
Abstract
An environmentally benign, simple, efficient, and convenient route is described for the synthesis of novel pyrazolo[1,5-a]pyrimidine derivatives under ultrasound irradiation assisted by KHSO4 in aqueous medium. 3-(4-Methoxyphenyl)-3-oxopropanenitrile reacted with hydrazine hydrate in refluxing ethanol to give 5-(4-methoxyphenyl)-1H-pyrazol-3-amine. Condensation of 3-aminopyrazoles with formylated active proton compounds furnished pyrazolopyrimidines in high to excellent yield. The chemical structure and regioselectivity of the synthesized compounds were confirmed by IR, 1H NMR, 13C NMR, and mass spectral data. X-ray crystallographic study of a selected compound was performed. Furthermore, these synthesized compounds were screened for their anti-inflammatory and anti-cancer activity and the results were promising. The major advantages of this protocol afford high yields, operational simplicity, short reaction times, and devoid of harsh reaction conditions.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
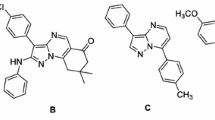
Pyrazolo[1,5-a]pyrimidines have received considerable attention due to their biological activities [1, 2], stimulating researchers to develop the chemistry for this class of compounds [3, 4]. Pyrazolo[1,5-a]pyrimidine framework has been identified as a privileged structure in drug discovery [5]. Zaleplon, Indiplon, and Ocinaplon are representative approved drugs containing the pyrazolo[1,5-a]pyrimidine nucleus. Prompted by these, we have recently reported facile synthetic routes to novel pyrazolo[1,5-a]pyrimidines of type Pypy-1 [6], Pypy-2 [6], Pypy-3 [7], and Pypy-4 [7] (Fig. 1). Also, recently our group have also reported [8] the synthesis of pyrazolo[1,5-a]pyrimidine derivatives bearing carboxamide group in position C-3 and studied their antibacterial properties.

Some recently synthesised pyrazolo[1,5-a]pyrimidines
In the light of the above, it was thought to design and synthesize new pyrazolo[1,5-a]pyrimidine derivatives by installing various groups to the pyrazolopyrimidine moiety and screen them for their biological activities. Several methods for the synthesis of pyrazolo[1,5-a]pyrimidines from 3(5)-aminopyrazoles are reported in the literature [1, 5, 9, 10] involving a refluxing solvent like ethanol [10, 11], DMF/acetic acid mixtures [12, 13], p-toluenesulfonic acid in o-dichlorobenzene [13], and by reaction in the presence of base [14–16]. These methods suffer from disadvantages such as low yields, prolonged reaction times, use of expensive and hazardous chemicals, and formation of regioisomers. Also, Mokhtar et al. [14] have reported the synthesis of pyrazolo[1,5-a]pyrimidines under solvent-free conditions using microwave irradiation in presence of Mg–Al hydrotalcite as solid catalyst.
The advancement of new, efficient, and green synthetic methodology still remains a challenging task in order to affect more comprehensive chemical space through the variation of different substituents. As a part of our interest [6, 7] aimed at synthesizing novel biologically active pyrazolo[1,5-a]pyrimidine derivatives using green chemistry tools, we planned to focus on a facile, practical, rapid, and efficient ultrasonic assisted synthesis in aqueous medium. Ultrasound waves are known for their wide applications in various fields like life sciences, medical, cleaning, sonar, electronics, agriculture, oceanography, material science, etc. [17]. Ultrasound provides an alternative and convenient pathway for reactions to be carried out efficiently [18]. Utility of ultrasound arises due to cavitation process that involves the sequential formation, growth, and implosive collapse of millions of bubbles in the liquid [19]. This directly helps in shortening the time span of reactions and increasing the yields of products [18]. Ultrasound energy can activate reactant molecules that are capable of penetrating the atmosphere of the bubble [20]. The advantages of ultrasound-assisted chemical reactions include enhanced reaction rates, formation of purer products in high yields, milder reaction conditions and are considered a processing aid in terms of energy conservation and waste minimization [21, 22]. However, use of ultrasound in heterocyclic system is not fully explored [21, 23] and thus adopting this eco-friendly method for the synthesis of the desired pyazolopyrimidine derivatives would meet the green protocol, offering higher yields, shorter reaction time and milder reaction conditions.
In continuation with our previous work [6, 7] and prompted by the varied biological activities of pyrazolo[1,5-a]pyrimidine derivatives presented in the literature and also keeping in view the environmental concerns, we envisioned our approach towards the synthesis of 2-(4-methoxyphenyl)-7-substituted-pyrazolo[1,5-a]pyrimidines 4 (Scheme 2), 2-(4-methoxyphenyl)-7-heteroarylpyrazolo[1,5-a]pyrimidine 6 (Scheme 3), 2-(4-methoxyphenyl)-6-arylpyrazolo[1,5-a]pyrimidin-7-amines 8 (Scheme 4), 2-(4-methoxyphenyl)-6,7-substitutedpyrazolo[1,5-a]pyrimidines 11 (Scheme 5), and 2-(4-methoxyphenyl)-8,8-dimethyl-8,9-dihydropyrazolo[1,5-a]quinazolin-6(7H)-one 14 (Scheme 5) under ultrasound irradiation in aqueous media for the assembly of molecules containing the pyrazolo[1,5-a]pyrimidine ring.
Results and discussion
In order to synthesize the proposed pyrazolo[1,5-a]pyrimidine derivatives we required the precursor, 5-(4-methoxyphenyl)-1H-pyrazol-3-amine (2), which is obtained by the reaction of 3-(4-methoxyphenyl)-3-oxopropanenitrile (1) with hydrazine hydrate in refluxing ethanol (Scheme 1) by following the previously reported procedure [24]. 3-Aminopyrazole 2 was used for subsequent reaction without further purification.

Thus, when 3-aminopyrazole 2 was reacted with enaminone 3a in the presence of KHSO4 in aqueous medium in an ultrasonic bath, a product precipitated out in 96 % yield, the structure of which was established as 2-(4-methoxyphenyl)-7-phenylpyrazolo[1,5-a]pyrimidine (4a, Scheme 2) on the basis of spectral and analytical data. The reaction conditions could well be applied for the reactions of 2 with other enaminones 3 giving the pyrazolopyrimidines 4b–4e in 96–97 % overall yields (Scheme 2) in 1.5–8 min under identical conditions.

The same methodology was applied for the synthesis of 7-heteroaryl-2-(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidine (6) from enaminone of type 5 (Scheme 3). The products were obtained in 86–96 % yields. However, in the case of reaction of 2 with 5c, regioisomeric products 6c and 6d were formed, which were characterised with the help of spectral and analytical data.

Encouraged by the success of these investigations, the reactions of aminopyrazole 2 with enaminonitriles 7 (Scheme 4) were subsequently investigated and the expected 7-aminopyrazolopyrimidines 8 were obtained in 87–95 % overall yields in 5–13 min under similar conditions. The structures of these products also could be established with the help of spectral and analytical data.

