
Abstract
The "Shock and Kill" method is being considered as a potential treatment for eradicating HIV-1 and achieving a functional cure for acquired immunodeficiency syndrome (AIDS). This approach involves using latency-reversing agents (LRAs) to activate human immunodeficiency virus (HIV-1) transcription in latent cells, followed by treatment with antiviral drugs to kill these cells. Although LRAs have shown promise in HIV-1 patient research, their widespread clinical use is hindered by side effects and limitations. In this review, we categorize and explain the mechanisms of these agonists in activating HIV-1 in vivo and discuss their advantages and disadvantages. In the future, combining different HIV-1 LRAs may overcome their respective shortcomings and facilitate a functional cure for HIV-1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acquired immunodeficiency syndrome (AIDS), was first identified in 1981 and is now one of the three major infectious diseases in the world [1]. Human immunodeficiency virus 1 (HIV-1), which causes AIDS, is a retrovirus that contains two copies of the viral single-stranded RNA genome within the core of the virus particle. There are long terminal repeats (LTRs) at each end of the HIV-1 genome, and the genes between the LTRs encode the viral structural proteins Gag, Pol, and Env, as well as regulatory proteins required for viral replication, which include the HIV-1 transcriptional transactivator (Tat), viral structural protein expression regulator (Rev), helper protein negative factor (Nef), viral infection factor (Vif), viral protein U (Vpu), and viral protein R (Vpr).
HIV-1 is a single-stranded RNA virus whose genomic RNA is reverse transcribed to DNA, which then integrates into the genome of CD4+ T cells [2]. During this process, some CD4+ T cells carrying HIV-1 DNA become quiescent. The level of viral transcription in the latent HIV-1 reservoir is extremely low, and almost no virus particles are produced. In the absence of viral proteins, infected cells remain in a stable dormant state for a long time, and their presence is not recognized by the immune system. However, the integrated proviruses still have the ability to replicate and can be activated to produce infectious virions when highly active antiretroviral therapy (HAART) is interrupted. AIDS is very difficult to cure due to the latency reservoir established during the infection. The majority of latent proviruses are believed to be present in resting memory CD4+ T cells, but some proviruses can persist in macrophages [3, 4], dendritic cells [5,6,7], astrocytes, and hematopoietic stem cells [8, 9].
Achieving a functional cure for AIDS has required solutions that can eliminate the latent virus. However, the most commonly used treatment, HAART, is not able to achieve this goal. Although it has been shown to significantly reduce the viral load in the peripheral blood to an undetectable level [10], it cannot completely eradicate latent HIV-1. As a result, patients receiving HAART may have to treat HIV-1 as a chronic disease, and those who stop taking the drugs may experience a resurgence of HIV-1 [11].
To address the problem of the latent HIV-1 viral reservoir, a "Shock and Kill" strategy has been proposed. In this approach, gene expression of the HIV-1 provirus is induced using latency reversing agents (LRAs), and HIV-1-bearing cells are subsequently eliminated by HAART [12]. In this review, we classify and summarize the mechanisms and roles of LRAs for a better understanding of the functions of these molecules in the process of AIDS treatment.
In general, LRAs can be divided into four categories: (1) small-molecule inhibitors that affect histone modifications, (2) DNA methylation inhibitors, (3) small molecules targeting transcriptional regulatory complexes, and (4) small-molecule inhibitors of the NF-κB pathway. Each of these is discussed separately below.
Small-molecule inhibitors that affect histone modifications
Histone deacetylase inhibitors
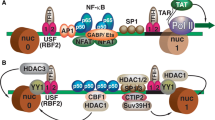
The function of histone deacetylase (HDAC) is to remove acetylation from lysine residues in histones. There are four types of HDAC (I-IV) that differ in their size, number of active centers, cellular localization, and homology to yeast HDAC proteins [13]. It has been reported that the regulation of chromatin function is associated with HIV-1 proliferation ability, and histone acetyltransferase and deacetylase are two chromatin-modifying enzymes that activate latent HIV-1 and are considered targets for treating HIV-1 infection [14]. HDACs can inhibit gene expression through deacetylation [15]. HDAC inhibitors typically interfere with the deacetylase activity of HDAC by blocking its catalytic domain [15, 16], resulting in an increase in histone acetylation levels, overcoming the inhibitory effect of HDACs and stimulating viral transcription in latently infected cells [17, 18] (Fig. 1). HDAC inhibitors can activate latent HIV-1 in vitro, induce the expression of transcripts and antigens, and reduce the reservoir of latent HIV-1 [19, 20]. Acetylation of the nucleosome 1 (NUC-1) protein has been recognized as a critical step in transcription initiation and subsequent activation of the virus [21]. For example, a reversible HDAC inhibitor, trichostatin A (TSA), has been shown to activate latent viruses in HIV-1-infected cell lines [22]. When cells are exposed to TSA, HDAC1 is translocated from NUC-1, increasing histone H4 acetylation, and activating the expression of latent virus [23]. CC-4a (Fig. 1) is another HDAC inhibitor that has anti-deacetylase ability and can activate HIV-1 transcription.

