
Abstract
Here, we report the complete genome sequence of chrysanthemum mosaic-associated virus (ChMaV), a putative new member of the genus Emaravirus. The ChMaV genome comprises seven negative-sense RNA segments (RNAs 1, 2, 3a, 3b, 4, 5, and 6), and the amino acid sequences of its RNA-dependent RNA polymerase (RNA1), glycoprotein precursor (RNA2), nucleocapsid protein (RNA3), and movement protein (RNA4) showed the closest relationship to pear chlorotic leaf spot-associated virus. Phylogenetic analysis showed that it clusters with emaraviruses whose host plants originate from East Asia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The viruses of the genus Emaravirus have multipartite RNA genomes, each of which encodes a single protein. Of these, RNA1, RNA2, RNA3, and RNA4 encode RNA-dependent RNA polymerase (RdRp), glycoprotein precursor (GP), nucleocapsid protein (NP), and movement protein (MP), respectively [1]. Since 2007, more than 20 emaraviruses have been identified [2,3,4], and some of these viruses are known to be transmitted by a specific eriophyid mite [5,6,7]. Chrysanthemum is one of the most important ornamental crops worldwide. In Japan, flowers are produced not only for ornamental purposes but also as food. It is believed that domestic chrysanthemum cultivars were introduced into Japan from China in the fifth to eighth centuries.
The occurrence of leaf chlorotic ringspot or mosaic symptoms of chrysanthemum, known as “Mon-mon” disease, has been observed since the 1960s. It has long been believed that the symptoms are a kind of physiological abnormality caused by infestation with an eriophyid mite, Paraphytoptus kikus [8,9,10]. However, using reverse transcription polymerase chain reaction (RT-PCR) with newly developed emaravirus-specific degenerate primers in combination with a high-throughput sequencing (HTS) approach (see below), we obtained a partial nucleotide sequence from symptomatic leaves showing sequence similarity to the RNA1 of emaraviruses and found a correlation between the symptoms and detection of the emaravirus-like sequence. The putative emaravirus was tentatively named "chrysanthemum mosaic-associated virus" (ChMaV) [11].
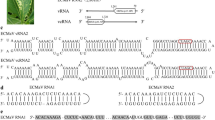
To determine the complete nucleotide sequence of ChMaV, symptomatic leaves of chrysanthemum (cv. Kin-nishiki, an edible flower variety) were collected from a greenhouse in Toyohashi, Aichi, in 2018. Total RNA was extracted from one leaf sample exhibiting representative mosaic symptoms, using the rapid CTAB method [12], and this sample was subjected to HTS as described previously [7]. Out of a total of 127,707 contigs (maximum length, 14,620 nt; minimum length, 125 nt; mean length, 409 nt) derived from 33,954,546 paired-end, 150-bp reads, sequences containing open reading frames (ORFs) encoding proteins with amino acid sequence similarity to emaravirus proteins were identified by BLASTx and DELTA-BLAST searches [13, 14]. Both searches detected seven contigs whose length and amino acid sequences showed the highest similarity to the following emaravirus proteins: 7,144 nt, 2,054 nt, 1,307 nt, 911 nt, 1,353 nt, 1,164 nt, and 1,761 nt; 44.4% to pear chlorotic leaf spot-associated virus (PCLSaV) P1 [15], 33.0% to PCLSaV P2, 40.4% to PCLSaV P3, 38.9% to PCLSaV P3, 59.3% to PCLSaV P4, 24.8% to perilla mosaic virus (PerMV) P5 [7], and 28.6% to rose rosette virus (RRV) P5 [16], respectively. The complete nucleotide sequences of these RNAs were determined via direct sequencing using specific primers designed based on the sequences of the contigs (Supplementary Table S1), and the terminal sequences were determined by 5′ and 3′ rapid amplification of cDNA ends using 5′ or 3′ full RACE Core Sets, (Takara Bio). The seven RNAs contained conserved 13- and 11- nt sequences at the 5′ and 3′ terminus (5′-AGUAGUGUUCUCC......AACACACUACU-3′), respectively, which is a common feature of the RNA segments of emaraviruses [1]. Based on the standard nomenclature for emaravirus RNAs and protein homologs, these seven RNAs were named RNA1, RNA2, RNA3a, RNA3b, RNA4, RNA5, and RNA6 (Fig. 1). To identify other possible segments, RT-PCR was performed with the primers BamHI-ChMaV-5′ter-11mer-fw and BamHI-ChMaV-3′ter-11mer-rv (Supplementary Table S1), which contain a BamHI site followed by the conserved 11-nt sequence at the 5′- and 3′-terminus, respectively, and the amplification products were cloned and sequenced. Of the 48 clones sequenced, 44 were from previously determined RNA segments (30 from RNA3a, seven from RNA3b, three from RNA4, two from RNA5, one from RNA6, and one that included sequences from both RNA5 and RNA6), and the others did not encode proteins; thus, no other emaravirus-like segments were found.