Further to examine the generality of this green methodology, we finally investigated the reaction of 3-aminopyrazole 2 with formylated active proton compounds 10 and 13 (Scheme 5) which were synthesised following the methods reported from our group [25]. It was utterly pleasing to observe that the reactions went to completion within 6–9 min and the precipitated products were isolated in 92–94 % yields. A summary of the synthesized pyrazolo[1,5-a]pyrimidines is presented in Table 1.

The structures of the synthesised compounds were confirmed by their spectral data (IR, 1H NMR, 13C NMR, and mass spectrometry) and by the X-ray analysis of a selected compound. The 1H NMR spectra of compounds 4a–4e showed doublet for the C5–H and C6–H protons at around 8.43 and 6.84 ppm with a coupling constant of 4 Hz, confirming the formation of regioselective products with substituent at C7–H [26]. The C2–H proton of compound 4a and 4d appeared as singlet at 6.99 and 6.83 ppm, respectively, whereas in the case of compounds 4b, 4c, and 4e it gets buried with the aromatic protons. The other protons due to aromatic groups resonated in their usual range. In the 13C NMR spectra of these products, the common methoxy carbon resonated around 55.4 ppm.
The IR spectra of compounds 8 showed distinct absorption peak due to NH2 group at around 3400 cm−1. In the 1H NMR spectra, the disappearance of doublets due to C-5 and C-6 proton groups and the appearance of a singlet due to C-5–H proton indicated the substitution at C6–H position. The C3–H proton resonated as singlets at around 6.84 ppm in compound 8a and 8c and that for 8b appeared as multiplet at 6.72–6.73 ppm. The C5–H proton resonated as singlet at 8.11 ppm in compound 8b whereas it appeared as multiplet in the range 8.02–8.09 ppm in compound 8a and 8c. The NH2 group protons at C-7 position appeared as singlet at 7.36 ppm in compound 8c and in compounds 8a and 8b it appeared as multiplet. In the 13C NMR spectra the peaks due to methoxy carbon appeared at about 55.2 ppm and C-7 carbon with NH2 substitution at 160.0 ppm.
For compound 6, the IR spectra showed absorption bands close to 1617, 1563, and 1460 cm−1 corresponding to C=N, C=C, and N–N groups respectively. In the 1H NMR spectra, the signal of C3–H proton gets mixed with those of aromatic protons. The C5–H, C6–H protons for compound 6a, 6b appeared as doublets in the range 8.55–8.80 and 6.91–6.96 ppm respectively with coupling constant of 4 Hz. In case of regioisomeric products 6c and 6d, clear distinctions of the substitution at C5–H or C6–H were made with the help of the coupling constant. Product 6c, showed doublet for C5–H proton at 8.57 ppm with coupling constant 4.12 Hz and C6–H proton remains obscured with the aromatic protons, whereas in product 6d, the C5–H and C6–H protons resonated as multiplet at 8.71–8.73 ppm. The methoxy protons in all cases appeared as sharp singlet around 3.87 ppm. In the 13C NMR spectra, the methoxy carbon appeared at 55.4 ppm.
The IR spectra for compound 11a showed carbonyl stretching value at 1636 cm−1, whereas in the case of compounds 11b and 11c, it occurs at higher wave number 1700 and 1711 cm−1 respectively, being an ester group. In the 1H NMR spectra, the signal of C3–H proton in compound 11a and 11b remains buried with the aromatic proton and in case of 11c it showed a sharp singlet at 6.95 ppm. The C5–H protons resonated as singlets at around 8.83 ppm. The methyl protons at C-7 appeared as singlet close to 3.25 ppm. For compound 11a, the methyl protons bonded to the carbonyl carbon gave singlet at 3.22 ppm. In compound 11b, the methyl protons appeared at 3.97 ppm, whereas in the case of 11c, the methyl protons gave triplet at about 1.44 ppm and the methylene proton resonated as quartet in the range 4.40–4.45 ppm. In the 13C NMR spectra, as expected the methoxy carbon appeared at 55.4 ppm. The carbonyl carbon for compound 11a gave a signal at 196.2 ppm and in 11b and 11c, the ester carbon appeared close to 165 ppm.
The IR spectra of compound 14 gave signal at 1622 cm−1 showing the presence of the carbonyl group. The 1H NMR spectra of compound 14 clearly showed the formation of the cyclised product with methyl group and methylene group of the dimedone being seen as sharp singlet at 1.10 ppm and as multiplet at 2.45–2.49 ppm respectively. The C5–H and C3–H protons appeared as singlet at 8.58 and 6.45 ppm, respectively. The methoxy proton appeared as singlet at 3.87 ppm like in other cases. In the 13C NMR, the carbonyl, methyl, and two methylene carbons of dimedone group appeared at 191.2, 28.6, 45.0, and 51.3 ppm, respectively. The common methoxy carbon appeared at 55.6 ppm.
Finally, the confirmation and regioselectivity of the structure was done with the help of X-ray crystal structure of 2-(4-methoxyphenyl)-6-phenylpyrazolo[1,5-a]pyrimidin-7-amine (8a, Fig. 2) as a model. Suitable crystals for X-ray data analysis were obtained by crystallization from ethyl acetate.

ORTEP representation of compound 8a drawn at 30 % probability
Crystal structure of 8a
X-ray data for compound 8a were collected with Bruker Nonius SMART APEX II CCD diffractometer. Yellow crystals of 8a suitable for single X-ray diffraction measurements were grown by the slow crystallisation in ethyl acetate. The crystallographic data for the structure were deposited to the Cambridge Crystallographic Data Center (CCDC No. 978087). Compound 8a (C19H16N4O, M r = 316.36, D x = 1.313 Mg m−3) crystallizes in a triclinic cell (space group P-1) with a = 6.9154(3) Å, b = 7.2935(3) Å, c = 16.2670(8) Å, α = 80.896(3)°, β = 80.960(3)°, γ = 87.389(3)°, V = 799.89(6) Å3, Z = 8, cubic, light yellow, T = 293 K, MoKα radiation, λ = 0.71073 Å, 10,138 measured, 3131 independent, 2225 observed reflections, R int = 0.026, phi and ω scans, refinement on F 2, R[F 2 > 2σ(F 2)] = 0.043, wR(F 2) 0.125, 233 parameters, ∆ρ max = 0.25 e Å−3, ∆ρ min = −0.37 e Å−3. The molecular graphic was performed using ORTEP-3 and displacement ellipsoids are drawn at 30 % probability level (Fig. 2).
Selected bond lengths and bond angles are mentioned in Tables 2 and 3. The C–C bond distances in the phenyl ring and pyrazolopyrimidine core range between 1.377–1.399 and 1.384–1.412 Å, respectively. The bond distances of N2–N3, N3–C10, C10–N4 are 1.360, 1.389, 1.352 Å, respectively. The substituents at C2, C6, and C7 are at a bond distance of 1.468, 1.485 and, 1.344 Å, i.e., C13–C12, C7–C4, and C9–N1, respectively.