Small-molecule compounds that modify histone methylation and acetylation activate latent HIV transcription. Histone methyltransferase inhibitors (HMT) reduce the level of histone methylation, which significantly activates latent HIV. BIX01294, chaetocin, and HMT inhibitors (HMTi) inhibit G9α, SUV39H1, and HMT, respectively, to facilitate the transcription of pre-viruses. Histone deacetylase inhibitors (HDACi) can inhibit the deacetylation of histones. With the accumulation of histone acetylation, the chromatin structure becomes loose, thus increasing the recruitment of transcription factors such as YY1, CTIP-2, p50-p50 homodimer, and CBF-1 to the HIV-1 5’ LTR. CC-4a is a novel selective inhibitor of histone deacetylase I that shows promise in reactivating latent HIV-1 and has low cytotoxicity
HDAC inhibitors can potentially be used in HIV-1 treatment to clear the latent reservoir [24, 25]. Clinical trials have demonstrated the efficacy of HDAC inhibitors [26] such as vorinostat, panobinostat, romidepsin (RMD), and valproic acid. Of these, RMD shows the highest potency and specificity against class I HDACs [27], and resistance experiments in monocytes have demonstrated the possibility of reducing the HIV-1 reservoir [17]. In a study using an in vitro T-cell model of HIV-1 latency, a sixfold increase in the amount of cellular HIV-1 RNA was observed after exposure to 40 nM RMD for 4 hours [28]. In preclinical models, the HDAC inhibitor entinostat (MS275) has proven successful in causing alterations in HDAC activity and pro-inflammatory cytokine expression levels in mice [29]. However, clinical trials have associated RMD with numerous serious adverse cardiac events [30]. Analysis of patient electrocardiograms revealed that the severity of the adverse effects was primarily influenced by the drug dosage and duration of administration. However, it was also observed that long-term oral administration of low doses of the drug might mitigate this toxicity [31]. In addition, HDAC inhibitors have many other drawbacks, including toxicity to normal cells, resulting in thrombocytopenia and neutropenia, as well as causing nausea, diarrhea, and fatigue [32,33,34]. Furthermore, the effectiveness of HDAC inhibitors in HIV-1 treatment is influenced by various factors, including pharmacokinetics, concentration, and exposure time [19].
Histone methylation inhibitors
Histone methylation not only participates in the structure of chromatin but also plays a critical role in regulating gene expression (Fig. 1). The histone methyltransferase (HMT) Suv39H1 mainly participates in the trimethylation of Lys9 of histone H3 (H3K9me3), leading to the silencing of HIV-1 transcription [35, 36]. An inhibitor of SUV39H1, chaetocin, has shown promise as a drug for reducing the HIV-1 viral reservoir and has been shown to produce a 25-fold increase in latent HIV-1 expression [37, 38] (Fig. 1). The combination of chaetocin and the HDAC inhibitor TSA has been shown to have a strong synergistic effect on HIV-1 expression [37]. Chaetocin does not activate T cells and can therefore be used without causing inflammation and related cytotoxic effects [37]. However, precise control of the concentration of chaetocin is crucial, because if used improperly, it can have deleterious effects on non-target cells [39]. H3K9me3 chromatin immunoprecipitation analysis showed that when SUV39H1 was incubated with HIV-1-infected cells, chaetocin could alter the HIV-1 LTR and significantly reduce trimethylation at H3K9 [40] (Fig. 1). Chaetocin can also promote the recombination of LTR chromatin, leading to reactivation of HIV-1 [37]. G9α is an important enzyme that is responsible for H3K9 dimethylation (H3K9me2). BIX01294, a small-molecule inhibitor of G9α (Fig. 1), when administered peripherally to mice, causes a reduction in H3K9 methylation [41] and activation of viral transcription. Histone methylation inhibitors, especially chaetocin, have shown positive effects in the treatment of HIV-1/AIDS [38].
DNA methylation inhibitors
DNA methylation is a critical epigenetic process that is involved in chromatin structure and gene regulation. Changes in methylation modifications are one of the epigenetic alterations caused by the integration of HIV-1 DNA into the host genome [42]. DNA methyltransferase inhibitors show promise in reversing abnormal DNA methylation processes, which makes them a potential target for reversing HIV-1 latency. By inhibiting DNA methyltransferases, these inhibitors can reactivate HIV-1 and potentially eliminate latent HIV-1 reservoirs. DNA methylation usually occurs at CpG sites, especially around transcription start sites [43] (Fig. 2). Studies have shown that the HIV-1 promoter is often affected by DNA methylation [44], and modification of DNA methylation proteins can affect the interaction between the virus and key transcription factors in the local epigenome, which may inhibit HIV-1 gene expression [45]. Therefore, DNA methyltransferase inhibitors can partially reactivate HIV-1 gene expression [46]. One inhibitor, 5-azacytidine (5-AzaC), a nucleoside analogue of cytosine, is phosphorylated by deoxycytidine kinase (Fig. 2). Administration of low doses of 5-AzaC to clinical patients that result in only minimal levels of DNA methylation [47] nevertheless leads to reactivation of HIV-1 [48]. DNA methyltransferase inhibitors may have significantly different effects on the activation of latent HIV-1, depending on the chromosomal location of the provirus and the epigenetic and transcriptional environment in the cell. MG98 is a specific inhibitor of human DNA methyltransferase 1 (DNMT1) that is generally well tolerated and easy to administer by intermittent intravenous infusion. However, in clinical trials, MG98 was found to cause side effects, including fever, chills, fatigue, and weakness [49,50,51].