Schematic representation of the organization of chrysanthemum mosaic-associated virus (ChMaV) genomic RNAs. Open reading frames are represented by rectangles, and those with amino acid sequence similarity to one another have the same color. The 13- and 11-nucleotide-conserved sequences at the 5′ and 3′ terminus on each segment are represented by blue and yellow boxes, respectively. Drawings are not to scale.
We previously reported that a partial nucleotide sequence of RNA1 was specifically detected in five symptomatic leaves, but not in five asymptomatic leaves, by RT-PCR [11]. Primer sets targeting RNAs 2, 3a, 3b, 4, 5, and 6 were designed and used for diagnosis (Supplementary Table S1). All of these RNAs were specifically detected in the five symptomatic leaves, whereas no RNAs were detected in the five asymptomatic leaf samples (data not shown), indicating that all of the segments identified are indeed associated with the mosaic symptoms.
RNA1 is 7,093 nt long and contains an ORF (nt positions 7,029 to 121) that encodes P1, a putative RdRp of 2,302 aa. The presence of the seven amino acid sequence motifs (Pre-A, F, and A–E) that are conserved in emaravirus RdRps was confirmed, as reported previously [11]. RNA2 is 2,054 nt long and contains an ORF (nt 1,984 to 83) that encodes P2, a putative GP of 633 aa. As with the P2 of other emaraviruses, ChMaV P2 contains a phlebovirus glycoprotein motif (G490CYSCTQG).
Like some other emaraviruses, the genome of ChMaV contains two derivatives of RNA3: RNA3a and RNA3b. RNA3a and RNA3b are 1,390 and 1,215 nt long, respectively, and each encodes an NP: P3a (ORF at nt 1,316 to 525) and P3b (nt 1,148 to 357), both of which are 263 amino acids in length. The amino acid sequence identity between ChMaV P3a and P3b is 48.3%, which is much lower than that between the P3s of High Plains wheat mosaic virus (HPWMoV) (88.9%) and PerMV (83.0%) [7, 17] (Fig. 2C). Of the three conserved motifs in P3 of emaraviruses [18], ChMaV P3a and P3b contain “N148RLA” and “G169XEX”, but “NX2SXNX3A” is missing. The lack of NX2SXNX3A is also observed in the P3s of PCLSaV, PerMV, CjaEV1, and CjaEV2 [7, 15, 19].

Phylogenetic relationships of chrysanthemum mosaic-associated virus (ChMaV), emaraviruses, and selected viruses of the order Bunyavirales. Analyses were performed using the amino acid sequence alignments for a RNA-dependent RNA polymerase (RdRp), b glycoprotein precursor (GP), c nucleocapsid protein (NP), and d movement protein (MP). The numbers at each node represent bootstrap values in percent, and those < 60% were omitted. The scale bars represent the number of residue substitutions per site. The GenBank accession numbers of the proteins used for phylogenetic analysis are shown. Sequences of maize mosaic virus (MMV), a member of the genus Alphanucleorhabdovirus, order Mononegavirales, rice stripe virus (RSV, genus Tenuivirus, family Phenuiviridae), and tomato spotted wilt virus (TSWV, genus Orthotospovirus, family Tospoviridae), of the order Bunyavirales, were used as outgroups. The members of the genus Emaravirus included actinidia chlorotic ringspot-associated virus (AcCRaV), actinidia emaravirus 2 (AcEV-2), aspen mosaic-associated virus (AsMaV), alfalfa ringspot-associated virus (ARaV), blackberry leaf mottle-associated virus (BLMaV), Camelia japonica-associated emaravirus (CjaEV) 1 and CjaEV2, European mountain ash ringspot-associated virus (EMARaV), fig mosaic virus (FMV), High Plains wheat mosaic virus (HPWMoV), lilac chlorotic ringspot-associated virus (LiCRaV), jujube yellow mottle-associated virus (JYMaV), pear chlorotic leaf spot-associated virus (PCLSaV), perilla mosaic virus (PerMV), pistacia virus B (PiVB), blue palo verde broom virus (PVBV), pigeonpea sterility mosaic virus (PPSMV-1), pigeonpea sterility mosaic virus 2 (PPSMV-2), raspberry leaf blotch virus (RLBV), rose rosette virus (RRV), redbud yellow ringspot-associated virus (RYRSaV), and ti ringspot-associated virus (TiRSaV). The subgroups of emaraviruses based on the trees for NP and MP are indicated by solid lines. The proteins of ChMaV are indicated by arrowheads.