The packing structure of the compound is stabilized by several non-bonded interactions and exists as inversion dimer as in Fig. 3a. Each dimer is connected with other molecule through intermolecular H-bonding between N1–H1–N4 with bond distance of 2.924 Å in a parallel fashion Fig. 3b. The respective chains of dimer run along in an anti-parallel fashion. The torsion angles for C8–C7–C4–C5, C11–C12–C13–C14, C9–C7–C4–C5, and N2–C12–C13–C14 are −40.3 (2), −20.4 (3), 139.9 (2), and 156.8 (2), respectively.

Packing diagram of compound 8c showing a the existence as inversion dimmer in a unit cell and b chains of molecules connected through intermolecular H-bonding between N1–H1/H2–N4, shown by the light dotted lines
Biological activities
The synthesised compounds were tested for their anti-cancer and anti-inflammatory activities.
Anti-cancer activity (MTT-based cytotoxic assay)
The metabolically active viable cells cleave MTT and produce purple formazan which can be quantified at 570 nm [27]. This assay is based on the ability of the enzyme succinate tetrazolium reductase, which is active only in viable cells to cleave MTT to purple formazan product [27]. Since the amount of formazan generated depends on cellular metabolism, this cleavage assay can be used to measure cell metabolism and viability and to quantitate cytotoxicity [28]. This assay was used to screen the anticancer activities of the compounds in cultured CHO K1 cell lines to metabolize 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT). The result shows that there is a varying degree of metabolism of MTT by these cells on exposure to these compounds (Fig. 4; Table 4). Compound 6c showed the highest reduction in metabolism of MTT by these cells as compared to normal unexposed cells, followed by 11b, 6a, 14, 4a, 6d, 11c, 8c, 8b, 4c, 6b, and 8a. Hence these compounds may be thought to have cytotoxic effects on CHO K1 cell lines.

The cytotoxic effects of the test compounds on treated mice were compared against the control using the MTT based assay. CHO K1 cells were grown in microtiter plates and treated with 20 mm3 of 10 mg/cm3 test compounds (in 10 % DMSO). Their ability to metabolise MTT was measured by taking the absorbance at 570 nm. Cells exposed to 10 % DMSO were used as control
Anti-inflammatory test
Percentage inhibition in paw diameter
Acute inflammation is characterised by tissue swelling (edema), inflamed tissue (erythema), heat, and pain which are triggered by the infiltration of tissues by serum and leukocytes [29]. Agents that can reduce vascular permeability help in reduction of the edema and serve as good anti-inflammatory agents [30]. Measuring the paw diameter of the mice after treatment with the compounds is therefore, a good physical parameter. On measuring the paw diameter at different intervals of time, it was found that some of these compounds showed a decrease in the diameter. The ability of these test compounds to reduce the paw edema was calculated as percentage inhibition of paw diameter [31]. Table 5 indicates that compounds 4a, 4b, 4d, 4e, 6b, 8c, 11a, and 11b resulted in inhibition of paw edema. Compounds 6a, 6d, and 8b showed the same inhibition of paw diameter at 24 h as compared to the controlled untreated group.
Nitric oxide assay
This assay was used to assess the potentiality of the test compounds as anti-inflammatory agents. As nitric oxide is produced by inducible nitric oxide synthase (iNOS) in activated macrophages during inflammation, the compounds/agents that can lower the production of nitric oxide can serve as good biological markers of inflammation and therefore agents that can lower down the production of nitric oxide may have anti-inflammatory activity [32]. In this study, it was found that the paw exudates of the mice inflamed with FCA showed a reduction in the concentration of nitric oxide when treated with the test compounds 11a, 11b, 6c, 6d, 6a, 4e, 6b, 8a, and 8c (Fig. 5).

Concentration of nitric oxide in paw exudates of the different treatment groups in comparison to the control (untreated) mice. The edema was induced by injection of FCA into the plantar side of the left hind paw of the mice. The mice (except the control) were then treated with the compounds (50 mg/kg body weight) via intraperitoneal injection. After 24 h, the paws were then excised, weighed, homogenized and centrifuged at 12,000 rpm in 10 % ice-cold normal saline. The supernatant was collected and used for nitric oxide assay. Absorbance was recorded at 520 nm and the concentration of nitric oxide was calculated using the standard nitrite curve. Values are mean ± SD of three readings
Nitric oxide concentration in blood was also determined in response to the test compounds (Fig. 6). The results presented in Fig. 6 showed varying level of reduction in NO concentration. Compound 11c showed highest reduction followed by 6c, 8b, 14, and 11a. Compounds 4d, 4e, 6b, 6d, and 8a showed almost equal degrees of reduction, while compounds 4a and 4c showed no reduction.

Concentration of nitric oxide in whole blood of the different treatment groups in comparison to the control (untreated) mice. The edema was induced by injection of FCA into the plantar side of the left hind paw of the mice. These mice (except the control) were then treated with the compounds (50 mg/kg body weight) via intraperitoneal injection. After 24 h, blood was collected by retro-orbital bleeding and used for nitric oxide assay. Absorbance was recorded at 520 nm and the concentration of nitric oxide was calculated using the standard nitrite curve. Values are mean ± SD of three readings
Differential leukocyte count in the blood
During an inflammatory response, the activation and stimulation of a large number of inflammatory cells such as basophils, eosinophils, neutrophils, and monocytes are triggered. Agents that can reduce the number of such inflammatory cells show anti-inflammatory action [30]. The differential leukocyte count in peripheral blood was done in order to determine any alteration in the number of the different leukocytes. This count was expressed as a percentage of the total leukocytes. Most of the compounds resulted in lower counts of both basophils and eosinophils as compared to control mice given in Fig. 7. The percentage of neutrophils were found to be lower in mice treated with compound 4a, 4b, 4c, 6c, 6d, 8a, and 11c, of which compounds 6d and 11c resulted in the highest reduction in neutrophils. A high percentage of neutrophils is indicative of inflammation or bacterial infection [33].

Percentage counts of the different leukocytes in control and treated mice. The blood from the mice was collected 24 h after FCA injection through retro-orbital bleeding. The blood smear was then stained with Wright’s stain and cells were counted under a microscope
From the results obtained, there is an indication that while most of the compounds showed slight anti-inflammatory effect, the test compounds 11a, 11b, 6c, 6d, and 4e, however, showed potent anti-inflammatory effect with 11a showing the highest anti-inflammatory activity. Thus these compounds may further be explored for potential use as anti-inflammatory agents.
Experimental
Melting points were recorded by open capillary method. The IR spectra were recorded on a Perkin-Elmer 983 spectrometer (Perkin-Elmer). 1H and 13C NMR spectra were recorded on a JEOL JNM-ECS 400 taking Me4Si as the internal standard in CDCl3. In the NMR spectral data, the abbreviations d, dd, s, m, and t, stand for doublet, double-doublet, singlet, multiplet, and triplet, respectively. The X-ray diffraction data were collected at 296 K with Mo Kα radiation (λ = 0.71073 Å) using a Bruker Nonius SMART APEX II CCD diffractometer equipped with a graphite monochromator. The structures were solved by direct methods (SHELXS97) and refined by full-matrix least-squares based on F 2. All calculations were carried out using WinGX system version 1.80.05. All the non-H atoms were refined in the anisotropic approximation: H-atoms were located at calculated positions. The electron spray mass spectra were recorded on a THERMO Finnigan LCQ advantage max ion trap mass spectrometer. Ultrasound irradiation was carried out in an EQUITRON digital ultrasonic cleaner-2.5 l, model 8425.025.424 at 170 W and 50 Hz. Formylated active proton compounds were synthesized by our previously reported procedure [25].
