
Abstract
Deep-sequencing analysis of double-stranded RNA extracted from a mosaic-diseased pigeonpea plant (Cajanus cajan L., family Fabaceae) revealed the complete sequence of six emaravirus-like negative-sense RNA segments of 7009, 2229, 1335, 1491, 1833 and 1194 nucleotides in size. In the order from RNA1 to RNA6, these genomic RNAs contained ORFs coding for the RNA-dependent RNA polymerase (RdRp, p1 of 266 kDa), the glycoprotein precursor (GP, p2 of 74.5 kDa), the nucleocapsid (NC, p3 of 34.9 kDa), and the putative movement protein (MP, p4 of 40.7 kDa), while p5 (55 kDa) and p6 (27 kDa) had unknown functions. All RNA segments showed distant relationships to viruses of the genus Emaravirus, and in particular to pigeonpea sterility mosaic virus (PPSMV), with which they shared nucleotide sequence identity ranging from 48.5 % (RNA3) to 62.5 % (RNA1). In phylogenetic trees constructed from the sequences of the proteins encoded by RNA1, RNA2 and RNA3 (p1, p2 and p3), this new viral entity showed a consistent grouping with fig mosaic virus (FMV) and rose rosette virus (RRV), which formed a cluster of their own, clearly distinct from PPSMV-1. In experimental greenhouse trials, this novel virus was successfully transmitted to pigeonpea and French bean seedlings by the eriophyid mite Aceria cajani. Preliminary surveys conducted in the Hyderabad region (India) showed that the virus in question is widespread in pigeonpea plants affected by sterility mosaic disease (86.4 %) but is absent in symptomless plants. Based on molecular, biological and epidemiological features, this novel virus is the second emaravirus infecting pigeonpea, for which the provisional name pigeonpea sterility mosaic virus 2 (PPSMV-2) is proposed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pigeonpea (Cajanus cajan, L.) is a major pulse crop grown on over 5 million ha with an annual production of over 3 million tonnes [12]. Nearly 90 % of its cultivation takes place in India and Nepal, followed by Africa (6 %), the Caribbean (2 %) and other Southeast Asian countries [19].
Sterility mosaic disease (SMD) is one of the most important constraints to pigeonpea production and a threat to its cultivation in the Indian subcontinent (Nepal, India, Bangladesh) and in Myanmar [37]. SMD has variable symptomatology, within which three major syndromes are recognized: (i) severe mosaic and sterility (partial or complete cessation of flower production); (ii) mild mosaic with partial sterility, and (iii) chlorotic ringspots with no apparent sterility [17, 22].
Although the SMD agent has long remained unknown, its viral nature was hypothesized based on the type of symptoms, transmission by mites (Aceria cajani) [20] and the presence of 100- to 150-nm quasi-spherical double membrane-bound bodies (DMBBs) in thin-sectioned plant tissues [22]. Comparable DMBBs have been identified as particles of the putative causal agents of a number of diseases, including fig mosaic [2, 7, 8, 29], ringspot of European mountain ash [32], and rosetting of rose [27], to which the names fig mosaic virus (FMV), European mountain ash ringspot-associated virus (EMARaV) and rose rosette virus (RRV) were assigned, respectively. Similar DMBBs structures were observed in the cells of high-plain diseased plants of maize (Zea mays L.) and wheat (Triticum aestivum L.) infected with wheat mosaic virus (WMoV) [16, 38, 40], and of Cercis spp. infected with redbud yellow ringspot-associated virus (RYRSaV) [25, 26]. Besides particle morphology, all of these viruses, plus raspberry leaf blotch virus (RLBV) [31] and blackberry leaf mottle-associated virus (BLMaV) [14] share common biological, molecular and epidemiological traits, i.e., type of symptoms (mosaic and ring spots), multipartite genome, and type of vectors (eriophyid mites) [33]. These features constituted the basis for the establishment of the genus Emaravirus [34], of which pigeonpea sterility mosaic virus (PPSMV) is a putative member [9].
The different types of symptoms associated with SMD may not simply depend on the host susceptibility and/or the virulence of PPSMV strains. In fact, during a survey of pigeonpea fields in Hyderabad (India), a few plants showing mosaic and stunting typical of SMD were negative when tested by RT-PCR for the presence of PPSMV. Thus, further studies were carried out, which, as reported hereinafter, identified an emaravirus clearly different from PPSMV (which will be referred to as PPSMV-1), for which the name pigeonpea sterility mosaic virus 2 (PPSMV-2) is proposed and used throughout this paper.