DNA methyltransferase (DNMT) inhibitors (DNMTi) affect the transcription of HIV-1 by acting on DNA methylation. They can inhibit the aggregation of DNMT on the HIV-1 LTR, preventing the methylation of two CpG islands near the HIV-1 transcription initiation site, thus relieving the transcriptional suppression caused by the high methylation in the promoter region of HIV-1. 5-aza-2'-deoxycytidine (5-aza-CdR) is also an inhibitor of DNMT
Small molecules targeting transcriptional regulatory complexes
The 7SK snRNP signaling pathway is an important target of current HIV treatment strategies.
The transcription elongation factor P-TEFb was first identified as a regulator of HIV transcription, but subsequently, many other viral and host proteins have been found to interact with P-TEFb [52, 53], mainly by regulating its activity. For example, hexamethylene bisacetamide (HMBA) was originally developed as a treatment for leukemia, but subsequent research has shown that it may also act as a P-TEFb agonist in the treatment of HIV-1 infection. This drug activates the Akt signaling pathway, promoting the release of P-TEFb due to the phosphorylation of HEXIM. Subsequently, Tat recruits active P-TEFb to the vicinity of the trans-activation response (TAR) element, which phosphorylates the carboxy-terminal domain (CTD) of RNA polymerase II and promotes transcriptional elongation of HIV genes (Fig. 3) [54, 55].

Small-molecule compounds affect the activation of HIV-1 by acting on transcriptional regulatory complexes. Dithiothreitol not only causes the degradation of PTEN but also increases the phosphorylation of Akt, causing HEXIM1 to dissociate from the 7SK SNP complex, releasing P-TEFb. HMBA activates Akt through the PI3K pathway, and the phosphorylation of HEXIM-1 can also release P-TEFb. Tat recruits active P-TEFb to the vicinity of the trans-activation response (TAR) element and promotes phosphorylation of the RNA polymerase II (RNA Pol-II) carboxy-terminal domain (CTD), the dissociation of NELF, and activation of DSIF, thereby stimulating transcription elongation. Since JQ1 inhibits the competition between Brd4 and Tat for P-TEFb, it also stimulates transcription
Upregulation of P-TEFb activity can have negative effects on cells [56, 57]. For example, abnormally upregulated P-TEFb may participate in other signaling pathways [58, 59], affecting the cell’s response to DNA damage [58, 60]. This suggests that P-TEFb may play a role in maintaining genome stability.
Bromodomain and extraterminal domain (BET) inhibitors are used to treat HIV-1 infection because they block the binding of bromodomain-containing proteins to P-TEFb. JQ1 is a well-known BET inhibitor that has been shown to reactivate latent HIV-1 provirus [61, 62] (Fig. 3). The mechanism of action of JQ1 involves inhibiting the binding of Brd4 to P-TEFb, thereby initiating gene transcription of the HIV-1 provirus.
BET inhibitors have also shown potential in regulating immune responses, preventing inflammation, and controlling cytokine synthesis [63,64,65]. Several BET inhibitors have been observed to exhibit HIV-1 reactivation properties and are currently undergoing clinical trials. For instance, RVX-208 and PFI-1 are considered potential candidates for anti-HIV-latency therapy. In a study investigating the ability of these two BET inhibitors to activate latent HIV-1 in latently infected Jurkat T cells in vitro as well as in patient-derived resting CD4+ T cells in vivo [66], neither RVX-208 nor PFI-1 elicited widespread and robust T cell activation. At present, I-BET-151 remains the only BET inhibitor being tested for HIV-1 activation in vivo [67]. In general, due to their potency and low toxicity, BET inhibitors hold significant promise as potential candidates for future therapy against reactivated latent HIV-1.
The PTEN inhibitor disulfiram releases HEXIM1 from the 7sk snRNP and recruits P-TEFb from its transcriptionally inactive form to the HIV-1 promoter through the Akt signaling pathway [68, 69] (Fig. 3), stimulating transcriptional elongation and virus production. Disulfiram is a safe and well-tolerated drug that can maintain a low viral load [70, 71]. However, due to significant inter-individual differences in the pharmacokinetics and pharmacodynamics of disulfiram, it is difficult to determine the appropriate dose. In addition, there are significant inter-individual differences in the plasma levels of disulfiram and its metabolites. Participants in a clinical study were able to sustain low levels of virus within two months after disulfiram treatment [70], indicating that higher levels of drug exposure in vivo may have long-term effects on HIV-1 production.
Small-molecule inhibitors of the NF-κB pathway
In the classical NF-κB signaling pathway, when IκB is phosphorylated and degraded [72], most of the released p65/p50 is recruited to the nucleus, promoting gene transcription [73, 74].
Protein kinase C (PKC) agonists are commonly used as drugs to reverse HIV-1 latency by stimulating the classical NF-κB signaling pathway [75, 76] (Fig. 4). Drugs such as prostaglandins and bryostatins induce transcription from the dormant HIV-1 provirus by releasing active NF-κB and promoting its entry into the nucleus to bind to the HIV promoter region. For example, bryostatin-170 isolated from marine invertebrates activates IκBα kinase [77], leading to the phosphorylation and degradation of IκBα. PKC can stimulate P-TEFb activity, thereby reactivating latent HIV-1 with minimal cell toxicity [75]. Recent data from clinical and preclinical trials are highly encouraging and provide strong support for further investigation of PKC agonists as safe and effective LRAs in patients. In a non-human primate model of AIDS, the administration of certain PKC agonists resulted in the reactivation of latent simian immunodeficiency virus (SIV) without any apparent toxicity observed in vivo [78]. Likewise, in a humanized mouse model of AIDS, a synthetic bryostatin analogue not only demonstrated the ability to reactivate latent HIV-1 but also exhibited a potential killing effect against the virus [79]. Although PKC agonists have proven to be highly effective in reversing HIV-1 latency, their clinical application has been hindered by their side effects and cytotoxicity [76, 80, 81], which would have to be controlled before they can be used in killing strategies against HIV-1 (see Table 1).

