods when compared with the standard drug, ascorbic acid. The 5′-(1H-indol-3-yl)-3′-p-tolyl-1H-pyrazole-1′-carbothioamide (5b) and 5′-(1H-indol-3-yl)-3′-(p-methoxyphenyl)-1H-pyrazole-1′-carbothioamide (5c) also showed good radical scavenging activity. However, 4′,5′-dihydro-5′-(1H-indol-3-yl)-3′-p-tolylpyrazole-1′-carbothioamide (4b) and 4′,5′-dihydro-5′-(1H-indol-3-yl)-3′-(p-methoxyphenyl)pyrazole-1′-carbothioamide (4c) displayed least activity, while 3′-(p-chlorophenyl)-4′,5′-dihydro-5′-(1H-indol-3-yl)pyrazole-1′-carbothioamide (4d), 3′-(p-bromophenyl)-4′,5′-dihydro-5′-(1H-indol-3-yl)pyrazole-1′-carbothioamide (4e) and 4′,5′-dihydro-5′-(1H-indol-3-yl)-3′-(p-nitrophenyl)pyrazole-1′-carbothioamide (4f) showed no activity. The structure–activity relationship of the compounds revealed that the compounds having indole in combination with pyrazole and thiazole moieties showed greater radical scavenging activity. Moreover, compounds with indole and pyrazole moieties displayed higher antioxidant activity than those with indole and pyrazoline. It was observed that electron donating groups (CH3, OCH3) on the phenyl ring enhanced the activity when compared with electron withdrawing groups (Cl, Br, NO2). Furthermore, the free radical scavenging activity of the compounds 7b and 7c was measured at different concentrations, and monitored the change in absorbance at 10, 20 and 30 min in DPPH method (Table 4). It was perceived that at these 10 min intervals, the values are very close and the results exemplified that the antioxidant activity is independent of time.

The in vitro antioxidant activity of 4, 5 and 7 in DPPH method

The in vitro antioxidant activity of 4, 5 and 7 in H2O2 method

The in vitro antioxidant activity of 4, 5 and 7 in NO method
Statistical analysis
All experiments are performed in triplicate (n = 3), and an analysis of variance (ANOVA) test (Microsoft Excel) is used to compare the mean values across compounds and concentrations. The results represented means ± standard deviation (SD) of three replicated determinations. The descriptive analysis is supported by the statistical analysis. The following inferences are supported through ANOVA analysis (Tables 5, 6, and 7). Since the p-value of rows (compounds) is less than 0.05 (α) can’t reject the null hypothesis, and so conclude (with 95% confidence) that there is significant difference in radical scavenging activity exhibited by the compounds for different concentrations. Since the p-value of columns (concentrations) less than 0.05 (α) can’t reject the null hypothesis, and so conclude (with 95% confidence) that there is significant difference in radical scavenging activity displayed across the compounds. Besides, the p-value (interactions) less than 0.05 indicates that there are significant differences in the interaction between compounds and concentrations.
Conclusion
A new class of bis and tris heterocycles–pyrazolyl indoles and thiazolyl pyrazolyl indoles were prepared from the Michael acceptor (E)-3-(1H-indol-3-yl)-1-arylprop-2-en-1-one by ultrasound irradiation technique, and tested for antioxidant activity. The thiazolyl pyrazolyl indoles derivatives and indolyl pyrazoles showed higher radical scavenging activity. Among all the tested compounds, 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-p-tolyl-1H-pyrazol-5′-yl)-1H-indole (7b) and 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-(p-methoxyphenyl)-1H-pyrazol-5′-yl)-1H-indole (7c) displayed promising antioxidant activity when compared with the standard drug, ascorbic acid. It was also noticed that the electron donating groups (CH3, OCH3) on the phenyl ring exhibited higher radical scavenging activity than those with electron withdrawing groups (Cl, Br, NO2).
Experimental protocols
All the chemicals were purchased from commercial sources and used without further purification. Ultrasonication was performed in a Bandelin Sonorex RK 102H ultrasonic bath operating at frequency of 35 kHz. Melting points were determined in open capillaries on a Mel-Temp apparatus and are uncorrected. The homogeneity of the compounds was checked by thin-layer chromatography (TLC) (silica gel H, BDH, hexane/ethyl acetate, 3:1). The IR spectra were recorded on a Thermo Nicolet IR 200 FT-IR spectrometer as KBr pellets and the wave numbers are given in cm−1. The 1H NMR spectra were recorded in DMSO-d 6 on a Jeol JNM λ-400 MHz spectrometer. The 13C NMR spectra were recorded in DMSO-d 6 on a Jeol JNM spectrometer operating at λ-100 MHz. High-resolution mass spectra were recorded on Micromass Q-TOF micromass spectrometer using electrospray ionization. All chemical shifts are reported in δ (ppm) using Tetramethylsilane as an internal standard. The microanalyses were performed on a Perkin–Elmer 240 C elemental analyzer. The temperature was measured by flexible probe throughout the reaction. The starting compound (E)-3-(1H-indol-3-yl)-1-arylprop-2-en-1-one (3) was prepared as per the literature procedure (Faritha et al. 2014).