Materials and methods
Plant source material
A symptomatic PPSMV-1-negative pigeonpea plant (accession HYD2, germplasm line ICP 8863) from an experimental plot of the International Crops Research Institute for the Semi-Arid Tropics (ICRISAT, Hyderabad, Telangana, India) was used as virus source for molecular analysis.
In addition, 85 samples were collected from seven pigeonpea fields at Hyderabad to assess the presence of PPSMV-1 and PPSMV-2 by RT-PCR. Samples consisted of fresh leaves from 10 symptomless and 75 symptomatic plants showing mottling, line patterns, chlorotic ringspots, stunting, and flower sterility.
Extraction of double-stranded RNA (dsRNA) and total nucleic acids (TNA)
Leaf tissues (30 g) from infected plants were used to recover dsRNAs by phenol/chloroform extraction and chromatography through a cellulose CF-11 column (Whatman, USA) in the presence of 17 % ethanol, according to a protocol [5] modified by Elbeaino et al. [7]. Further purification was done by chromatography through a second column of cellulose CC41 (Whatman, USA), followed by digestion with RQ1-DNase (60 ng/ml) (Promega, USA) and RNase A (25 ng/µl). Extracts were then passed through Micro Bio-Spin 30 Columns in RNase-Free Tris (Bio-Rad Laboratories, USA) and analyzed by electrophoresis in a 1.5 % TBE agarose gel at 120 V for 90 min [7].
TNAs were extracted from leaf tissues according to Foissac et al. [13]. Briefly, 100 mg of tissue was ground in liquid nitrogen and homogenized in 1 ml of extraction buffer (6 M guanidine isothiocyanate, 0.2 M sodium acetate, 1 M potassium acetate, 0.025 mM EDTA, 2.5 % PVP-40), with the addition of 6 M sodium iodide, 0.15 M sodium sulphite, 150 µl ethanol and 25 µl silica particles (1 g/ml pH 2.0). After stripping by heat treatment in sterile water (70 °C for 3 min) and centrifugation (3 min at 16 000 g), TNAs were recovered and stored at -20 °C until use.
Deep sequencing
The sequence of the emaraviral genome present in the HYD2 accession was determined by using a next-generation sequencing approach with the Illumina technology. A TruSeq RNA Sample Prep Kit v2 (Illumina) was used with total nuclease-treated dsRNA to synthesize a cDNA library. Reverse-transcribed templates were sequenced in a run of 50-bp single reads at the University of Bari, Department of Soil Sciences, Plants and Food (http://www.selge.uniba.it) using a HiScan SQ apparatus (Illumina, San Diego, USA). The Velvet Software 1.2.08 [43], with a kmer of 23, was used for de novo assembly of all the previously filtered reads into larger contigs. BLASTX (e-value with cut-off from 10-6 to 10-2) and BLASTP (http://www.ncbi.nlm.nih.gov/) were used for sequence homology screening of the assembled contigs. The sequences of genomic RNA segments were reconstructed using CLC software (CLCbio, Aarhus, Denmark) by aligning homologous contigs of the most closely related sequences found in the GenBank database and sequences generated from RT-PCRs using virus-specific primers. Once assembled, all viral segments were re-sequenced through several RT-PCR amplifications, using a battery of sense- and antisense-specific primers designed based on deep-sequencing-generated reads to ensure at least fourfold nucleotide sequence coverage.
PCR amplification of 5′ and 3′ RNA termini
Two strategies for PCR amplification and sequencing of the 5′ and 3′ termini of the PPSMV-2 RNA segments were adopted. The first approach encompassed the use of the RNA-Ligase-Mediated Rapid Amplification of cDNA Ends (RLM-RACE) method [4]. Briefly, 200 ng of total nuclease-treated dsRNA, and TNA extracts were denatured by treatment with 20 mM methyl mercuric hydroxide, and the 3′ end of each RNA strand was ligated to 200 ng of the primer Adapt-Link [(5′-UUU)RNA(AACCGCATCCTTCTC-3′)DNA] using T4 RNA ligase according to the manufacturer’s instructions (Ambion, USA). Primer-ligated denatured dsRNAs were precipitated with ethanol, re-suspended in double-distilled sterile water, and then mixed with the primer Adapt/comp (5′-GAGAAGGATGCGGTT-3′) for cDNA synthesis, using 20 U of reverse transcriptase enzyme (Life Technologies, Italy) at 50 °C for 1 h. PCR amplifications were done using the downstream primer Adapt/comp and sense and/or antisense PPSMV-2-specific primers (designed to be close to the 5′/3′ termini) and the conditions reported by Coutts and Livieratos [4].