Small-molecule inhibitors affect the latency of HIV-1 by acting on the NF-κB signaling pathway. TNF-α is recognized by TNF-α receptor and gradually recruits TRADD, TNF receptor-associated factor TRAF, and receptor-interacting protein (RIP), leading to activation of the NF-κB signaling pathway. NF-κB binds to the NF-κB binding site in the LTR promoter, thereby stimulating transcription. In addition, PKC agonists can also promote the entry of NF-κB into the nucleus. CC-4a as a novel HDAC inhibitor that can release IKB-α from the IKB-α/NF-κB complex and activate proviruses through the NF-κB signaling pathway
Tumor necrosis factor (TNF) appears to be a major driver of HIV-1 transcription early in the disease [82]. While TNF is recognized by its receptor protein (Fig. 4), the TNFR-associated death domain (TRADD) interacts with the cytoplasmic death domain of TNFR1 through its own death domain to affect the secretion of growth factors and cell proliferation. Then, TNFR2 interacts with the TRAF protein, and receptor interaction proteins (RIPs) are recruited, activating several signaling cascades and leading to the activation of transcription factors to favor the binding of p65/p50 NF-κB complexes to binding sites present in the HIV-1 LTR promoter [83]. Moreover, the close interaction between TNF and HIV-1 Nef persists throughout the course of the disease. Studies have shown that Nef interacts directly with proteins of the TRAF family (TRAF2, TRAF5, and TRAF6) to stimulate HIV-1 replication in individual cells and primary macrophages [84, 85]. Although TNF has been used to reduce the viral repertoire, it has limited ability to reactivate viruses and is virulent [86, 87].
The high degree variability of HIV-1 virus makes achieving a functional cure through a single treatment plan difficult. However, combining HAART with LRAs is a promising approach to help HIV-1 patients overcome the disease.
To overcome the challenges of curing HIV-1 infections, researchers are exploring various strategies to target viral reservoirs in T cells, adipose tissue, bone marrow, the central nervous system, and gut-associated lymphoid tissue [88]. One approach is the use of small molecules that can reactivate latent HIV-1 infection in different anatomical locations [89,90,91]. However, much research is still needed to optimize the use of these drugs and to evaluate their safety and efficacy.
The "Shock and Kill" strategy involves using a combination of small-molecule LARs, which can enhance treatment specificity and reduce cellular toxicity, and HAART. Although no single LRA is perfect, using different agents in combination can help to mitigate the drawbacks of each drug.
Activation of proviruses activates the immune system, which contributes to the "Shock and Kill" strategy. Several LRAs have been documented to cause widespread activation of proviruses. For instance, chidamide, a benzamide-based HDAC inhibitor, has been demonstrated to successfully reactivate latent HIV-1 in cellular models and in primary CD4+ T cells from HIV-1-infected individuals [92, 93]. Such findings highlight the potential of LRAs in aiding HIV-1 eradication strategies by activating latent viral reservoirs and rendering them vulnerable to immune recognition and elimination.
Through oral administration of chidamide and using HIV-1 DNA and HIV-1 RNA levels in patients for reference, researchers have observed a notable enhancement in the HIV-1-specific cellular immune response accompanied by a modest 37.7% reduction in cell-associated HIV-1 DNA levels [94]. Initially, at the initial dose, the researchers did not observe a significant increase in cell-associated HIV-1 RNA. However, as the dose was increased, changes became apparent. It is important to emphasize that individual LRAs have a tendency to over-activate the immune system, which can lead to various side effects. Combining multiple LRAs might help to minimize these side effects.
In summary, the combination of HAART and LRAs represents a promising approach to HIV-1 treatment. However, given the significant challenges posed by the variability of the virus, it is crucial to continue developing new and more-effective therapies.