General procedure for the synthesis of 2-(4-methoxyphenyl)-7-substituted-pyrazolo[1,5-a]pyrimidines 4a-4e
To a solution of 3-amino-1H-pyrazole 2 (1 mmol) and enaminones 3 (1 mmol) in 2.5 cm3 ethanol was added a solution of KHSO4 (2 mmol) in 2.5 cm3 water and the resulting mixture was irradiated in ultrasonic bath at 60 °C. Within 30–60 s a precipitate started appearing and then reaction went to completion within 1.5–16 min (monitored by TLC). At the end of the reaction, the reaction mixture was allowed to cool and the precipitated product was collected by filtration, washed with water–ethanol (3 × 1 cm3), and finally dried over calcium chloride in a desiccator to give practically pure product in 96–97 % yields. Further purification for analytical purposes was achieved by column chromatography (silica gel, 10 % EtOAc-hexane).
2-(4-Methoxyphenyl)-7-phenylpyrazolo[1,5-a]pyrimidine (4a, C19H15N3O)
Yellow solid (292 mg, 96 %); m.p.: 138–140 °C; 1H NMR (400 MHz, CDCl3): δ = 3.85 (s, 3H, OCH3), 6.86 (d, 1H, J = 4 Hz, C6–H), 6.97 (d, 2H, J = 8.7 Hz, phenyl), 6.99 (s, 1H, C3–H), 7.57–7.58 (m, 3H, phenyl), 7.94 (d, 2H, J = 8.7 Hz, phenyl), 8.16–8.18 (m, 2H, phenyl), 8.46 (d, 1H, J = 4 Hz, C5–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 55.4 (OCH3), 93.0 (CH, pyrazole), 106.0 (2, CH-phenyl), 114.2 (CH-pyrimidine), 125.6 (Cq), 128.0 (2, CH-phenyl), 128.6 (2, CH-phenyl), 129.3 (CH-phenyl), 129.5 (2, CH-phenyl), 131.1 (Cq), 146.4 (Cq), 148.7 (Cq), 151.1 (Cq), 155.9 (CH-pyrimidine), 160.4 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1462 (C=C), 1533 (N–N), 1615 (C=N) cm−1; MS (ESI): m/z = 301 ([M]+).
2-(4-Methoxyphenyl)-7-(p-tolyl)pyrazolo[1,5-a]pyrimidine (4b, C20H17N3O)
Yellow solid (302 mg, 96 %); m.p.: 137–139 °C; 1H NMR (400 MHz, CDCl3): δ = 2.47 (s, 3H, CH3), 3.85 (s, 3H, OCH3), 6.84 (d, 1H, J = 4 Hz, C6–H), 6.96–6.98 (m, 3H, 2H-phenyl, 1H–C3–H), 7.38 (d, 2H, J = 8 Hz, CH-phenyl), 7.94 (d, 2H, J = 8 Hz, phenyl), 8.09 (d, 2H, J = 8 Hz, phenyl), 8.43 (d, 1H, J = 4 Hz, C5–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 21.6 (CH3), 55.4 (OCH3), 92.8 (CH-pyrazole), 106.5 (2, CH-phenyl), 114.1 (CH-pyrimidine), 125.0 (Cq), 128.0 (2, CH-phenyl), 128.2 (2, CH-phenyl), 129.3 (2, CH-phenyl), 129.5 (Cq), 141.6 (Cq), 146.5 (Cq), 148.7 (Cq), 151.1 (Cq), 155.8 (CH-pyrimidine), 160.4 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1460 (C=C), 1534 (N–N), 1612 (C=N) cm−1; MS (ESI): m/z = 315 ([M+H]+).
2,7-Bis(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidine (4c, C20H17N3O2)
Yellow solid (318 mg, 96 %); m.p.: 150 °C; 1H NMR (400 MHz, CDCl3): δ = 3.85 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 6.83 (d, 1H, J = 4 Hz, C6–H), 6.96–6.99 (m, 3H, 2H-phenyl, 1H–C3–H), 7.08 (d, 2H, J = 9 Hz, CH-phenyl), 7.95 (d, 2H, J = 8.7 Hz, phenyl), 8.22 (d, 2H, J = 8.7 Hz, phenyl), 8.42 (d, 1H, J = 4 Hz, C5–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 55.4 (OCH3), 55.5 (OCH3), 92.7 (CH, pyrazole), 106.0 (CH, pyrimidine), 114.0 (2, CH-phenyl), 114.1 (2, CH-phenyl), 123.3 (Cq), 125.7 (Cq), 128.0 (2, CH-phenyl), 131.3 (2, CH-phenyl), 146.1 (Cq), 148.6 (CH, pyrimidine), 151.2 (Cq), 155.7 (Cq), 160.4 (Cq), 161.9 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1465 (C=C), 1526 (N–N), 1616 (C=N) cm−1; MS (ESI): m/z = 331 ([M]+).
7-(4-Chlorophenyl)-2-(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidine (4d, C19H14ClN3O)
Yellow solid (325 mg, 97 %); m.p.: 158–159 °C; 1H NMR (400 MHz, CDCl3): δ = 3.80 (s, 3H, OCH3), 6.86 (d, 1H, J = 4.1 Hz, C6–H), 6.96–7.01 (m, 3H, 2H-phenyl, 1H–C3–H), 7.55 (d, 2H, J = 8.7 Hz, CH-phenyl), 7.93 (d, 2H, J = 9.1 Hz, phenyl), 8.14 (d, 2H, J = 8.7 Hz, phenyl), 8.47 (d, 1H, J = 4 Hz, C5–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 55.3 (OCH3), 92.9 (CH, pyrazole), 106.5 (CH, pyrimidine), 114.1 (2, CH-phenyl), 125.1 (Cq), 127.9 (2, CH-phenyl), 128.8 (2, CH-phenyl), 129.2 (Cq), 130.8 (2, CH-phenyl), 137.2 (Cq), 145.3 (Cq), 148.4 (CH, pyrimidine), 150.5 (Cq), 156.0 (Cq), 160.4 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1463 (C=C), 1529 (N–N), 1615 (C=N) cm−1; MS (ESI): m/z = 335 ([M+H]+).
2-(4-Methoxyphenyl)-7-(4-nitrophenyl)pyrazolo[1,5-a]pyrimidine (4e, C19H14N4O3)
Orange solid (332 mg, 97 %); m.p.: 173–174 °C; 1H NMR (400 MHz, CDCl3): δ = 3.76 (s, 3H, OCH3), 7.01–7.04 (m, 2H, phenyl), 7.28–7.31 (m, 2H, C6–H, C3–H), 7.96 (d, 2H, J = 8 Hz, phenyl), 8.44–8.49 (m, 4H, phenyl), 8.61 (d, 1H, J = 4.1 Hz, C5–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 55.7 (OCH3), 93.5 (CH, pyrazole), 108.9 (CH, pyrimidine), 114.7 (2, CH-phenyl), 124.0 (2, CH-phenyl), 125.2 (Cq), 128.2 (2, CH-phenyl), 131.4 (2, CH-phenyl), 137.2 (Cq), 143.4 (Cq), 149.0 (CH, pyrimidine), 150.0 (Cq), 151.1 (Cq), 155.4 (Cq), 160.6 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1482 (C=C), 1515 (N–N), 1636 (C=N) cm−1; MS (ESI): m/z = 347 ([M+H]+).