The second approach consisted of a PCR assay conducted on randomly reverse-transcribed dsRNAs recovered from infected pigeonpea plants, in which two primers designed based on two 13-nucleotide stretches that are conserved in the 5′/3′ genomic RNA ends of all emaraviruses were used [8, 9, 27, 32]. These primers were used for RT-PCR to amplify additional RNA segments of PPSMV possibly not discovered by the deep sequencing. PCR conditions and thermal cycling were as described by Elbeaino et al. [9].
RT-PCR detection
TNAs (500 ng) were randomly reverse transcribed by adding 4 μl of 5x M-MLV buffer (50 mM Tris-HCl, pH 8.3, 75 mM KCl, 3 mM MgCl2), 2 μl of 10 mM DTT, 0.5 μl of 10 mM dNTPs, and 200 units of Moloney murine leukaemia virus (M-MLV) reverse transcriptase (Life Technologies, Italy) in a final volume of 20 μl, for 1 h at 39 °C. A battery of sense and antisense primers specific for all RNA segments of both PPSMV-1 and PPSMV-2 were designed (Table 2) for RT-PCR assays. The primers designed based on RNA1 and RNA2 sequences were utilized to detect both viruses in the 85 collected samples, whereas those designed based on the other PPSMV-2 RNA sequences were used to distinguish these viruses in singly infected samples. Random-primed cDNA (2.5 µl) was added to 5x Taq polymerase buffer (Promega, USA) containing MgCl2 to a final concentration of 1 mM, 0.2 mM dNTPs, 0.2 µM each specific primer, and 0.2 µl of Taq polymerase (5 unit/µl) in a final volume of 25 µl. PCR, with all primers, consisted of an initial denaturation at 94 °C for 4 min, followed by 35 cycles at 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s. The final extension was at 72 °C for 7 min. Amplification products were visualized in a 1.2 % TBE agarose gel.
Cloning, sequencing and computer-assisted analysis
All amplified products were ligated in StrataClone™ PCR Cloning vector pSC-A (Stratagene, USA), cloned in Escherichia coli DH5α or SoloPACK cells, and custom sequenced (Primm, Milan, Italy). Nucleotide and protein sequences were analysed with the assistance of the DNA Strider 1.1 program [28]. Multiple alignments of nucleotide and amino acid sequences were obtained using the default options of CLUSTALX 1.8 [41]. Search for homologies with proteins from the Protein Information Resources database (PIR, release 47.0) was done with the FASTA [36], BLASTX and BLASTP [1] programs. The protein function was investigated using the software for the prediction of glycosylation sites (NetNGlyc 1.0 vers.) [10], transmembrane helices (TMHMM 2.0 vers.), and cleavage sites (SignalP 3.0, TragetP vers.) [11], and the SLP-Local program was used for the prediction of the subcellular location of proteins [30]. Phylogenetic analysis was conducted on aligned amino acid sequences using “MUSCLE” [6], and trees were generated by the maximum-likelihood and maximum-parsimony methods [18], applying the JTT matrix and pairwise gap deletion options implemented in MEGA6 [39]. Branches with less than 70 % bootstrap support were collapsed.
Virus transmission by the leaf-stapling technique
A pigeonpea plant grown in an isolated cage, infected with both PPSMV-1 and PPSMV-2, and infested only by Aceria cajani, as ascertained by microscopy, was used as a donor for virus transmission trials in a greenhouse using the “leaf-stapling” technique [35]. Small pieces from symptomatic leaves carrying at least 20 individual mites were stapled onto young leaves of 15 healthy seedlings each of pigeonpea (line ICP8863) and Phaseolus vulgaris cv. Topcrop that had been tested by RT-PCR to ascertain the absence of both emaraviruses. Diseased leaflets were folded on the primary leaves of test plants in such a way that their lower surface came in contact with the leaf blade, thus facilitating the migration of viruliferous mites from the inoculum source to the recipient host. Starting one week post-inoculation, the plants were observed daily for symptom development. The presence of PPSMVs in the stapled leaves and in the leaves above them was assessed at weekly intervals by PCR using virus-specific primers (Table 1).