RNA4 is 1,303 nt long and contains an ORF (nt 1,233 to 310) that encodes the P4 of 307 amino acids with a predicted molecular mass of 35.4 kDa, a putative MP. ChMaV P4 contains a conserved D110XR motif, which is present in the MPs of other emaraviruses [20]. However, the most conserved WKT motif is substituted by Y174KV, which is similar to YKT in the P4 of PCLSaV [15].
RNA5 is 1,154 nt long and contains an ORF (nt 1,098 to 574) that encodes P5, a 174-amino acid protein of 20.1 kDa. ChMaV P5 exhibits amino acid sequence similarity to the P5 protein of PerMV and the P7 protein of HPWMoV, a suppressor of RNA silencing [21, 22]. RNA6 is 1,707 nt long and contains an ORF (nt 1,624 to 164) that encodes P6, a 486-amino-acid protein of 57.1 kDa. ChMaV P6 exhibits amino acid sequence similarity to raspberry leaf blotch virus (RLBV) P5 and RRV P7, whose functions are unknown [16, 23].
The ranges of amino acid sequence identity of P1 to P4 of ChMaV to the corresponding proteins of other emaraviruses, calculated by SDT v.1.2 [24], were as follows: P1, 31.3% (PerMV) to 45.0% (PCLSaV); P2, 22.4% (TiRSaV) to 32.6% (PCLSaV); P3a, 21.6% (PPSMV-1) to 39.8% (PCLSaV); P3b, 21.0% (PPSMV-1) to 36.4% (PCLSaV); and P4, 19.3% (EMARaV) to 59.6% (PCLSaV). ChMaV P5 shared the highest sequence identity (30.4%) with PerMV P5 (BBM96182), followed by P7 of Camellia japonica-associated emaravirus 1 (CjaEV1) (QHG11079) (29.1%), HPWMoV P7 (AIK23038) (27.8%), and PCLSaV P5 (QKY77007) and CjaEV1 P8 (QHG11080) (26.5%). ChMaV P6 shared the highest identity with RRV P7 (QJR96844) (28.4%), followed by its orthologous proteins [16] RRV P5 (QIB98227) (25.4%), HPWMoV P6 (AIK23037) (24.8%), HPWMoV P5 (AIK23036) (24.7%), PPSMV-1 P5 (CCW28369) (23.7%), and RLBV P5 (CBZ42028) (22.5%). Following the demarcation criterion for the genus Emaravirus that the amino acid sequences of the relevant gene products of RNA1 (RdRp), RNA2 (GP), and RNA3 (NP) differ by >25% [1], we concluded that ChMaV is a member of a distinct species of the genus.
The amino acid sequences of proteins P1 to P4 of ChMaV, known emaraviruses, and viruses belonging to the order Bunyavirales were aligned using MAFFT [24], and phylogenetic trees were reconstructed by the neighbor-joining method implemented in MEGA X [25] with 1,000 bootstrap replicates (Fig. 2). P1 to P4 of ChMaV consistently segregated together with those of PCLSaV and formed a cluster, namely, subgroup III, with PerMV, CjaEV1, and CjaEV2 (Fig. 2). Interestingly, these viruses were found in Japan or China [7, 15, 27, 28], and their host plants (Chrysanthemum morifolium, Pyrus pyrifolia, Perilla frutescens, and Camelia japonica) also originate from or are widely distributed in East Asia [29,30,31,32]. These observations indicate that subgroup III represents a unique genetic cluster of known and unknown emaraviruses of East Asian origin.