General procedure for the synthesis of 7-heteroaryl-2-(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidines 6a–6d
A mixture of aminopyrazole 2 (1 mmol) and enaminone 5a or 5b (1 mmol) in presence of KHSO4 (2 mmol) was irradiated under the influence of ultrasound for 7–25 min in 5 cm3 of EtOH:H2O (1:1) mixture resulting in the formation of a precipitated product. After the completion of the reaction monitored by TLC, the precipitate was collected by filtration, washed repeatedly with ethanol–water (1:1) to ensure complete removal of acid and dried to give practically pure pyrazolopyrimidine 6 in 94–96 % yields. Further purification was achieved by column chromatography (silica gel, 20 % EtOAc-hexane).
In case of the reaction between 2 and 5c under similar conditions, two regioisomeric products 6c and 6d were isolated in 41 and 45 % yield, respectively. The products 6c (R f = 0.2) and 6d (R f = 0.15) were successfully isolated by subjecting the reaction mixture to column chromatography using silica gel (60–120 mesh) and 15 % EtOAc-hexane as eluent.
2-(4-Methoxyphenyl)-7-(pyridin-4-yl)pyrazolo[1,5-a]pyrimidine (6a, C18H14N4O)
Yellow solid (96 %); m.p.: 133 °C; 1H NMR (400 MHz, CDCl3): δ = 3.87 (s, 3H, OCH3), 6.96 (d, 1H, J = 4 Hz, C6–H), 6.99–7.04 (m, 3H, 2H-phenyl, 1H–C3–H), 7.94 (d, 2H, J = 8.7 Hz, CH-phenyl), 8.17 (d, 2H-pyridine, J = 4 Hz), 8.53 (d, 1H, C5–H, J = 4 Hz), 8.90 (s, br, pyridine) ppm; 13C NMR (100 MHz, CDCl3): δ = 55.4 (OCH3), 93.7 (CH, pyrazole), 107.3 (CH, pyrimidine), 114.3 (2, CH-phenyl), 123.6 (2, CH pyridine), 125.1 (Cq), 128.0 (2, CH-phenyl), 139.5 (Cq), 142.8 (Cq), 148.7 (CH-pyrimidine), 149.6 (2, CH-pyridine), 151.7 (Cq), 156.3 (Cq), 160.6 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1460 (C=C), 1533 (N–N), 1618 (C=N) cm−1; MS (ESI): m/z = 303 ([M+H]+).
2-(4-Methoxyphenyl)-7-(pyridin-3-yl)pyrazolo[1,5-a]pyrimidine (6b, C18H14N4O)
Yellow solid (94 %); m.p.: 154–155 °C; 1H NMR (400 MHz, CDCl3): δ = 3.89 (s, 3H, OCH3), 6.91 (d, 1H, J = 4 Hz, CH-pyrimidine), 6.96–7.01 (m, 3H, 2H-phenyl, 1H–C3–H), 7.56 (dd, 1H–5′H, J = 4 Hz, 8 Hz), 7.93 (d, 2H, J = 8 Hz, phenyl), 8.49 (d, 1H–4′H, J = 4 Hz), 8.65 (d, 1H–6′H, J = 8 Hz), 8.80 (d, 1H, J = 4 Hz, C5–H), 9.33 (s, 1H–2′H) ppm; 13C NMR (100 MHz, CDCl3): δ = 55.4 (OCH3), 93.4 (CH, pyrazole), 106.9 (CH, pyrimidine), 114.2 (2, CH-phenyl), 123.4 (5′-CH pyridine), 125.2 (4′-CH pyridine), 127.6 (Cq), 128.0 (2, CH-phenyl), 137.3 (Cq), 143.0 (Cq), 148.8 (2′-CH pyridine), 149.6 (Cq), 151.1 (6′-CH pyridine), 151.4 (Cq), 156.1 (CH, pyrimidine), 160.6 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1465 (C=C), 1563 (N–N), 1617 (C=N) cm−1; MS (ESI): m/z = 302 ([M]+).
2-(4-Methoxyphenyl)-7-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine (6c, C18H14N4O)
Yellow solid (41 %); m.p.: 175–177 °C; 1H NMR (400 MHz, CDCl3): δ = 3.87 (s, 3H, OCH3), 7.00–7.04 (m, 3H, 2H-phenyl, 1H–C3–H), 7.46–7.50 (m, 1H–5′H), 7.72–7.75 (m, 1H–4′H), 7.98–8.00 (m, 3H, 2H-phenyl, C6–H), 8.57 (d, 1H, C5–H, J = 4.1 Hz), 8.82–8.83 (m, 1H–3′H), 9.32–9.34 (m, 1H–6′H) ppm; 13C NMR (100 MHz, CDCl3): δ = 55.4 (OCH3), 93.0 (CH, pyrazole), 107.6 (CH, pyrimidine), 114.2 (2CH-phenyl), 125.5 (3′-CH pyridine), 125.6 (Cq), 128.0 (2, CH-phenyl), 136.9 (5′-CH pyridine), 143.6 (Cq), 145.0 (4′-CH-pyridine), 148.2 (CH-pyrimidine), 148.8 (6′-CH-pyridine), 149.8 (Cq), 151.4 (Cq), 155.8, 160.5 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1460 (C=C), 1566 (N–N), 1617 (C=N) cm−1; MS (ESI): m/z = 303 ([M+H]+).
2-(4-Methoxyphenyl)-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine (6d, C18H14N4O)
Yellow solid (45 %); m.p.: 165–166 °C; 1H NMR (400 MHz, CDCl3): δ = 3.87 (s, OCH3), 6.95–7.02 (m, 3H, 1H–C3–H, 2 CH-phenyl), 7.37–7.40 (m, 1H–5′H), 7.85–7.97 (m, 4H, 2 CH-phenyl, 2H–3′, 4′–H), 8.51 (d, 1H–6′H, J = 8.2 Hz), 8.71–8.73 (m, 2H, C6, C7) ppm; 13C NMR (100 MHz, CDCl3): δ = 55.4 (OCH3), 93.4 (CH, pyrazole), 105.3 (CH, pyrimidine), 114.3 (2, CH-phenyl), 121.7 (3′-CH pyridine), 124.9 (Cq), 125.4 (5′-CH pyridine), 127.9 (2, CH-phenyl), 134.9 (Cq), 137.2 (4′-CH pyridine), 149.2 (CH-pyrimidine), 149.4 (6′-CH-pyridine), 154.0 (Cq), 155.0 (Cq), 156.9 (Cq), 160.5 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1463 (C=C), 1526 (N–N), 1616 cm−1; MS (ESI): m/z = 303 ([M+H]+).