Results
Deep sequencing and genome analysis
Six dsRNA bands, with size ranging from 1.2 kbp to approximately 7 kbp, were obtained from accession HYD2 (Fig. 1). A similar and consistent pattern of six dsRNA bands was also obtained subsequently from four other mosaic-diseased pigeonpea plants infected with PPSMV-2 (partial data shown in Fig. 1). No dsRNAs were extracted from healthy pigeonpea plants.

Electrophoresis of dsRNAs extracted from pigeonpea accessions HYD2 and HYD7 infected with PPSMV-2 (lanes 2 and 3) and from accession TAN17 infected with PPSMV-1 (lane 4) showing six and five bands, respectively. Lane 5, healthy pigeonpea plant, lane 1, DNA ladder
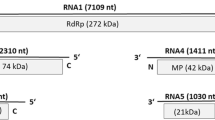
Deep sequencing of dsRNA extracts from accession HYD2 gave a total of 2,837,355 single reads. After adapter removal and size selection (21-51 nt), a data set of 283,360 short reads was obtained and subjected to de novo assembly, generating a total of 63,258 contigs, ranging in size from 30 to 643 nt with an N50 value of 121 nt. Using the resultant contigs for a BLAST search of the NCBI virus database, with a cutoff e-value of 10-6 for BLASTX, we identified 612 contigs with amino acid sequence similarity to genes encoded by different emaraviruses. No additional emaravirus-related sequences were identified when contigs without BLASTX matches at an e-value cutoff of 10-6 were queried again using an e-value of 10-2. Moreover, no additional contigs with similarity to genes encoded by viruses of different genera emerged from the analysis of the cDNA library. All sequences derived from deep-sequencing contigs and PCR clones were compared, and used to generate the full genome sequence of PPSMV-2, which consisted of six large RNA fragments, with a length of ca. 7009, 2229, 1335, 1491, 1833, and 1194 nt, which, after BLAST analysis, were identified as RNA1 to RNA6 (Fig. 2).

Schematic representation of the organization of the six PPSMV-2 genomic RNAs. The terminal 13 nucleotides conserved at the 5′ and 3′ termini are indicated as black boxes on each segment. Letters (A–F) represent the conserved motifs in the RdRp (RNA1) gene. Expression products of each RNA (p1 to p6) are represented as dark gray boxes. The protein function and estimated molecular weight of each segment are indicated. The figure is not drawn to scale
RNA1 contains a single large open reading frame (ORF1) between nt 46 and 6,930 (acc. no. HF912243). ORF1 encodes a 266-kDa polypeptide (p1) of 2,294 amino acids (aa) and has 30.9 % to 72.1 % sequence identity to other emaraviral RNA-dependent RNA polymerases (RdRps), with the highest identity to FMV (Table 2). PPSMV-2 RdRp contains all of the typical conserved motifs common to the members of the family Bunyaviridae: motifs A (DASKWS1129-1134) and C (SDDS1255-1258), which are part of the palm domain of the replicase [3] and are involved in binding of divalent metal cations; motif B (QGNLNHLSS1214-1222), which is thought to be involved in RNA binding with the Gly1215 residue allowing for mobility in the peptide backbone; motif E (EFLST1313-1317), which is likely to be involved in cap-snatching in bunyaviruses as well as in possible endonuclease activity [27].
RNA2 encodes a single 74.3-kDa polypeptide of 649 aa, between nt 48 and 1997 (acc. no. HF912244). The RNA2-encoded polyprotein shares 21.3 % to 57.1 % identity with the glycoprotein precursors (GP) of all emaraviruses (Table 2), again sharing the highest identity with FMV. The following essential GP elements were identified in p2 by computer-assisted analysis: three putative transmembrane helices at aa positions 115-137, 173-190 and 583-602 and five putative glycosylation sites at aa positions 65, 204, 331, 393, 438. A potential cleavage site was predicted between AKA190-192 and DD193-194, which would release two single glycoproteins: Gn (22.1 kDa) and Gc (52.2 kDa). Like FMV, RRV, EMARaV and PPSMV-1, PPSMV-2 p2 contains a phlebovirus glycoprotein motif (GCYDCQ478-483).