References
Elbeaino T, Digiaro M, Mielke-Ehret N, Muehlbach HP, Martelli GP (2018) ICTV virus taxonomy profile: Fimoviridae. J Gen Virol 99:1478–1479. https://doi.org/10.1099/jgv.0.001143
Mielke N, Muehlbach HP (2007) A novel, multipartite, negative-strand RNA virus is associated with the ringspot disease of European mountain ash (Sorbus aucuparia L.). J Gen Virol 88:1337–1346. https://doi.org/10.1099/vir.0.82715-0
Elbeaino T, Digiaro M, Martelli GP (2009) Complete nucleotide sequence of four RNA segments of fig mosaic virus. Arch Virol 154:1719–1727. https://doi.org/10.1007/s00705-009-0509-3
Mielke-Ehret N, Mühlbach HP (2012) Emaravirus: a novel genus of multipartite, negative strand RNA plant viruses. Viruses 4:1515–1536. https://doi.org/10.3390/v4091515
Kulkarni NK, Kumar PL, Muniyappa V, Jones AT, Reddy DVR (2002) Transmission of Pigeon pea sterility mosaic virus by the eriophyid mite, Aceria cajani (Acari: Arthropoda). Plant Dis. 86:1297–1302. https://doi.org/10.1094/PDIS.2002.86.12.1297
Skare JM, Wijkamp I, Rezende J, Michels G, Rush C, Scholthof KB, Scholthof HB (2003) Colony establishment and maintenance of the eriophyid wheat curl mite Aceria tosichella for controlled transmission studies on a new virus-like pathogen. J Virol Methods 108:133–137. https://doi.org/10.1016/s0166-0934(02)00257-4
Kubota K, Usugi T, Tomitaka Y, Shimomoto Y, Takeuchi S, Kadono F, Yanagisawa H, Chiaki Y, Tsuda S (2020) Perilla mosaic virus is a highly divergent emaravirus transmitted by Shevtchenkella sp. (Acari: Eriophyidae). Phytopathology 110:1352–1361. https://doi.org/10.1094/PHYTO-01-20-0013-R
Nemoto H, Kobayashi M, Osawa T, Yamashita S, Doi Y (1980) Studies on Eriophyoid mites I. Two eriophyid mites injurious to fig and chrysanthemum in Japan. Jpn J Appl Entomol Z 24:49-53
Chinose S (1981) A new species of the genus Paraphytoptus (Acari, Eriophydae) on chrysanthemum in Japan. Proc Jpn Soc Syst Zool 21:23–26
Yamashita S (1997) Current problems on eriophyid mites in Japan: their injurious diseases and transmitted viruses. Plant Prot 51:471–476 ((in Japanese))
Kubota K, Chiaki Y, Yanagisawa H, Yamasaki J, Horikawa H, Tsunekawa K, Morita Y (2021) Novel degenerate primer sets for the detection and identification of emaraviruses reveal new chrysanthemum species. J Virol Methods 288:113992. https://doi.org/10.1016/j.jviromet.2020.113992
Gambino G, Perrone I, Gribaudo I (2008) A rapid and effective method for RNA extraction from different tissues of grapevine and other woody plants. Phytochem Anal 19:520–525. https://doi.org/10.1002/pca.1078
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL (2012) Domain enhanced lookup time accelerated BLAST. Biol Direct 7:12. https://doi.org/10.1186/1745-6150-7-12
Liu H, Wang G, Yag Z, Wang Y, Zhang Z, Li L, Waqas M, Hong N (2020) Identification and characterization of a pear chlorotic leaf spot-associated virus, a novel emaravirus associated with a severe disease of pear trees in China. Plant Dis 104:2786–2798. https://doi.org/10.1094/PDIS-01-20-0040-RE
Di Bello PL, Ho T, Tzanetakis IE (2015) The evolution of emaraviruses is becoming more complex: seven segments identified in the causal agent of rose rosette disease. Virus Res 210:241–244. https://doi.org/10.1016/j.virusres.2015.08.009
Tatineni S, McMechan AJ, Wosula EN, Wegulo SN, Graybosch RA, French R, Hein GL (2014) An eriophyid mite-transmitted plant virus contains eight genomic RNA segments with unusual heterogeneity in the nucleocapsid protein. J Virol 88:11834–11845. https://doi.org/10.1128/JVI.01901-14
Elbeaino T, Digiaro M, Uppala M, Sudini H (2015) Deep sequencing of dsRNAs recovered from mosaic-diseased pigeonpea reveals the presence of a novel emaravirus: pigeonpea sterility mosaic virus 2. Arch Virol 160:2019–2029. https://doi.org/10.1007/s00705-015-2479-y
Peracchio C, Forgia M, Chiapello M, Vallino M, Turina M, Ciuffo M (2020) A complex virome including two distinct emaraviruses associated with virus-like symptoms in Camellia japonica. Virus Res 286:197964. https://doi.org/10.1016/j.virusres.2020.197964