Change history
16 February 2024
A Correction to this paper has been published: https://doi.org/10.1007/s00705-024-05972-1
References
Menéndez-Arias L, Delgado R (2022) Update and latest advances in antiretroviral therapy. Trends Pharmacol Sci 43(1):16–29
Chen J, Zhou T, Zhang Y, Luo S, Chen H, Chen D, Li C, Li W (2022) The reservoir of latent HIV. Front Cell Infect Microbiol 12:945956
Kumar A, Abbas W, Herbein G (2014) HIV-1 latency in monocytes/macrophages. Viruses 6(4):1837–1860
Kruize Z, Kootstra NA (2019) The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front Microbiol 10:2828
Balan S, Saxena M, Bhardwaj N (2019) Dendritic cell subsets and locations. Int Rev Cell Mol Biol 348:1–68
Yin X, Chen S, Eisenbarth SC (2021) Dendritic Cell Regulation of T Helper Cells. Annu Rev Immunol 39:759–790
Yu HJ, Reuter MA, McDonald D (2008) HIV traffics through a specialized, surface-accessible intracellular compartment during trans-infection of T cells by mature dendritic cells. PLoS Pathog 4 (8), e1000134
Carter CC, McNamara LA, Onafuwa-Nuga A, Shackleton M, Riddell Jt, Bixby D, Savona MR, Morrison SJ, Collins KL (2011) HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe 9(3):223–234
Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell Jt, Bixby D, Savona MR, Collins KL (2010) HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med 16(4):446–451
Bandera A, Gori A, Clerici M, Sironi M (2019) Phylogenies in ART: HIV reservoirs, HIV latency and drug resistance. Curr Opin Pharmacol 48:24–32
Baba M (2004) [Recent progress in anti-HIM-1 research]. Uirusu 54(1):59–66
Deeks SG (2012) Shock and kill. Nature 487(7408):439–440
Boateng AT, Abaidoo-Myles A, Bonney EY, Kyei GB (2022) Isoform-Selective Versus Nonselective Histone Deacetylase Inhibitors in HIV Latency Reversal. AIDS Res Hum Retroviruses 38(8):615–621
Varier RA, Kundu TK (2006) Chromatin modifications (acetylation/ deacetylation/ methylation) as new targets for HIV therapy. Curr Pharm Design 12(16):1975–1993
Archin NM, Kirchherr JL, Sung JA, Clutton G, Sholtis K, Xu Y, Allard B, Stuelke E, Kashuba AD, Kuruc JD, Eron J, Gay CL, Goonetilleke N, Margolis DM (2017) Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J Clin Investig 127(8):3126–3135
Glozak MA, Seto E (2007) Histone deacetylases and cancer. Oncogene 26(37):5420–5432
Vandergeeten C, Quivy V, Moutschen M, Van Lint C, Piette J, Legrand-Poels S (2007) HIV-1 protease inhibitors do not interfere with provirus transcription and host cell apoptosis induced by combined treatment TNF-alpha + TSA. Biochem Pharmacol 73(11):1738–1748
Lin S, Zhang Y, Ying H, Zhu H (2011) HIV-1 reactivation induced by apicidin involves histone modification in latently infected cells. Curr HIV Res 9(4):202–208
Newhard W, Patel M, Cassaday J, Ballard J, Squadroni B, Wu G, Liu J, Yu W, Kozlowski J, Zuck P, Howell B, Hazuda D, Vargo R, Barnard R (2021) In Vitro Pharmacokinetic/Pharmacodynamic Modeling of HIV Latency Reversal by Novel HDAC Inhibitors Using an Automated Platform. SLAS discovery: advancing life sciences R & D 26(5):642–654
Garrido C, Tolstrup M, Søgaard OS, Rasmussen TA, Allard B, Soriano-Sarabia N, Archin NM, Margolis DM (2019) In-vivo administration of histone deacetylase inhibitors does not impair natural killer cell function in HIV + individuals. AIDS 33(4):605–613
Kiefer HL, Hanley TM, Marcello JE, Karthik AG, Viglianti GA (2004) Retinoic acid inhibition of chromatin remodeling at the human immunodeficiency virus type 1 promoter. Uncoupling of histone acetylation and chromatin remodeling. J Biol Chem 279(42):43604–43613
Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401(6749):188–193
He G, Margolis DM (2002) Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol Cell Biol 22(9):2965–2973
Iveland TS, Hagen L, Sharma A, Sousa MML, Sarno A, Wollen KL, Liabakk NB, Slupphaug G (2020) HDACi mediate UNG2 depletion, dysregulated genomic uracil and altered expression of oncoproteins and tumor suppressors in B- and T-cell lines. J translational Med 18(1):159
Bose P, Dai Y, Grant S (2014) Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol Ther 143(3):323–336
Jeng MY, Ali I, Ott M (2015) Manipulation of the host protein acetylation network by human immunodeficiency virus type 1. Crit Rev Biochem Mol Biol 50(4):314–325
Beliakova-Bethell N, Mukim A, White CH, Deshmukh S, Abewe H, Richman DD, Spina CA (2019) Histone deacetylase inhibitors induce complex host responses that contribute to differential potencies of these compounds in HIV reactivation. J Biol Chem 294(14):5576–5589
McMahon DK, Zheng L, Cyktor JC, Aga E, Macatangay BJ, Godfrey C, Para M, Mitsuyasu RT, Hesselgesser J, Dragavon J, Dobrowolski C, Karn J, Acosta EP, Gandhi RT, Mellors JW (2021) Phase 1/2 Randomized, Placebo-Controlled Trial of Romidespin in Persons With HIV-1 on Suppressive Antiretroviral Therapy. J Infect Dis 224(4):648–656