RNA3 encodes a 34.9-kDa polypeptide (p3) of 314 aa, between nt 102 and 1,046 (acc. no. HF912245). BLASTP analysis of the RNA3-encoded polypeptide (ORF3) of PPSMV-2 showed identities ranging from 16 % to 80 % to comparable emaraviral nucleocapsid proteins (NC), once again sharing the highest identity with FMV (Table 2). PPSMV-2 p3 contains three stretches of amino acids (NVVSFNKACA130-139, NRLA178-181 and GYEF199-202) that are well conserved in all emaraviruses [8, 9] and are likely to be involved in RNA binding [27]. By analogy to other emaraviruses, in which RNA3 encodes the nucleocapsid protein, this segment has been conventionally referred to as RNA3, although it is smaller than RNA4 and RNA5.
RNA4 encodes a 40.7-kDa polypeptide (p4) of 375 aa, between nt 83 and 1168 (acc. no. HF912246). A database search showed the highest identity of this polypeptide with those encoded by RNA4 of FMV (75 %) and RRV (61.2 %) (Table 2). The SLP-Local (Subcellular Location Predictor) program predicted the expression product of RNA4 to be a possible membrane-located protein lacking the transmembrane helices that are likely to be involved in virus cell-to-cell movement, similar to comparable movement proteins encoded by FMV, PPSMV-1, RLBV, WMoV and RRV RNA4 [8, 9, 15, 31, 40, 42]. However, this segment contained a conserved motif (FDPNFQQFITGSL148-160), whose function is unknown.
RNA5 encodes a 55-kDa polypeptide (p5) of 473 aa, the translation of which initiates at an AUG at position 106-108 and terminates at a UAA stop codon at position 1525-1527 (acc. no. HG939489). This polypeptide shares different levels of sequence identity with its homologues in PPSMV-1, FMV, WMoV and RLBV, the only viruses with a fifth RNA segment in their genomes. No amino acid sequence motif was identified in this segment that might indicate its function.
RNA6 encodes a 27-kDa polypeptide (p6) of 238 aa, the translation of which initiates at an AUG at position 69-71 and terminates at a UGA stop codon at position 783-785 (acc. no. HG939490). This polypeptide shares 23.9 % identity with the p6 of FMV. It is noteworthy that p6 has not been found in EMARaV, RRV, RYRSaV and PPSMV-1. Like RNA5, there is no hint of the possible function of this segment.
Sequence analysis of 5′ and 3′ RNA termini
Like those of other emaraviruses, the 5′ and 3′ termini of all RNA segments of PPSMV-2 possess untranslated regions (UTRs) that extend from 46 to 106 nt and from 79 to 409 nt, respectively, and exhibit a conserved stretch of 13 nt at their termini. Primers designed based on these stretches and used in RT-PCR were able to amplify DNA fragments of different sizes whose sequences coincided with those of the segments generated from the deep-sequencing library, with the exception of RNA1, which failed to be amplified, presumably because of its large size (7009 bp). In this case, the complete sequence of RNA1 was reconstructed by sequencing eight RT-PCR amplicons obtained by using internal primers designed based on deep-sequencing-generated clones. No sequence of any additional segment was detected.
Phylogenetic analysis
The phylogenetic trees constructed with predicted amino acid sequences of RNA1, RNA2 and RNA3 of PPSMV-2 and of the comparable polypeptides encoded by the corresponding RNA segments of known emaraviruses showed the consistent grouping of PPSMV-2 with FMV and RRV, which formed a cluster of their own, clearly distinct from PPSMV-1 (Fig. 3).

Phylograms constructed with predicted amino acid sequences of RNA1 (RNA-dependent RNA polymerase, p1), RNA2 (glycoprotein precursor, p2) and RNA3 (nucleocapsid, p3) of PPSMV-2 together with orthologues from other members of the genus Emaravirus. Sequences from the following viruses were used to construct the phylogenetic trees: European mountain ash ringspot-associated virus (EMARaV), fig mosaic virus (FMV), rose rosette virus (RRV), redbud yellow ringspot-associated virus (RYRSaV), raspberry leaf blotch virus (RLBV), wheat mosaic virus (WMoV), pigeonpea sterility mosaic virus 1 (PPSMV-1). The tospoviruses impatiens necrotic spot virus (INSV, acc. no. X93218; NC_003616; NC_003624) and tomato spotted wilt virus (TSWV, acc. no. D10066; S48091; NC_002051) were used as outgroups. The accession numbers of the emaraviruses are shown in Table 2
The phylogenetic trees constructed with RdRp, GP, and NC proteins of PPSMV-2 and of all emaraviruses yielded similar topologies by generating two (or three) main clades, suggesting that emaraviruses could have evolved into two (or three) distinct lineages (Fig. 3). The first group included only WMoV and RLBV, while the second comprised all the others, which in turn could be subdivided into two subgroups: subgroup a, comprising PPSMV-2 together with FMV, RRV and PPSMV-1, and subgroup b, including EMARaV and RYRSaV. Although PPSMV-1 and PPSMV-2 are two emaraviral agents that infect the same crop, these two viruses are distinct from each other, since PPSMV-2 was more closely related to FMV and RRV than to PPSMV-1.