General procedure for the synthesis of 4′,5′-dihydro-5′-(1H-indol-3-yl)-3′-arylpyrazole-1′-carbothio amide (4a–f)
To an equimolar (1 mmol) mixture of compound 3 and thiosemicarbazide, ethanol (3 ml) and sodium hydroxide (1.5 mmol) were added. It was sonicated for 1–2 h at room temperature. After completion of the reaction (monitored by TLC), the contents of the flask were poured onto crushed ice. The separated solid was collected by filtration and purified by recrystallization from 2-propanol.
4′,5′-Dihydro-5′-(1H-indol-3-yl)-3′-phenylpyrazole-1′-carbothioamide (4a)
m. p. 150–152 oC; yield 78%; IR (KBr) (cm−1): 3437, 3329 (NH2), 3237 (NH), 1571 (C=N), 1332 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 3.02 (dd, 1H, HX, J AX = 6.7 Hz, J MX = 10.8 Hz), 3.75 (dd, 1H, HM, J AM = 12.5 Hz, J MX = 10.8 Hz), 4.87 (dd, 1H, HA, J AM = 12.5 Hz, J AX = 6.7 Hz), 5.53 (bs, 2H, NH2), 6.88–7.53 (m, 10H, Ar–H & C2–H), 10.08 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 45.1 (C-4′), 66.8 (C-5′), 112.3 (C-8), 118.7 (C-3), 120.4 (C-5), 124.6 (C-7), 126.2 (C-6), 127.6 (C-2), 128.4 (C-4), 130.1 (C-3′′ and C-5′′), 132.7 (C-2′′ and C-6′′), 133.5 (C-4′′), 134.8 (C-1′′), 135.3 (C-9), 157.2 (C-3′), 176.0 (C=S); MS (m/z): 343.0982 [M + Na]. Anal. calcd. for C18H16N4S: C, 67.47; H, 5.03; N, 17.49%; found: C, 67.55; H, 5.01; N, 17.63%.
4′,5′-Dihydro-5′-(1H-indol-3-yl)-3′-p-tolylpyrazole-1′-carbothioamide (4b)
m. p. 133–135 oC; yield 74%; IR (KBr) (cm−1): 3445, 3337 (NH2), 3233 (NH), 1576 (C=N), 1336 (C=S); 1H NMR (400 MHz, DMSO-d 6) : δ 2.36 (s, 3H, Ar–CH3), 3.09 (dd, 1H, HX, J AX = 6.6 Hz, J MX = 10.7 Hz), 3.79 (dd, 1H, HM, J AM = 12.7 Hz, J MX = 10.7 Hz), 4.91 (dd, 1H, HA, J AM = 12.7 Hz, J AX = 6.6 Hz), 5.71 (bs, 2H, NH2), 6.91–7.58 (m, 9 H, Ar–H & C2–H), 10.12 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 25.5 (Ar–CH3), 46.2 (C-4′), 67.8 (C-5′), 112.8 (C-8), 119.2 (C-3), 120.8 (C-5), 125.1 (C-7), 126.8 (C-6), 128.2 (C-2), 128.9 (C-4), 130.6(C-3′′&C-5′′), 133.5(C-2′′&C-6′′), 135.6 (C-1′′), 136.4 (C-9), 141.2 (C-4′′), 158.1 (C-3′), 177.2 (C=S); MS (m/z): 357.1136 [M + Na]. Anal. calcd. for C19H18N4S: C, 68.23; H, 5.42; N, 16.75%; found: C, 68.18; H, 5.43; N, 16.85%.
4′,5′-Dihydro-5′-(1H-indol-3-yl)-3′-(p-methoxyphenyl)pyrazole-1′-carbothioamide (4c)
m. p. 147–148 oC; yield 76%; IR (KBr) (cm−1): 3430, 3326 (NH2), 3235 (NH), 1568 (C=N), 1329 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 3.04 (dd, 1H, HX, J AX = 6.2 Hz, J MX = 10.4 Hz), 3.72 (dd, 1H, HM, J AM = 12.4 Hz, J MX = 10.4 Hz), 3.81 (s, 3H, Ar–OCH3), 4.80 (dd, 1H, HA, J AM = 12.4 Hz, J AX = 6.2 Hz), 5.41 (bs, 2H, NH2), 6.86–7.52 (m, 9H, Ar–H & C2–H), 9.15 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 44.9 (C-4′), 56.8 (Ar–OCH3), 66.2 (C-5′), 111.9 (C-8), 118.4(C-3′′ and C-5′′), 120.1 (C-3), 124.2 (C-5), 125.8 (C-7), 127.3 (C-6), 128.1 (C-2), 132.5 (C-1′′), 134.5 (C-4), 138.0 (C-2′′ and C-6′′), 135.1 (C-9), 155.2 (C-4′′), 156.8 (C-3′), 175.6 (C=S); MS (m/z): 373.1084 [M + Na]. Anal. calcd. for C19H18N4OS: C, 65.12; H, 5.18; N, 15.99%; found: C, 65.21; H, 5.17; N, 16.18%.