Zhang H, Li X, Zhang Q, Yang F, Chu X, Zhang D, Wang L, Gong Z (2017) Role of histone deacetylase expression levels and activity in the inflammatory responses of patients with chronic hepatitis B. Mol Med Rep 15(5):2744–2752
Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M (2006) Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res 12(13):3997–4003
Molife R, Fong P, Scurr M, Judson I, Kaye S, de Bono J (2007) HDAC inhibitors and cardiac safety. Clin Cancer Res 13 (3), 1068; author reply 1068-9
Bai M, Cui M, Li M, Yao X, Wu Y, Zheng L, Sun L, Song Q, Wang S, Liu L, Yu C, Huang Y (2021) Discovery of a novel HDACi structure that inhibits the proliferation of ovarian cancer cells in vivo and in vitro. Int J Biol Sci 17(13):3493–3507
Kroesen M, Gielen P, Brok IC, Armandari I, Hoogerbrugge PM, Adema GJ (2014) HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 5(16):6558–6572
Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL (2010) Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals (Basel Switzerland) 3(9):2751–2767
du Chéné I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M (2007) Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. Embo j 26(2):424–435
Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O (2007) Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. Embo j 26(2):412–423
Bernhard W, Barreto K, Saunders A, Dahabieh MS, Johnson P, Sadowski I (2011) The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett 585(22):3549–3554
Bouchat S, Gatot JS, Kabeya K, Cardona C, Colin L, Herbein G, De Wit S, Clumeck N, Lambotte O, Rouzioux C, Rohr O, Van Lint C (2012) Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS 26(12):1473–1482
Rombo R, Weiher H, Schmidt-Wolf IG (2016) Effect of chaetocin on renal cell carcinoma cells and cytokine-induced killer cells. German medical science: GMS e-journal 14, Doc04
Imai K, Togami H, Okamoto T (2010) Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J Biol Chem 285(22):16538–16545
Chase KA, Feiner B, Ramaker MJ, Hu E, Rosen C, Sharma RP (2019) Examining the effects of the histone methyltransferase inhibitor BIX-01294 on histone modifications and gene expression in both a clinical population and mouse models. PLoS ONE 14 (6), e0216463
Zhang X, Justice AC, Hu Y, Wang Z, Zhao H, Wang G, Johnson EO, Emu B, Sutton RE, Krystal JH, Xu K (2016) Epigenome-wide differential DNA methylation between HIV-infected and uninfected individuals. Epigenetics 11(10):750–760
van der Wijst MG, Venkiteswaran M, Chen H, Xu GL, Plösch T, Rots MG (2015) Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics 10(8):671–676
Pierard V, Guiguen A, Colin L, Wijmeersch G, Vanhulle C, Van Driessche B, Dekoninck A, Blazkova J, Cardona C, Merimi M, Vierendeel V, Calomme C, Nguyên TL, Nuttinck M, Twizere JC, Kettmann R, Portetelle D, Burny A, Hirsch I, Rohr O, Van Lint C (2010) DNA cytosine methylation in the bovine leukemia virus promoter is associated with latency in a lymphoma-derived B-cell line: potential involvement of direct inhibition of cAMP-responsive element (CRE)-binding protein/CRE modulator/activation transcription factor binding. J Biol Chem 285(25):19434–19449
Bouchat S, Delacourt N, Kula A, Darcis G, Van Driessche B, Corazza F, Gatot JS, Melard A, Vanhulle C, Kabeya K, Pardons M, Avettand-Fenoel V, Clumeck N, De Wit S, Rohr O, Rouzioux C, Van Lint C (2016) Sequential treatment with 5-aza-2'-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol Med 8(2):117–138
Abdel-Hameed EA, Ji H, Shata MT (2016) HIV-Induced Epigenetic Alterations in Host Cells. Adv Exp Med Biol 879:27–38
Oran B, de Lima M, Garcia-Manero G, Thall PF, Lin R, Popat U, Alousi AM, Hosing C, Giralt S, Rondon G, Woodworth G, Champlin RE (2020) A phase 3 randomized study of 5-azacitidine maintenance vs observation after transplant in high-risk AML and MDS patients. Blood Adv 4(21):5580–5588
Fernandez G, Zeichner SL (2010) Cell line-dependent variability in HIV activation employing DNMT inhibitors. Virol J 7:266
Stewart DJ, Donehower RC, Eisenhauer EA, Wainman N, Shah AK, Bonfils C, MacLeod AR, Besterman JM, Reid GK (2003) A phase I pharmacokinetic and pharmacodynamic study of the DNA methyltransferase 1 inhibitor MG98 administered twice weekly. Annals of oncology: official journal of the European Society for Medical Oncology 14(5):766–774
Winquist E, Knox J, Ayoub JP, Wood L, Wainman N, Reid GK, Pearce L, Shah A, Eisenhauer E (2006) Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: a National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Invest New Drugs 24(2):159–167
Plummer R, Vidal L, Griffin M, Lesley M, de Bono J, Coulthard S, Sludden J, Siu LL, Chen EX, Oza AM, Reid GK, McLeod AR, Besterman JM, Lee C, Judson I, Calvert H, Boddy AV (2009) Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin Cancer Res 15(9):3177–3183