PCR detection and survey
All 75 symptomatic pigeonpea plants tested by RT-PCR, using sets of primers designed based on both RNA1 and RNA2 of PPSMV-1 and PPSMV-2, proved to be infected by at least one of the two emaraviruses. In particular, 59 samples (78.7 %) were multiply infected, while single infections by PPSMV-1 and PPSMV-2 were found in 10 (13.3 %) and 6 (8.0 %) plants, respectively. The 10 symptomless pigeonpea plants were negative. All primers designed based on the other genome segments of PPSMV-2 (Table 1) amplified only the six PPSMV-2-infected samples but none of those hosting PPSMV-1 (data not shown). The sequences of the six RNA segments partially amplified from the six singly infected PPSMV-2 plants showed nucleotide sequence identities ranging from 97 % to 100 %, with the lowest values found in RNA3.
Field observations showed that PPSMV-2-infected plants of different cultivars consistently exhibited mosaic (Fig. 4a) and stunting, and, in a single case, sterility, whereas chlorotic ringspots and line patterns of the leaves, but no apparent decrease in vigour, were shown by PPSMV-1-infected plants (Fig. 4b). When both viruses were present in the same plant, these plants displayed more-severe symptoms, i.e., chlorosis, leaf deformation, mosaic and sterility typical of SMD (Fig. 4c and d).

(a) Mottling and mosaic symptoms observed on a pigeonpea plant infected with PPSMV-2; (b) line patterns and ringspots symptoms observed on a pigeonpea plant infected with PPSMV-1; (c) severe mosaic, chlorotic ringspots and deformation symptoms observed on a pigeonpea plant infected with PPSMV-1 and PPSMV-2; (d) severe mosaic, stunting and sterility symptoms observed on a pigeonpea plant infected with PPSMV-1 and PPSMV-2; (e) stapling technique applied to pigeonpea and French bean seedlings in a greenhouse; (f) chlorosis and mild mottling observed on French bean seedlings ca 20 days post-inoculation
Virus transmission by the leaf-stapling technique
Aceria cajani mites succeeded in transmitting PPSMV-2 onto most of the inoculated pigeonpea and French bean seedlings (80 % and 93.3 %, respectively), which showed chlorotic mottling and mild mosaic symptoms 15-20 days post-inoculation (Fig. 4e and f). PCR testing two weeks post-inoculation revealed the presence of both PPSMV-1 and PPSMV-2 in both inoculated and newly formed leaves of test plants.
Discussion
The present study has revealed the presence of a new emaravirus (PPSMV-2) that infects pigeonpea and is clearly distinct from the known causal agent of SMD in India (PPSMV-1). PPSMV-2 is molecularly distinct from PPSMV-1 [9] and has a larger number of RNA genomic segments (six instead of five). As with other emaraviruses, four of these segments code for known functional proteins (an RNA-dependent RNA polymerase [RNA1], a putative glycoprotein precursor [RNA2], a putative nucleocapsid protein [RNA3], and a putative movement protein [RNA4]), whereas the proteins encoded by RNA5 and RNA6 have unknown functions. Nevertheless, their viral nature is supported by their similarity to the corresponding RNAs of other emaraviruses (PPSMV-1, FMV, WMoV and RLBV) and by the overall organisation of the viral genome, which corresponds to that of emaraviruses [7, 8, 32]. The recent finding of a new emaravirus (WMoV) with eight RNA segments [40] raises questions about the number of RNAs that compose an emaraviral genome. Even though, in principle, the presence of additional RNA segments in the PPSMV-2 genome cannot be excluded, we are confident that the techniques used in this study (deep sequencing, repeated dsRNA analysis, use of 5′/3′-terminal virus-specific primers) would have detected them.