Yu C, Karlin DG, Lu Y, Wright K, Chen J, MacFarlane S (2013) Experimental and bioinformatic evidence that raspberry leaf blotch emaravirus P4 is a movement protein of the 30K superfamily. J Gen Virol 94:2117–2128. https://doi.org/10.1099/vir.0.053256-0
Gupta AK, Hein GL, Graybosch RA, Tatineni S (2018) Octapartite negative-sense RNA genome of High Plains wheat mosaic virus encodes two suppressors of RNA silencing. Virology 518:152–162. https://doi.org/10.1016/j.virol.2018.02.013
Gupta AK, Hein GL, Tatineni S (2019) P7 and P8 proteins of High Plains wheat mosaic virus, a negative-strand RNA virus, employ distinct mechanisms of RNA silencing suppression. Virology 535:20–31. https://doi.org/10.1016/j.virol.2019.06.011
Lu Y, McGavin W, Cock PJA, Schnettler E, Yan F, Chen J, MacFarlane S (2015) Newly identified RNAs of raspberry leaf blotch virus encoding a related group of proteins. J Gen Virol 96:3432–3439. https://doi.org/10.1099/jgv.0.000277
Muhire BM, Varsani A, Martin DP (2014) SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 9:e108277. https://doi.org/10.1371/journal.pone.0108277
Katoh K, Standley DM (2013) MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 1:1547–1549. https://doi.org/10.1093/molbev/msy096
Zhang S, Yang L, Ma L, Tian X, Li R, Zhou C, Cao M (2020) Virome of Camellia japonica: discovery of and molecular characterization of new viruses of different taxa in camellias. Front Microbiol 11:945. https://doi.org/10.3389/fmicb.2020.00945
Wu X, Liu J, Cheng X (2020) First report of Camellia japonica associated emaravirus 1 associated with camellia leaf ringspot and flower color-breaking disease in China. Plant Dis. https://doi.org/10.1094/PDIS-03-20-0657-PDN
Su J, Jiang J, Zhang F, Liu Y, Ding L, Chen S, Chen F (2019) Current achievements and future prospects in the genetic breeding of chrysanthemum: a review. Hort Res 6:109
Xue L, Liu Q, Hu H, Song Y, Fan J, Bai B, Zhang M, Wang R, Qin M, Li X, Wu J (2018) The southwestern origin and eastward dispersal of pear (Pyrus pyrifolia) in East Asia revealed by comprehensive genetic structure analysis with SSR markers. Tree Genet Genomes 14:48
Ahmed HM (2019) Ethnomedicinal, phytochemical and pharmacological investigations of Perilla frutescens (L.) Britt. Molecules 24:102
Chung MY, Epperson BK, Gi Chung M (2003) Genetic structure of age classes in Camellia japonica (Theaceae). Evolution 57:62
Acknowledgements
We are grateful to A. Suzuki for sampling, and Y. Matsumura, Y. Narita, J. Sato, and M. Tawa for their assistance in preparing the biological samples and laboratory work. This study was supported by the Fund for Objective Basic Research provided by CARC/NARO.
Funding
This study was supported by a Grant-in-Aid for Development of Practical Technologies to Implement New Agricultural, Forestry, and Fishery Policies administered by the Ministry of Agriculture, Forestry, and Fisheries of Japan (27001C) and the Fund for Objective Basic Research provided by CARC/NARO.
Author information
Authors and Affiliations
Contributions
Conceptualization, KK; methodology, KK; formal analysis and investigation, YC, JY, KK, HY, and FK; writing—original draft preparation, KK; writing—review and editing, KK; funding acquisition, KK, HY, and YM; resources, HH and KT.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Availability of data and material
The nucleotide sequence reported here is available in the DDBJ/ENA/GenBank database under accession numbers LC576445–LC576451.
Additional information
Handling Editor: Massimo Turina.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kubota, K., Yanagisawa, H., Chiaki, Y. et al. Complete nucleotide sequence of chrysanthemum mosaic-associated virus, a novel emaravirus infecting chrysanthemum. Arch Virol 166, 1241–1245 (2021). https://doi.org/10.1007/s00705-021-04979-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-04979-2