3′-(p-Chlorophenyl)-4′,5′-dihydro-5′-(1H-indol-3-yl)pyrazole-1′-carbothioamide (4d)
m. p. 156–158 oC; yield 80%; IR (KBr) (cm−1): 3452, 3342 (NH2), 3236 (NH), 1580 (C=N), 1338 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 3.11 (dd, 1H, HX, J AX = 6.4 Hz, J MX = 10.8 Hz), 3.82 (dd, 1H, HM, J AM = 12.8 Hz, J MX = 10.8 Hz), 4.94 (dd, 1H, HA, J AM = 12.8 Hz, J AX = 6.4 Hz), 5.79 (bs, 2H, NH2), 6.93–7.64 (m, 9H, Ar–H & C2–H), 10.17 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 46.5 (C-4′), 68.1 (C-5′), 112.9 (C-8), 120.1 (C-3), 121.3 (C-5), 125.6 (C-7), 127.2 (C-6), 128.4 (C-2), 129.3 (C-4), 130.8 (C-3′′&C-5′′), 133.8 (C-2′′&C-6′′), 135.7 (C-1′′), 136.6 (C-9), 137.4 (C-4′′), 158.4 (C-3′), 177.8 (C=S); MS (m/z): 377.0592 [M + Na]. Anal. calcd. for C18H15ClN4S: C, 60.92; H, 4.26; N, 15.79%; found: C, 60.99; H, 4.28; N, 15.95%.
3′-(p-Bromophenyl)-4′,5′-dihydro-5′-(1H-indol-3-yl)pyrazole-1′-carbothioamide (4e)
m. p. 162–164 oC; yield 77%; IR (KBr) (cm−1): 3436, 3332 (NH2), 3230 (NH), 1577 (C=N), 1333 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 3.07 (dd, 1H, HX, J AX = 6.5 Hz, J MX = 10.6 Hz), 3.76 (dd, 1H, HM, J AM = 12.6 Hz, J MX = 10.6 Hz), 4.89 (dd, 1H, HA, J AM = 12.6 Hz, J AX = 6.5 Hz), 5.62 (bs, 2H, NH2), 6.89–7.55 (m, 9H, Ar–H & C2–H), 10.19 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 45.8 (C-4′), 67.2 (C-5′), 112.5 (C-8), 118.9 (C-3), 120.6 (C-5), 124.8 (C-7), 126.5 (C-6), 127.9 (C-2), 128.7 (C-4′′), 130.4 (C-4), 132.9 (C-2′′&C-6′′), 133.8 (C-3′′&C-5′′), 135.3 (C-1′′), 135.8 (C-9), 157.6 (C-3′), 176.4 (C=S); MS (m/z): 422.2953 [M + Na]. Anal. calcd. for C18H15BrN4S: C, 54.14; H, 3.79; N, 14.03%; found: C, 54.24; H, 3.80; N, 14.21%.
4′,5′-Dihydro-5′-(1H-indol-3-yl)-3′-(p-nitrophenyl)pyrazole-1′-carbothioamide (4f)
m. p. 171–173 oC; yield 82%; IR (KBr) (cm−1): 3459, 3348 (NH2), 3242 (NH), 1581 (C=N), 1341 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 3.14 (dd, 1H, HX, J AX = 6.8 Hz, J MX = 10.9 Hz), 3.85 (dd, 1H, HM, J AM = 12.9 Hz, J MX = 10.9 Hz), 4.98 (dd, 1H, HA, J AM = 12.9 Hz, J AX = 6.8 Hz), 5.84 (bs, 2H, NH2), 6.95–7.67 (m, 9H, Ar–H & C2–H), 10.20 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 46.8 (C-4′), 68.4 (C-5′), 113.1 (C-8), 120.3 (C-3), 121.5 (C-5), 125.8 (C-7), 127.6 (C-3′′&C-5′′), 128.6 (C-6), 129.5 (C-2), 131.2 (C-4), 134.2 (C-2′′&C-6′′), 138.7 (C-9), 140.2 (C-1′′), 141.3 (C-4′′), 158.9 (C-3′), 178.2 (C=S); MS (m/z): 388.0839 [M + Na]. Anal. calcd. for C18H15N5O2S: C, 59.16; H, 4.14; N, 19.17%; found: C, 59.28; H, 4.18; N, 19.40%.
General procedure for the synthesis of 5′-(1H-indol-3-yl)-3′-aryl-1H-pyrazole-1′-carbothioamide (5a–f)
A solution of compound 4 (1 mmol) and chloranil (1.2 mmol) in xylene (10 ml) was subjected to ultrasound irradiation for 4–5 h at 60 oC. Then it was treated with 5% NaOH solution. The organic layer was separated and repeatedly washed with water. It was dried over an. Na2SO4 and the solvent was removed under reduced pressure. The resultant solid was recrystallized from 2-propanol.