Zaborowska J, Isa NF, Murphy S (2016) P-TEFb goes viral. BioEssays 38(Suppl 1):S75–85
Franco LC, Morales F, Boffo S, Giordano A (2018) CDK9: A key player in cancer and other diseases. J Cell Biochem 119(2):1273–1284
Chen D, Wang H, Aweya JJ, Chen Y, Chen M, Wu X, Chen X, Lu J, Chen R, Liu M (2016) HMBA Enhances Prostratin-Induced Activation of Latent HIV-1 via Suppressing the Expression of Negative Feedback Regulator A20/TNFAIP3 in NF-κB Signaling. Biomed Res Int 2016. 5173205
Contreras X, Barboric M, Lenasi T, Peterlin BM (2007) HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog 3(10):1459–1469
Cheng Y, Jin Z, Agarwal R, Ma K, Yang J, Ibrahim S, Olaru AV, David S, Ashktorab H, Smoot DT, Duncan MD, Hutcheon DF, Abraham JM, Meltzer SJ, Mori Y (2012) LARP7 is a potential tumor suppressor gene in gastric cancer. Lab Invest 92(7):1013–1019
Dey A, Chao SH, Lane DP (2007) HEXIM1 and the control of transcription elongation: from cancer and inflammation to AIDS and cardiac hypertrophy. Cell Cycle 6(15):1856–1863
Claudio PP, Cui J, Ghafouri M, Mariano C, White MK, Safak M, Sheffield JB, Giordano A, Khalili K, Amini S, Sawaya BE (2006) Cdk9 phosphorylates p53 on serine 392 independently of CKII. J Cell Physiol 208(3):602–612
Simone C, Bagella L, Bellan C, Giordano A (2002) Physical interaction between pRb and cdk9/cyclinT2 complex. Oncogene 21(26):4158–4165
Fujinaga K (2020) P-TEFb as A Promising Therapeutic Target. Molecules 25 (4)
Gohda J, Suzuki K, Liu K, Xie X, Takeuchi H, Inoue JI, Kawaguchi Y, Ishida T (2018) BI-2536 and BI-6727, dual Polo-like kinase/bromodomain inhibitors, effectively reactivate latent HIV-1. Sci Rep 8(1):3521
Darcis G, Kula A, Bouchat S, Fujinaga K, Corazza F, Ait-Ammar A, Delacourt N, Melard A, Kabeya K, Vanhulle C, Van Driessche B, Gatot JS, Cherrier T, Pianowski LF, Gama L, Schwartz C, Vila J, Burny A, Clumeck N, Moutschen M, De Wit S, Peterlin BM, Rouzioux C, Rohr O, Van Lint C (2015) An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1 + JQ1 and Ingenol-B + JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog 11(7):e1005063
Sánchez-Ventura J, Amo-Aparicio J, Navarro X, Penas C (2019) BET protein inhibition regulates cytokine production and promotes neuroprotection after spinal cord injury. J Neuroinflamm 16(1):124
Domínguez-Andrés J, Ferreira AV, Jansen T, Smithers N, Prinjha RK, Furze RC, Netea MG (2019) Bromodomain inhibitor I-BET151 suppresses immune responses during fungal-immune interaction. Eur J Immunol 49(11):2044–2050
Salahong T, Schwartz C, Sungthong R, Are BET (2021) Inhibitors yet Promising Latency-Reversing Agents for HIV-1 Reactivation in AIDS Therapy? Viruses 13 (6)
Lu P, Shen Y, Yang H, Wang Y, Jiang Z, Yang X, Zhong Y, Pan H, Xu J, Lu H, Zhu H (2017) BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci Rep 7(1):16646
Li G, Zhang Z, Reszka-Blanco N, Li F, Chi L, Ma J, Jeffrey J, Cheng L, Su L (2019) Specific Activation in Vivo of HIV-1 by a Bromodomain Inhibitor from Monocytic Cells in Humanized Mice under Antiretroviral Therapy. J Virol 93:12
Doyon G, Zerbato J, Mellors JW, Sluis-Cremer N (2013) Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 27(2):F7–f11
Xing S, Bullen CK, Shroff NS, Shan L, Yang HC, Manucci JL, Bhat S, Zhang H, Margolick JB, Quinn TC, Margolis DM, Siliciano JD, Siliciano RF (2011) Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4 + T cell model without inducing global T cell activation. J Virol 85(12):6060–6064
Spivak AM, Andrade A, Eisele E, Hoh R, Bacchetti P, Bumpus NN, Emad F, Buckheit R 3rd;, McCance-Katz EF, Lai J, Kennedy M, Chander G, Siliciano RF, Siliciano JD, Deeks SG (2014) A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin Infect diseases: official publication Infect Dis Soc Am 58(6):883–890
Lee SA, Elliott JH, McMahon J, Hartogenesis W, Bumpus NN, Lifson JD, Gorelick RJ, Bacchetti P, Deeks SG, Lewin SR, Savic RM (2019) Population Pharmacokinetics and Pharmacodynamics of Disulfiram on Inducing Latent HIV-1 Transcription in a Phase IIb Trial. Clin Pharmacol Ther 105(3):692–702
Deng L, Zeng Q, Wang M, Cheng A, Jia R, Chen S, Zhu D, Liu M, Yang Q, Wu Y, Zhao X, Zhang S, Liu Y, Yu Y, Zhang L, Chen X (2018) Suppression of NF-κB Activity: A Viral Immune Evasion Mechanism. Viruses 10:8
Khan SZ, Gasperino S, Zeichner SL (2019) Nuclear Transit and HIV LTR Binding of NF-κB Subunits Held by IκB Proteins: Implications for HIV-1 Activation. Viruses 11 (12)
Fernandez G, Zaikos TD, Khan SZ, Jacobi AM, Behlke MA, Zeichner SL (2013) Targeting IκB proteins for HIV latency activation: the role of individual IκB and NF-κB proteins. J Virol 87(7):3966–3978