General procedure for the synthesis of 2-(4-methoxyphenyl)-6-substituted-pyrazolo[1,5-a]pyrimidin-7-amines 8a–8c
A mixture of aminopyrazole 2 (1 mmol) and enaminonitriles 7 (1 mmol) in presence of KHSO4 (2 mmol) was irradiated under the influence of ultrasound for 5–15 min in 5 cm3 of EtOH:H2O (1:1) mixture resulting in the formation of a precipitated product. After the completion of reaction monitored by TLC the precipitate was collected by filtration, washed repeatedly with ethanol–water (1:1) to ensure complete removal of acid and dried to give practically pure pyrazolopyrimidines 8 in 85–92 % yield. Further purification was achieved by column chromatography (silica gel, 10 % EtOAc-hexane).
2-(4-Methoxyphenyl)-6-phenylpyrazolo[1,5-a]pyrimidin-7-amine (8a, C19H16N4O)
Off white solid (87 %); m.p.: 190–192 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ = 3.82 (s, 3H, OCH3), 6.86 (s, 1H, C3–H), 7.04 (d, 2H-phenyl, J = 8 Hz), 7.37–7.41 (t, 1H-phenyl), 7.46–7.54 (m, 6H, 4H-phenyl, 2H–NH2) 8.04 (d, 2H-phenyl, J = 8 Hz), 8.08–8.09 (m, 1H, C5–H) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ = 55.6 (OCH3), 91.3 (CH, pyrazole), 102.3 (2CH-phenyl), 114.4 (Cq), 114.6 (2CH-phenyl), 125.9 (2CH-phenyl), 128.0 (CH-phenyl), 129.5 (2CH-phenyl), 134.4 (Cq), 145.2 (Cq), 149.7 (CH-pyrimidine), 150.6 (Cq), 154.6 (Cq), 160.0 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1458 (C=C), 1519 (N–N), 1623 (C=N), 3444 (NH) cm−1; MS (ESI): m/z = 317 ([M+H]+).
2,6-Bis(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-amine (8b, C20H18N4O2)
Yellow solid (95 %); m.p.: 240–241 °C; 1H NMR (600 MHz, DMSO-d 6 ): δ = 3.81 (s, 6H, OCH3), 6.84 (s, 1H, C3–H), 7.03–7.07 (m, 4H, ArH), 7.36 (s, 2H, NH2), 7.44 (d, 2H, J = 8 Hz), 8.02–8.04 (m, 3H, C5–H, 2H–ArH) ppm; 13C NMR (150 MHz, DMSO-d 6 ): δ = 55.1 (OCH3), 90.6 (CH, pyrazole), 101.0 (Cq), 114.0 (2CH-phenyl), 114.4 (2CH-phenyl), 116.9 (Cq), 126.2 (Cq), 127.5 (2CH-phenyl), 130.4 (2CH-phenyl), 144.7 (Cq), 149.3 (CH-pyrimidine), 149.9 (Cq), 154.0 (Cq), 158.5 (Cq), 159.7 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1460 (C=C), 1507 (N–N), 1624 (C=N), 3436 (NH) cm−1; MS (ESI): m/z = 347 ([M+H]+).
6-(4-Chlorophenyl)-2-(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidin-7-amine (8c, C19H15ClN4O)
Brown solid (91 %); m.p. 261–262 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ = 3.86 (s, 3H, OCH3), 6.72–6.73 (m, 1H, C3–H), 6.99 (d, 2H, ArH, J = 8.4 Hz), 7.50–7.70 (m, 6H, ArH), 7.97–7.98 (m, 2H, NH2), 8.11 (s, C5–H) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ = 55.2 (OCH3), 90.6 (CH, pyrazole), 114.2 (2CH-phenyl), 116.9 (Cq), 127.9 (Cq), 129.3 (2, CH-phenyl), 131.3 (2, CH-phenyl), 133.3 (2, CH-phenyl), 133.3 (Cq), 141.6 (Cq), 146.1 (Cq), 149.9 (Cq), 154.4 (CH-pyrimidine), 160.5 (Cq), 166.8 (Cq), 106.9 (CH, pyrimidine), 114.2 (2, CH-phenyl), 123.4 (CH pyridine), 125.2 (CH-pyridine), 127.6 (Cq), 128.0 (2, CH-phenyl), 137.3 (Cq), 143.0 (Cq), 148.8 (CH-pyridine), 149.6 (Cq), 151.1 (CH-pyridine), 151.4 (Cq), 156.1, 160.6 (Cq) ppm; IR (KBr): \(\overline{\nu }\) = 1462 (C=C), 1519 (N–N), 1614 (C=N), 3417 (NH) cm−1; MS (ESI): m/z = 351 ([M+H]+).
General procedure for the synthesis of 6-acetyl/carboalkoxy-7-methyl-N-phenylpyrazolo[1,5-a]pyrimidines 11a–11c
In order to synthesize the target pyrazolo[1,5-a]pyrimidine 11, formylated active proton compounds of type 10 were required. This was synthesized by the irradiation of acyclic active proton compounds 9 (1 mmol) with DMF–DMA in microwave digester for 5 min. The reaction mixture (monitored by TLC) was evaporated to dryness under reduced pressure. To this, aminopyrazole 2 (1 mmol) as synthesized in Scheme 1 was added and dissolved in 5 cm3 of ethanol:water mixture (1:1). KHSO4 (2 mmol) was then added and subjected to ultrasound irradiation for 6–12 min resulting in the formation of a precipitated product. After the completion of reaction (monitored by TLC), the precipitate was collected by filtration, washed repeatedly with ethanol–water (1:1) and dried over anhydrous CaCl2 to give practically pure products 11 in 92–94 % yield. Further purification was achieved by column chromatography (silica gel, 10 % EtOAc-hexane).
1-[2-(4-Methoxyphenyl)-7-methylpyrazolo[1,5-a]pyrimidin-6-yl]ethanone (11a, C16H15N3O2)
Yellow solid (98 %); m.p.: 196 °C; 1H NMR (400 MHz, CDCl3): δ = 2.70 (s, 3H, CH3), 3.22 (s, 3H, COCH3), 3.89 (s, 3H, OCH3), 6.98–7.02 (m, 3H, 2H-phenyl, 1H–C3–H), 7.98 (d, 2H, J = 8.7 Hz, phenyl), 8.83 (s, 1H, C5–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 15.4 (CH3), 29.7 (CH3), 55.4 (OCH3), 94.0 (CH-pyrazole), 114.2 (2, CH-phenyl), 117.0 (Cq), 124.7 (Cq), 128.2 (2, CH-phenyl), 142.5 (Cq), 148.7 (CH-pyrimidine), 155.6 (Cq), 158.3 (Cq), 160.9 (Cq), 196.2 (CO) ppm; IR (KBr): \(\overline{\nu }\) = 1460 (C=C), 1519 (N–N), 1623 (C=N), 1636 (C=O) cm−1; MS (ESI): m/z = 281 ([M]+).