5′-(1H-Indol-3-yl)-3′-phenyl-1H-pyrazole-1′-carbothioamide (5a)
m. p. 143–145 oC; yield 75%; IR (KBr) (cm−1): 3441, 3333 (NH2), 3245 (NH), 1628 (C=C), 1575 (C=N), 1337 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 5.56 (bs, 2H, NH2), 6.72 (s, 1H, C4′-H), 6.90–7.55 (m, 9H, Ar–H & C2–H), 10.16 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 99.6 (C-4′), 112.6 (C-8), 119.2 (C-5), 120.9 (C-7), 125.2 (C-6), 126.8 (C-2′′&C-6′′), 127.7 (C-3), 128.1 (C-2), 130.5 (C-4′′), 133.4 (C-3′′&C-5′′), 134.2 (C-1′′), 135.5 (C-4), 136.2 (C-9), 144.8 (C-5′), 157.7 (C-3′), 176.2 (C=S); MS (m/z): 341.0826 [M + Na]. Anal. calcd. for C18H14N4S: C, 67.90; H, 4.43; N, 17.60%; found: C, 68.00; H, 4.46; N, 17.79%.
5′-(1H-Indol-3-yl)-3′-p-tolyl-1H-pyrazole-1′-carbothioamide (5b)
m. p. 128–130 oC; yield 73%; IR (KBr) (cm−1): 3454, 3343 (NH2), 3251 (NH), 1631 (C=C), 1582 (C=N), 1343 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 2.37 (s, 3H, Ar-CH3), 5.73 (bs, 2H, NH2), 6.83 (s, 1H, C4′-H), 6.94–7.62 (m, 9H, Ar–H & C2–H), 10.18 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 24.7 (Ar-CH3), 100.7 (C-4′), 113.1 (C-8), 119.7 (C-5), 121.2 (C-7), 125.8 (C-6), 127.4 (C-2′′&C-6′′), 128.6 (C-3), 129.5 (C-2), 131.2 (C-3′′&C-5′′), 135.3 (C-1′′), 136.2 (C-4), 136.8 (C-9), 141.7 (C-4′′), 145.3 (C-5′), 158.5 (C-3′), 177.6 (C=S); MS (m/z): 355.0981 [M + Na]. Anal. calcd. for C19H16N4S: C, 68.65; H, 4.85; N, 16.85%; found: C, 68.76; H, 4.87; N, 17.15%.
5′-(1H-Indol-3-yl)-3′-(p-methoxyphenyl)-1H-pyrazole-1′-carbothioamide (5c)
m. p. 151–153 oC; yield 71%; IR (KBr) (cm−1): 3437, 3330 (NH2), 3248 (NH), 1626 (C=C), 1573 (C=N), 1336 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 3.87 (s, 3H, Ar-OCH3), 5.49 (bs, 2H, NH2), 6.65 (s, 1H, C4′-H), 6.88–7.54 (m, 9H, Ar–H & C2–H), 10.20 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 57.1 (Ar-OCH3), 99.1 (C-4′), 112.2 (C-8), 118.9 (C-2′′&C-6′′), 120.8 (C-5), 124.6 (C-7), 126.2 (C-6), 127.9 (C-3), 128.5 (C-1′′), 130.5 (C-2), 133.6 (C-3′′&C-5′′), 138.8 (C-4), 139.4 (C-9), 144.5 (C-5′), 155.4 (C-4′′), 157.1 (C-3′), 175.8 (C=S); MS (m/z): 371.0930 [M + Na]. Anal. calcd. for C19H16N4OS: C, 65.50; H, 4.63; N, 16.08%; found: C, 65.58; H, 4.64; N, 16.22%.
3′-(p-Chlorophenyl)-5′-(1H-indol-3-yl)-1H-pyrazole-1′-carbothioamide (5d)
m. p. 160–162 oC; yield 76%; IR (KBr) (cm−1): 3458, 3347 (NH2), 3253 (NH), 1634 (C=C), 1588 (C=N), 1344 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 5.81 (bs, 2H, NH2), 6.86 (s, 1H, C4′-H), 6.96–7.67 (m, 9H, Ar–H & C2–H), 10.22 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 101.4 (C-4′), 113.5 (C-8), 120.7 (C-5), 122.0 (C-7), 125.9 (C-6), 127.8 (C-3), 128.7 (C-2), 129.9 (C-2′′&C-6′′), 131.6 (C-3′′&C-5′′), 134.2 (C-1′′), 136.3 (C-4), 137.6 (C-9), 138.7 (C-4′′), 145.7 (C-5′), 158.7 (C-3′), 178.2 (C=S); MS (m/z): 375.0432 [M + Na]. Anal. calcd. for C18H13ClN4S: C, 61.27; H, 3.71; N, 15.88%; found: C, 61.40; H, 3.73; N, 16.12%.