Mbonye U, Leskov K, Shukla M, Valadkhan S, Karn J (2021) Biogenesis of P-TEFb in CD4 + T cells to reverse HIV latency is mediated by protein kinase C (PKC)-independent signaling pathways. PLoS Pathog 17 (9), e1009581
Okoye AA, Fromentin R, Takata H, Brehm JH, Fukazawa Y, Randall B, Pardons M, Tai V, Tang J, Smedley J, Axthelm M, Lifson JD, Picker LJ, Favre D, Trautmann L, Chomont N (2022) The ingenol-based protein kinase C agonist GSK445A is a potent inducer of HIV and SIV RNA transcription. PLoS Pathog 18 (1), e1010245
Gutiérrez C, Serrano-Villar S, Madrid-Elena N, Pérez-Elías MJ, Martín ME, Barbas C, Ruipérez J, Muñoz E, Muñoz-Fernández MA, Castor T, Moreno S (2016) Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 30(9):1385–1392
Gama L, Abreu CM, Shirk EN, Price SL, Li M, Laird GM, Pate KA, Wietgrefe SW, O'Connor SL, Pianowski L, Haase AT, Van Lint C, Siliciano RF, Clements JE (2017) Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 31(1):5–14
Marsden MD, Loy BA, Wu X, Ramirez CM, Schrier AJ, Murray D, Shimizu A, Ryckbosch SM, Near KE, Chun TW, Wender PA, Zack JA (2017) In vivo activation of latent HIV with a synthetic bryostatin analog effects both latent cell kick and kill in strategy for virus eradication. PLoS Pathog 13 (9), e1006575
Beans EJ, Fournogerakis D, Gauntlett C, Heumann LV, Kramer R, Marsden MD, Murray D, Chun TW, Zack JA, Wender PA (2013) Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc Natl Acad Sci USA 110(29):11698–11703
DeChristopher BA, Loy BA, Marsden MD, Schrier AJ, Zack JA, Wender PA (2012) Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat Chem 4(9):705–710
Clerici M, Galli M, Bosis S, Gervasoni C, Moroni M, Norbiato G (2000) Immunoendocrinologic abnormalities in human immunodeficiency virus infection. Ann N Y Acad Sci 917:956–961
Nakano H (2004) Signaling crosstalk between NF-kappaB and JNK. Trends Immunol 25(8):402–405
Khan KA, Abbas W, Varin A, Kumar A, Di Martino V, Dichamp I, Herbein G (2013) HIV-1 Nef interacts with HCV Core, recruits TRAF2, TRAF5 and TRAF6, and stimulates HIV-1 replication in macrophages. J Innate Immun 5(6):639–656
Herbein G (2016) HIV-1 Nef: An Intimate Interplay. EBioMedicine 13:25–26
Reuse S, Calao M, Kabeya K, Guiguen A, Gatot JS, Quivy V, Vanhulle C, Lamine A, Vaira D, Demonte D, Martinelli V, Veithen E, Cherrier T, Avettand V, Poutrel S, Piette J, de Launoit Y, Moutschen M, Burny A, Rouzioux C, De Wit S, Herbein G, Rohr O, Collette Y, Lambotte O, Clumeck N, Van Lint C (2009) Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: implications for treatment of latent infection. PLoS ONE 4 (6), e6093
Cummins NW, Badley AD (2010) Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis 1(11):e99
Fletcher CV, Dyavar SR, Acharya A, Byrareddy SN (2021) The Contributions of Clinical Pharmacology to HIV Cure Research. Clin Pharmacol Ther 110(2):334–345
Kim Y, Anderson JL, Lewin SR (2018) Getting the Kill into Shock and Kill: Strategies to Eliminate Latent HIV. Cell Host Microbe 23(1):14–26
Garcia-Vidal E, Badia R, Pujantell M, Castellví M, Felip E, Clotet B, Riveira-Muñoz E, Ballana E, Esté JA (2019) Dual effect of the broad spectrum kinase inhibitor midostaurin in acute and latent HIV-1 infection. Antiviral Res 168:18–27
Cary DC, Fujinaga K, Peterlin BM (2016) Molecular mechanisms of HIV latency. J Clin Investig 126(2):448–454
Yang W, Sun Z, Hua C, Wang Q, Xu W, Deng Q, Pan Y, Lu L, Jiang S (2018) Chidamide, a histone deacetylase inhibitor-based anticancer drug, effectively reactivates latent HIV-1 provirus. Microbes Infect 20(9–10):626–634
Kuai Q, Lu X, Qiao Z, Wang R, Wang Y, Ye S, He M, Wang Y, Zhang T, Wu H, Ren S, Yu Q (2018) Histone deacetylase inhibitor chidamide promotes reactivation of latent human immunodeficiency virus by introducing histone acetylation. J Med Virol 90(9):1478–1485
Li JH, Ma J, Kang W, Wang CF, Bai F, Zhao K, Yao N, Liu Q, Dang BL, Wang BW, Wei QQ, Kang WZ, Sun YT (2020) The histone deacetylase inhibitor chidamide induces intermittent viraemia in HIV-infected patients on suppressive antiretroviral therapy. HIV Med 21(11):747–757
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81802975), the Zhejiang Provincial Natural Science Foundation (Grant No. LY24H190004), and the Zhejiang Medical and Health Science and Technology Project (Grant No. 2019KY361, 2020KY101).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Communicated by Carolina Scagnolari
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Affiliations of authors updated. Funding infomation ammended.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhou, Z., Jiang, Y., Zhong, X. et al. Characteristics and mechanisms of latency-reversing agents in the activation of the human immunodeficiency virus 1 reservoir. Arch Virol 168, 301 (2023). https://doi.org/10.1007/s00705-023-05931-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00705-023-05931-2