Methyl 2-(4-methoxyphenyl)-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxylate (11b, C16H15N3O3)
Yellow solid (94 %); m.p.: 173 °C; 1H NMR (400 MHz, CDCl3): δ = 3.25 (s, 3H, CH3), 3.87 (s, 3H, OCH3-phenyl), 3.97 (s, 3H, CO–OCH3), 6.87–6.93 (m, 3H, 2H-phenyl, 1H–C3–H), 7.89 (d, 2H, J = 8.7 Hz, phenyl), 8.83 (s, 1H, C5–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 15.1 (CH3), 52.4 (OCH3), 55.3 (OCH3-phenyl), 94.2 (CH-pyrazole), 109.7 (Cq), 114.2 (2, CH-phenyl), 124.7 (Cq), 128.1 (2, CH-phenyl), 149.5 (Cq), 149.8 (CH-pyrimidine), 151.5 (Cq), 157.7 (Cq), 160.7 (Cq), 165.2 (CO) ppm; IR (KBr): \(\overline{\nu }\) = 1435 (C=C), 1519 (N–N), 1616 (C=N), 1700 (C=O) cm−1; MS (ESI): m/z = 297 ([M]+).
Ethyl 2-(4-methoxyphenyl)-7-methylpyrazolo[1,5-a]pyrimidine-6-carboxylate (11c, C17H17N3O3)
Yellow solid (92 %); m.p.: 129–130 °C; 1H NMR (400 MHz, CDCl3): δ = 1.42–1.46 (t, 3H, CH2CH3), 3.25 (s, 3H, CH3), 3.85 (s, 3H, OCH3), 4.40–4.45 (q, 2H, CH2CH3), 6.95 (s, 1H, C3–H), 7.00 (d, 2H-phenyl, J = 8 Hz), 7.97 (d, 2H-phenyl, J = 8 Hz), 8.91 (s, 1H, C6H) ppm; 13C NMR (100 MHz, CDCl3): δ = 14.4 (CH2CH3), 15.2 (CH3), 55.4 (OCH3), 61.5 (CH2CH3), 94.3 (C3-pyrazole), 110.1 (Cq), 114.3 (2CH-phenyl), 125.0 (Cq), 128.2 (2CH-phenyl), 149.7 (Cq), 149.8 (Cq), 151.4 (Cq), 157.8 (Cq), 160.8 (C6-pyrimidine), 164.9 (CO) ppm; IR (KBr): \(\overline{\nu }\) = 1457 (C=C), 1524 (N–N), 1603 (C=N), 1711 (C=O) cm−1; MS (ESI): m/z = 311 ([M]+).
2-(4-Methoxyphenyl)-8,8-dimethyl-8,9-dihydropyrazolo[1,5-a]quinazolin-6(7H)-one (14, C19H19N3O2)
A mixture of dimedone (1 mmol) and N,N-dimethylformamide dimethylacetal (1.5 mmol) was irradiated under MW at 850 W for 5 min (monitored by TLC). The mixture was evaporated to dryness under reduced pressure. To this reaction mixture (13, without isolation), aminopyrazole 2 (1 mmol) was added, dissolved in 5 cm3 ethanol–water (1:1) mixture and irradiated in ultrasound in presence of KHSO4 (2 mmol). The reaction was monitored by TLC and by the end of 9 min, the precipitate was collected in the same manner as mentioned. The product was obtained in 92 % yield. For further purification of the product, column chromatography was employed. Yellow solid (92 %); m.p.: 214 °C; 1H NMR (400 MHz, CDCl3): δ = 1.10 (s, 2CH3), 2.45–2.49 (m, 4H, 2CH2), 3.87 (s, 3H, OCH3), 6.45 (s, 1H, C3–H), 6.99–7.03 (m, 3H-phenyl), 7.65–7.67 (m, 1H-phenyl), 8.58 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ = 28.6 (2CH3), 31.6 (Cq), 45.0 (CH2), 51.3 (CH2), 55.6 (OCH3), 92.2 (CH, pyrazole), 114.7 (2 CH-phenyl), 119.2 (Cq), 128.1 (2CH-phenyl), 148.6 (Cq), 153.1 (Cq), 153.3 (Cq), 156.0 (CH-pyrimidine), 160.2 (Cq), 171.0 (Cq), 191.2 (CO) ppm; IR (KBr): \(\overline{\nu }\) = 1412 (C=C), 1499 (N–N), 1618 (C=N), 1622 (C=O) cm−1; MS (ESI): m/z = 321 ([M]+).
Anti-cancer assay
Chinese hamster ovary K1 (CHO K1) cell lines were procured from NCCS (Pune), Dulbecco’s modified Eagle medium/Nutrient mixture F-12, dimethyl sulfoxide, fetal bovine serum, sodium chloride, potassium chloride, disodium hydrogen phosphate, potassium dihydrogen phosphate, trypsin phosphate versene glucose (TPVG) solution, 3-(4,5-dimethylthiazoyl-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) were purchased from HIMEDIA. Antibiotic antimycotic solution was purchased from Sigma Aldrich.
The cytotoxicity of the test compounds was assessed using MTT assay, according to Freshney [34] with certain modifications. CHO K1 cells at a concentration of 0.5 × 10–103 in 100 mm3 growth medium (Dulbecco’s modified Eagle medium/Nutrient mixture F-12 with 10 % FBS) per well were seeded in a 96-well-microtiter plate. The cells were then incubated in the CO2 incubator at 37 °C. After 24 h, the medium was removed and 80 mm3 of the fresh medium was added. To six wells, 20 mm3 (10 mg/cm3 in 10 % DMSO) of test compounds (in triplicate) was added, while three wells were loaded with 20 mm3 of 10 % DMSO to serve as control. The plate was again incubated for 24 h and at the end of incubation period, the spent medium was replaced with 100 mm3 of the fresh medium and 20 mm3 of MTT (5 mg/cm3) reagent prepared in PBS was added to all the wells. The plates were then wrapped in aluminium foil and returned to the incubator for 2 h until an insoluble purple formazan product is formed. After 2 h, the medium was removed and 200 mm3 of 10 % DMSO was added to all the wells to dissolve the formazan product. Contents were mixed and the absorbance was read at 570 nm.
Anti-inflammatory assay
Griess reagent system (naphthylethylenediamine dihydrochloride, sulphanilamide, and nitrite standard) from promega (USA), Freund’s complete adjuvant (FCA) from GeNei (India), Wright’s stain, sodium chloride, disodium hydrogen phosphate dihydrate, potassium dihydrogen orthophosphate, dimethyl sulfoxide (DMSO) were procured from HIMEDIA (India), methanol was purchased from MERCK (India). Swiss albino mice of both sexes were purchased from Pasteur institute, Shillong, India. The animals were kept in a temperature controlled room under 12 h light and dark cycle. The mice were supplied with food and water ad libitum.