3′-(p-Bromophenyl)-5′-(1H-indol-3-yl)-1H-pyrazole-1′-carbothioamide (5e)
m. p. 155–157 oC; yield 79%; IR (KBr) (cm−1): 3446, 3339 (NH2), 3247 (NH), 1630 (C=C), 1578 (C=N), 1340; 1H NMR (400 MHz, DMSO-d 6): δ 5.65 (bs, 2H, NH2), 6.77 (s, 1H, C4′-H), 6.93–7.59 (m, 9H, Ar–H & C2–H), 10.25 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 100.2 (C-4′), 145.0 (C-5′), 112.9 (C-8), 119.3 (C-5), 121.3 (C-7), 125.4 (C-6), 127.1 (C-4′′), 128.3 (C-3), 129.2 (C-2), 130.8 (C-2′′&C-6′′), 133.2 (C-4), 134.7 (C-1′′), 136.8 (C-3′′&C-5′′), 137.2 (C-9), 157.9 (C-3′), 176.8 (C=S); MS (m/z): 420.2823 [M + Na]. Anal. calcd. for C18H13BrN4S: C, 54.42; H, 3.30; N, 14.10%; found: C, 55.52; H, 3.29; N, 14.28%.
5′-(1H-Indol-3-yl)-3′-(p-nitrophenyl)-1H-pyrazole-1′-carbothioamide (5f)
m. p. 166–168 oC; yield 83%; IR (KBr) (cm−1): 3464, 3350 (NH2), 3256 (NH), 1637 (C=C), 1586 (C=N), 1347 (C=S); 1H NMR (400 MHz, DMSO-d 6): δ 5.86 (bs, 2H, NH2), 6.90 (s, 1H, C4′-H), 6.98–7.71 (m, 9H, Ar–H & C2–H), 10.28 (bs, 1H, NH), 13C NMR (100 MHz, DMSO-d 6): δ 101.7 (C-4′), 114.3 (C-8), 121.2 (C-5), 122.4 (C-7), 125.9 (C-6), 128.1 (C-3′′&C-5′′), 129.6 (C-3), 130.2 (C-2), 131.7 (C-2′′&C-6′′), 135.1 (C-4), 136.8 (C-9), 137.2 (C-1′′), 141.6 (C-4′′), 146.2 (C-5′), 159.2 (C-3′), 178.6 (C=S); MS (m/z): 386.0673 [M + Na]. Anal. calcd. for C18H13N5O2S: C, 59.49; H, 3.61; N, 19.27%; found: C, 59.61; H, 3.63; N, 19.47%.
General procedure for the synthesis of 3-(1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-3′-aryl-1H-pyrazol-5′-yl)-1H-indole (7a–f)
A mixture of compound 5 (1 mmol) and p-chlorophenacyl bromide (6) (1 mmol) in ethanol (10 ml) was sonicated for 60–80 min. After completion of the reaction, the contents of the flask were cooled and filtered on a Buchner funnel. It was purified by column chromatography (silica gel 60–120 mesh) using ethyl acetate / hexane (1:3) as eluent.
3-(1-(4′′-(p-Chlorophenyl)thiazol-2′′-yl)-3′-phenyl-1H-pyrazol-5′-yl)-1H-indole (7a)
m. p. 175–176 oC; yield 70%; IR (KBr) (cm−1): 3250 (NH), 1629 (C=C), 1577 (C=N); 1H NMR (400 MHz, DMSO-d 6): δ 6.75 (s, 1H, C4′-H), 6.92–7.58 (m, 15H, Ar–H, C2–H, C5′′-H), 10.09 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 100.2 (C-4′), 112.5 (C-5′′), 112.8 (C-8), 119.6 (C-5), 122.4 (C-7), 126.5 (C-6), 127.2 (C-2′′&C-6′′), 127.9 (C-2), 128.8 (C-4′′), 129.6 (C-2′v&C-6′v), 130.8 (C-3′v&C-5′v), 131.5 (C-3′′&C-5′′), 132.4 (C-1′v), 133.1 (C-4), 134.7 (C-1′′), 135.5 (C-4′v), 136.7 (C-3), 137.2 (C-9), 145.1 (C-5′), 153.8 (C-4′′), 158.1 (C-3′), 160.3 (C-2′′); MS (m/z): 475.0749 [M + Na]. Anal. calcd. for C26H17ClN4S: C, 68.94; H, 3.78; N, 12.37%; found: C, 69.01; H, 3.79; N, 12.52%.
3-(1-(4′′-(p-Chlorophenyl)thiazol-2′′-yl)-3′-p-tolyl-1H-pyrazol-5′-yl)-1H-indole (7b)
m. p. 183–185 oC; yield 72%; IR (KBr) (cm−1): 3255 (NH), 1633 (C=C), 1584 (C=N); 1H NMR (400 MHz, DMSO-d 6): δ 2.39 (s, 3H, Ar-CH3), 6.84 (s, 1H, C4′-H), 6.97–7.64 (m, 14H, Ar–H, C2–H, C5′′-H), 10.79 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 24.9 (Ar-CH3), 100.8 (C-4′), 68.3 (C-5′), 113.8 (C-8), 120.1 (C-5), 123.2 (C-7), 126.9 (C-6), 127.8 (C-2′′&C-6′′), 128.6 (C-2), 129.7 (C-2′v&C-6′v), 129.9 (C-3′v&C-5′v), 130.2 (C-3′′&C-5′′), 131.8 (C-1′′), 132.4 (C-1′v), 133.8 (C-4), 134.0 (C-4′v), 135.6 (C-3), 136.2 (C-9), 137.8 (C-4′′), 145.6 (C-5′′), 155.1 (C-4′′′), 158.8 (C-3′), 161.5 (C-2′′); MS (m/z): 489.0907 [M + Na]. Anal. calcd. for C27H19ClN4S: C, 69.44; H, 4.10; N, 12.00%; found: C, 69.55; H, 4.12; N, 12.22%.
3-(1-(4′′-(p-Chlorophenyl)thiazol-2′′-yl)-3′-(p-methoxyphenyl)-1H-pyrazol-5′-yl)-1H-indole (7c)
m. p. 190–192 oC; yield 75%; IR (KBr) (cm−1): 3248 (NH), 1628 (C=C), 1579 (C=N); 1H NMR (400 MHz, DMSO-d 6): δ 3.89 (s, 3H, Ar-OCH3), 6.68 (s, 1H, C4′-H), 6.90–7.60 (m, 14H, Ar–H, C2–H, C5′′-H), 9.98 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 57.6 (Ar-O-CH3), 99.6 (C-4′), 112.1 (C-5′′), 111.6 (C-8), 119.2 (C-5), 121.9 (C-7), 126.1 (C-6), 126.9 (C-1′′), 127.1 (C-2), 128.2 (C-2′′&C-6′′), 128.9 (C-2′v&C-6′v), 130.2 (C-3′v&C-5′v), 130.8 (C-1′v), 132.1 (C-3′′&C-5′′), 132.7 (C-4), 134.1 (C-4′v), 135.2 (C-3), 136.3 (C-9), 145.6 (C-5′), 152.7 (C-4′′′), 157.4 C-3′), 156.1 (C-4′′), 160.2 (C-2′′); MS (m/z): 505.9747 [M + Na]. Anal. calcd. for C27H19ClN4OS: C, 67.14; H, 3.97; N, 11.60%; found: C, 67.27; H, 4.02; N, 11.85%.
3-(3′-(p-Chlorophenyl)-1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-1H-pyrazol-5′-yl)-1H-indole (7d)
m. p. 195–197 oC; yield 81%; IR (KBr) (cm−1): 3257 (NH), 1636 (C=C), 1585 (C=N); 1H NMR (400 MHz, DMSO-d 6): δ 6.92 (s, 1H, C4′-H), 6.98–7.69 (m, 14H, Ar–H, C2–H, C5′′-H), 10.91 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 1021.7 (C-4′), 113.1 (C-5′′), 114.2 (C-8), 120.4 (C-5), 123.5 (C-7), 127.7 (C-6), 128.4 (C-2), 129.3 (C-2′v&C-6′v), 130.4 (C-3′v&C-5′v), 130.7 (C-1′v), 132.5 (C-3′′&C-5′′), 131.5 (C-2′′&C-6′′), 132.8 (C-4), 134.4 (C-4′v), 134.9 (C-1′′), 135.9 (C-3), 136.8 (C-9), 138.2 (C-4′′), 146.3 (C-5′), 155.7, (C-4′′), 158.9 (C-3′), 161.8 (C-2′′); MS (m/z): 510.3917 [M + Na]. Anal. calcd. for C26H16Cl2N4S: C, 64.07; H, 3.31; N, 11.49%; found: C, 64.01; H, 3.34; N, 11.60%.
3-(3′-(p-Bromophenyl)-1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-1H-pyrazol-5′-yl)-1H-indole (7e)
m. p. 212–215 oC; yield 83%; IR (KBr) (cm−1): 3252 (NH), 1632 (C=C), 1583 (C=N); 1H NMR (400 MHz DMSO-d 6): δ 6.80 (s, 1H, C4′-H), 6.40–7.61 (m, 14H, Ar–H, C2–H, C5′′-H), 10.56 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 101.3 (C-4′), 112.7 (C-5′′), 113.2 (C-8), 119.9 (C-5), 122.8 (C-7), 126.7 (C-6), 127.4 (C-4′′), 128.2 (C-2), 129.3 (C-2′v&C-6′v), 129.8 (C-3′v&C-5′v), 131.4 (C-2′′&C-6′′), 131.9 (C-1′v), 132.8 (C-4), 133.7 (C-1′′), 135.1 (C-3′′&C-5′′), 135.8 (C-4′v), 136.2 (C-3), 137.3 (C-9), 145.9 (C-5′), 154.2 (C-4′′), 158.2 (C-3′), 161.1 (C-2′′); MS (m/z): 554.8426 [M + Na]. Anal. calcd. for C26H16BrClN4S: C, 58.72; H, 3.03; N, 10.53%; found: C, 58.81; H, 3.05; N, 10.69%.