Measurement of paw edema
This method according to Lai et al. [35] with certain modifications was used. Swiss albino mice aged between 10 and 12 weeks (three per group) were injected with 50 mm3 of Fruend’s complete Adjuvant (FCA) into plantar side of left hind paw, to induce inflammation. Diameter of paw edema was then measured at different intervals of time (0, 1, 2, 3, and 24 h) after the administration of the FCA using a calliper. The test compounds (50 mg/kg body weight in DMSO) were then administered 1 h after FCA injection. The percentage inhibition in the diameter of the paw edema was then calculated using the formula \(\frac{a - b}{a} \times 100\) [31], where ‘a’ and ‘b’ denote the mean increase in paw diameter of the control and the drug treated mice respectively. After 24 h, blood was collected by retro-orbital bleeding and stored for nitric oxide assay and differential WBC count. The paws were then excised, weighed and kept in ice-cold normal saline. These were then homogenized in 10 % ice-cold normal saline, centrifuged at 12,000 rpm and the supernatant was collected and stored at −20 °C for nitric oxide assay.
Nitric oxide assay
The amount of nitric oxide produced was calculated using Griess reaction. The sulfanilamide and naphthylethylenediamine dihyrochloride (NED) solution were allowed to equilibrate to room temperature for 15–30 min. To a 96-well microtiter plate, 50 mm3 of the sample was added in triplicate. To these wells, 50 mm3 of the sulfanilamide solution was then added and incubated for 5–10 min at room temperature protected from light. After this incubation period, 50 mm3 of the NED solution was added to all the wells and incubated again in the dark at room temperature. A purple colour develops which was further quantified at 520 nm. Three columns in the 96-well plate were used for the nitrite standard reference curve in which a six serial fold dilution of 100 µM nitrite solution (50 mm3/well) in triplicate was performed to generate the nitrite standard reference curve.
Differential WBCs count
Differential WBCs counts were performed according to the method described by Houwen [36]. Blood film was prepared on glass slides until it dry. The film was fixed in absolute methanol for 30 s. The slides were then stained with Wright’s stain for 2 min in a horizontal position after which Sorensen’s buffer (KH2PO4, Na2HPO4, pH 6.4) was added and mixed. This was allowed to stand for 3 min and then rinsed with distilled water and dried. The slides were then observed under a microscope and differential WBC counting was done.
References
Elnagdi MH, Elmoghayar MRH, Elgemeie GEH (1987) Adv Heterocycl Chem 41:319
Regan AC (2008) Pyrazolo[1,5-c]pyrimidine (73). In: Katritzky AR, Ramsden CA, Scriven EFV, Taylor RJK (eds) Comprehensive heterocyclic chemistry III, vol 11. Elsevier, Oxford, p 577
Youssef S (1997) Monatsh Chem 128:493
Kandeel ZE, Hafez EA, Sleim MA, Abdelatif FM, Elnagdi MH (1995) Heteroat Chem 6:305
Stepaniuk OO, Matviienko VO, Kondratov IS, Vitruk IV, Tolmachev AO (2012) Synthesis 45:925
Kalita U, Kaping S, Nellanant J, Helissey P, Vishwakarma JN (2014) Heteroletters 4:137
Devi AS, Kaping S, Vishwakarma JN (2015) Mol Divers 19:759
Kaping S, Boiss I, Singha LI, Helissey P, Vishwakarma JN (2015) Mol Divers. doi:10.1007/s11030-015-9639-6
Petrov AA, Emelina EE, Selivanov SI (2008) Russ J Org Chem 44:263
Quiroga J, Mejia D, Insuasty B, Abonia R, Nogueras M, Sanchez A, Cobo J (2002) J Heterocycl Chem 39:51
Aggarwal R, Sumran G, Garg N, Aggarwal A (2011) Eur J Med Chem 46:3038
Behbehani H, Ibrahim HM, Makhseed S, Mahmoud H (2011) Eur J Med Chem 46:1813
Compton DR, Carlson KE, Katzenellenbogen JA (2004) J Med Chem 17:5872
Mokhtara M, Saleha TS, Basahel SN (2012) J Mol Catal A 353–354:122
Shaaban MR, Saleh TS, Farag AM (2007) Heterocycles 71:1765
Behbehani H, Ibrahim HM, Makhseed S (2010) Arkivoc 2010(ii):267
Baluja S, Kachhadia N, Solanki A (2013) Open J Org Chem 1:1
Nagargoje D, Mandhane P, Shingote S, Badadhe P, Gill C (2012) Ultrason Sonochem 19:94
Singh BS, Lobo HR, Pinjari DV, Jarag KJ, Pandit AB, Shankarling GS (2013) Ultrason Sonochem 20:287
Jarag KJ, Pinjari DV, Pandit AB, Shankarling GS (2011) Ultrason Sonochem 18:617
Banitaba SH, Safari J, Khalili SD (2013) Ultrason Sonochem 20:401
Bazgir A, Ahadi S, Ghahremanzadeh R, Khavasi HR, Mirzaei P (2010) Ultrason Sonochem 17:447
Khosropour AR (2008) Ultrason Sonochem 15:659
Gopalsamy A, Ciszewski G, Shi M, Berger D, Hu Y, Lee F, Feldberg L, Frommer E, Kim S, Collins K, Wojciechowicz, Mallon R (2009) Bioorg Med Chem Lett 19:6890
Chanda K, Dutta MC, Karim E, Vishwakarma JN (2004) J Indian Chem Soc 81:791
Radl S, Blahovcova M, Tkadlecová M, Havlicek J (2010) Heterocycles 80:1359
Alam A, Imliwati L, Rapthap C, Singh V (2001) Indian J Exp Biol 39:201
Ferrari M, Fomasiero MC, Isetta AM (1990) J Immunol Methods 131:165
Singh A, Malhotra S, Subban R (2008) Int J Integr Biol 3:57
Khan FH (2009) The elements of Immunology. Pearson education, India
Ahmad F, Khan R, Rasheed S (1992) J Islam Acad Sci 5:111
Kang OH, Chae HS, Oh YC, Choi JG, Lee YS, Jang HJ, Kim JH, Kim YC, Sohn DH, Park H, Kwon DY (2008) Am J Chin Med 36:913
Mantovani NV, Germano G, Marchesi F, Locatelli M, Biswas SK (2011) Eur J Immunol 41:2522
Freshney RI (2010) Culture of animal cells: a manual of basic technique and specialized applications, 6th edn. Wiley, New Jersey, p 373
Lai SC, Peng WH, Huang SC, Ho YL, Huang JH, Lai Z, Chang Y (2009) Am J Chin Med 37:573
Houwen B (2000) Lab Hematol 6:1
Acknowledgments
Authors wish to thank Rev. Fr. Dr. Stephen Mavely, Vice Chancellor, Assam Don Bosco University for providing infrastructure for the execution of this work. Authors also wish to express their gratitude to IIT, Guwahati, Tezpur University, Tezpur, SAIF-NEHU, Shillong and SAIF-CDRI, Lucknow. Our thanks are also due to the Department of Biotechnology (DBT), Government of India for a research grant. SK thanks NER-BPMC-DBT, New Delhi for a research fellowship.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kaping, S., Kalita, U., Sunn, M. et al. A facile, regioselective synthesis of pyrazolo[1, 5-a]pyrimidine analogs in the presence of KHSO4 in aqueous media assisted by ultrasound and their anti-inflammatory and anti-cancer activities. Monatsh Chem 147, 1257–1276 (2016). https://doi.org/10.1007/s00706-015-1638-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1638-x