3-(3′-(p-Nitrophenyl)-1-(4′′-(p-chlorophenyl)thiazol-2′′-yl)-1H-pyrazol-5′-yl)-1H-indole (7f)
m. p. 206–208 oC; yield 85%; IR (KBr) (cm−1): 3260 (NH), 1638 (C=C), 1587 (C=N); 1H NMR (400 MHz, DMSO-d 6): δ 6.93 (s, 1H, C4′-H), 7.00–7.73 (m, 14H, Ar–H, C2–H, C5′′-H), 11.09 (bs, 1H, NH); 13C NMR (100 MHz, DMSO-d 6): δ 102.3 (C-4′), 113.6 (C-5′′), 114.8 (C-8), 120.8 (C-5), 123.0 (C-7), 123.9 (C-6), 128.2 (C-3′′&C-5′′), 128.9 (C-2), 129.5 (C-2′′&C-6′′), 130.8 (C-2′v&C-6′v), 131.2 (C-3′v&C-5′v), 132.7 (C-1′v), 133.7 (C-4), 134.6 (C-4′v), 135.5 (C-3), 136.4 (C-9), 137.3 (C-1′′), 142.4 (C-4′′), 146.7 (C-5′), 156.3 (C-4′′), 159.8 (C-3′), 162.3 (C-2′′); MS (m/z): 520.0601 [M + Na]. Anal. calcd. for C26H16ClN5O2S: C, 62.71; H, 3.24; N, 14.06%; found: C, 62.78; H, 3.23; N, 14.18%.
Antioxidant activity
The compounds 4, 5 and 7 were assayed for antioxidant property by DPPH (Burits and Bucar, 2000; Cuendet et al. 1997), H2O2 (Ruch et al. 1989) and nitric oxide (Green et al. 1982; Marcocci et al. 1994) methods.
2,2-Diphenyl-1-picrylhydrazyl radical scavenging activity
The hydrogen atom or electron donation ability of the compounds was measured from the bleaching of the purple colored methanol solution of DPPH radical. The spectrophotometric assay uses the stable radical DPPH as a reagent. To 4 ml of 0.004% (w/v) methanol solution of DPPH, 1 ml of various concentrations of the test compounds (50, 75 and 100 μg ml−1) in methanol were added. After a 30-min incubation period at room temperature, the absorbance was read against blank at 517 nm. Ascorbic acid was used as the standard. The percent of inhibition (I%) of free radical production from DPPH was calculated by the following equation
where A control is the absorbance of the control reaction (containing methanolic DPPH and ascorbic acid), A sample is the absorbance of the test compound (containing methanolic DPPH and test compound) and A blank is the absorbance of the blank (containing only methanolic DPPH). Tests were carried out in triplicate.
Hydrogen peroxide (H2O2) scavenging activity
A solution of H2O2 (40 mM) was prepared in phosphate buffer (pH 7.4). The 50, 75 and 100 μg ml−1 concentrations of the test compounds in 3.4 ml phosphate buffer were added to H2O2 solution (0.6 ml, 40 mM). The absorbance value of the reaction mixture was recorded at 230 nm. Ascorbic acid was used as the standard. The percent of scavenging of H2O2 was calculated by the following equation
Where A control is the absorbance of the control reaction (containing all reagents and ascorbic acid), A sample is the absorbance of the test compound (containing all reagents and test compound) and A blank is the absorbance of the blank (containing only reagents). Tests were carried out in triplicate.
Nitric oxide scavenging activity
NO radicals were generated from sodium nitroprusside. A volume of 1 ml of sodium nitroprusside (10 mM) and 1.5 ml of phosphate buffer saline (0.2 M, pH 7.4) were added to different concentrations (50, 75 and 100 μg/ml) of the test compounds and incubated for 150 min at 25 °C. After incubation, 1 ml of the reaction mixture was treated with 1 ml of Griess reagent (1% sulfanilamide, 2% H3PO4 and 0.1% naphthylethylenediamine dihydrochloride). The absorbance of the chromatophore was measured at 546 nm. Ascorbic acid was used as the standard. NO scavenging activity was calculated by the following equation
where A control was the absorbance of the control reaction (containing all reagents and Ascorbic acid), A sample was the absorbance of the test compound (containing all reagents and test compound) and A blank was the absorbance of the blank (containing only reagents). Tests were carried out in triplicate.
References
Abdel-Aziz M, El Din A, Abuo Rahma G, Hassan AA (2009) Synthesis of novel pyrazole derivatives and evaluation of their antidepressant and anticonvulsant activities. Eur J Med Chem 44:3480–3487
Ahuja P, Siddiqui N (2014) Anticonvulsant evaluation of clubbed indole-1,2,4-triazine derivatives: a synthetic approach. Eur J Med Chem 80:509–522
Altintop MD, Ozdemir A, Ilgin S, Atli O (2014) Synthesis and biological evaluation of new pyrazole-based thiazolyl hydrazone derivatives as potential anticancer agents. Lett Drug Des Discov 11:833–839
Burits M, Bucar F (2000) Antioxidant activity of Nigella sativa essential oil. Phytotherapy Research 14(5):323–328
Buyukbingol E, Suzen S, Klopman G (1994) Studies on the synthesis and structure-activity relationships of 5-(3′-indolyl)-2-thiohydantoin derivatives as aldose reductase enzyme inhibitors. Farmaco 49:443–447
Chen I, Safe S, Bjeldanes L (1996) Indole-3-carbinol and diindolylmethane as aryl hydrocarbon (Ah) receptor agonists and antagonists in T47D human breast cancer cells. Biochem Pharmacol 51:1069–1076
Chyan YJ, Poeggler B, Omar RA, Chain DG, Frangione B, Ghiso J, Pappolla MA (1999) Potent neuroprotective properties against the Alzheimer β-amyloid by anendogenous melatonin-related indole structure, indole-3-propionic acid. J Biol Chem 274:21937–21942
Cuendet M, Hostettmann K, Potterat O, Dyatmiko W (1997) Iridoid Glucosides with Free Radical Scavenging Properties from Fagraea blumei. Helvetica Chimica Acta 80(4):1144–1152
El-Sabbagh OI, Baraka MM, Ibrahim SM, Pannecouque P, Andrei G, Snoeck R, Balzarini J, Rashad AA (2009) Synthesis and antiviral activity of new pyrazole and thiazole derivatives. Eur J Med Chem 44:3746–3753
Faritha A, Jamal Abdul Nasser A, Parveen Ahamed A, Thajuddin N (2014) Synthesis, characterization and biological activity of certain pyrazole derivatives. J Chem Pharm Res 6:189–193
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Analytical Biochemistry 126(1):131–138
Marcocci L, Maguire JJ, Droylefaix MT, Packer L (1994) The Nitric Oxide-Scavenging Properties of Ginkgo Biloba Extract EGb 761. Biochemical and Biophysical Research Communications 201(2):748–755
Radwan MA, Ragab EA, Sabry NM, ElShenawy SM (2007) Synthesis and biologicalevaluation of new 3-substituted indole derivatives as potential anti inflammatory and analgesic agents. Bioorg Med Chem 15:3832–3841
Raffa D, Migliara O, Maggio B, Plescia F, Cascioferro S, Cusimano MG, Tringale G, Cannizzaro C, Plescia F (2010) Pyrazolobenzotriazinones derivatives as COX inhibitors: synthesis, biological activity and molecular modelling studies. Arch Pharm Chem Life Sci 10:631–638
Ruch RJ, Cheng S, Klaunig JE (1989) Prevention of cytotoxicity and inhibition of intercellular communication by antioxidant catechins isolated from Chinese green tea. Carcinogenesis 10(6):1003–1008
Siddiqui N, Arshad MF, Ahsan W, Alam MS (2009) Thiazoles: a valuable insight into the recent advances and biological activities. Int J Pharm Sci Drug Res 1:136
Sridhar R, Perumal PT, Etti S, Shanmugam G, Ponnuswamy MN, Prabavathy VR, Mathivanan N (2004) Design, synthesis and antimicrobial activity of 1H-pyrazole carboxylates. Bioorg Med Chem Lett 4:6035–6040
Suzen S, Buyukbingol E (1998) Evaluation of anti-HIV activity of 5- (2-phenyl-3-indolal)-2- thiohydantoin. Farmaco 53:525–527
Suzen S, Buyukbingol E (2000) Anti-cancer activity studies of indolalthiohydantoin (PIT) on certain cancer cell lines. Farmaco 55:246–248
Wu YJ, Yang BV (2011) Five-membered ring systems: with N and S (Se) atoms. In: Gordon G, John AJ (eds) Progress in heterocyclic chemistry, vol 22, ch 5.5. Elsevier, Great Britain, pp 259–307
Zhang MZ, Chen Q, Yang GF (2015) A review on recent developments of indole containing antiviral agents. Eur J Med Chem 89:421–441
Zhang MZ, Mulholland N, Beattie D, Irwin D, Gu YC, Chen Q, Yang GF, Clough J (2013) Synthesis and antifungal activity of 3-(1,3,4-oxadiazol-5-yl)indoles and 3-(1,3,4-oxadiazol- 5-yl)methylindoles. Eur J Med Chem 63:22–32
Zhou D, Zhou P, Evrard DA, Meagher K, Webb M, Harrison BL, Huryn DM, Golembieski J, Hornby GA, Schechter LE, Smith DL, Andree TH, Mewshaw RE (2008) Studies toward the discovery of the next generation of antidepressants. Part 6: dual 5-HT1A receptor and serotonin transporter affinity within a class of arylpiperazinyl-cyclohexyl indole derivatives. Bioorg Med Chem 16:6707–6723
Acknowledgements
The authors are grateful to University Grants Commission, New Delhi for the sanction of UGC-BSR fellowship. The authors are also thankful to Prof. Ch. Appa Rao, Department of Bio-chemistry (SVU), for providing facilities to carry out the antioxidant activity.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Ummadi, N., Gundala, S., Venkatapuram, P. et al. Synthesis and antioxidant activity of a new class of pyrazolyl indoles, thiazolyl pyrazolyl indoles. Med Chem Res 26, 1574–1584 (2017). https://doi.org/10.1007/s00044-017-1827-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-1827-8