At the biological level, the discovery of PPSMV-2 has revealed the complex nature of sterility mosaic, a disease whose variable symptomatology has been attributed to the diverse virulence of PPSMV-1 strains or serotypes (in particular P and C isolates) [23] and/or to the different susceptibility/resistance of cultivated and wild Cajanus spp. [24] rather than to the possible presence of additional causal agents. The presence of double emaravirus infections can now explain the wide range of symptoms observed in infected plants (severe systemic mosaic and sterility, systemic mild mosaic and partial sterility, localized chlorotic ringspots and no sterility) [23] and the complex pattern of dsRNA bands (5 to 7) previously reported for PPSMV [21].
Field observations, though preliminary, suggest that PPSMV-2 is associated with leaf mosaic, stunting and sterility symptoms, whereas PPSMV-1 induces chlorotic ringspots and line patterns of the leaves but no apparent reduction in plant vigour. A more severe SMD was shown by plants with mixed infections.
Successful “stapling transmission” experiments confirmed the efficacy of this technique for transferring PPSMV-1 and PPSMV-2 to healthy test plants (pigeonpea and French bean) by A. cajani under greenhouse conditions. However, it remains to be established if transmission occurs with a different efficiency for each virus or if other eriophyid mites have a role in their spread in nature. The specific sets of primers for specific identification of these two viruses by RT-PCR will provide a useful tool for addressing these and other questions.
Finally, the separate branching of PPSMV-2 and PPSMV-1 in the phylogenetic tree suggests that these two pigeonpea-infecting emaraviruses have followed two independent evolutionary lines. Furthermore, the molecular, biological and epidemiological features of PPSMV-2 seem to justify its inclusion in the genus Emaravirus as a member of a separate species.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402
Bradfute OR, Whitmoyer RE, Nault RL (1970) Ultrastructure of plant leaf tissue infected with mite-borne viral-like particles. Proc Electron Microsc Soc Am 258:178–179
Bruenn JA (2003) A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucl Acids Res 31:1821–1829
Coutts RHA, Livieratos IC (2003) A rapid method for sequencing the 5′- and 3′-termini of double-stranded RNA viral templates using RLM-RACE. J Phytopathol 151:525–527
Dodds JA (1993) DsRNA in diagnosis. In: Matthews REF (ed) Diagnosis of plant virus diseases. CRC Press, Boca Raton, pp 273–294
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32:1792–1797
Elbeaino T, Digiaro M, Alabdullah A, De Stradis A, Minafra A, Mielke N, Castellano MA, Martelli GP (2009) A multipartite single-stranded negative-sense RNA virus is the putative agent of fig mosaic disease. J Gen Virol 90:1281–1288
Elbeaino T, Digiaro M, Martelli GP (2009) Complete nucleotide sequence of four RNA segments of Fig mosaic virus. Arch Virol 154:1719–1727
Elbeaino T, Digiaro M, Uppala M, Sudini H (2014) Deep sequencing of Pigeonpea sterility mosaic virus discloses five RNA segments related to emaraviruses. Virus Res 188:27–31
Emanuelsson O, Nielsen H, Brunak S, Von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016
Emanuelsson O, Brunak S, Von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP, and related tools. Nat Protoc 2:953–971
FAO (2010) http://faostat.fao.org/site/567/default.aspx#ancor
Foissac X, Svanella-Dumas L, Dulucq MJ, Candresse T, Gentit P (2001) Polyvalent detection of fruit tree tricho, capillo and foveavirus by nested RT-PCR using degenerated and inosine containing primers (DOP RT-PCR). Acta Hort 550:37–44
Hassan M, Sabanadzovic S, Keller KE, Martin RR, Tzanetakis IE (2012) A putative new emaravirus associated with blackberry yellow vein disease. In: Proceedings of the 22nd international conference on virus and other transmissible diseases of fruit crops (ICVF). Petria 22:354–358
Ishikawa K, Maejima K, Komatsu K, Netsu O, Keima T, Shiraishi T, Okano Y, Hashimoto M, Yamaji Y, Namba S (2013) Fig mosaic emaravirus p4 protein is involved in cell-to-cell movement. J Gen Virol 94:682–686
Jensen SG, Lane LC, Seifers DL (1996) A new disease of maize and wheat in the High Plains. Plant Dis 80:1387–1390
Jones AT, Kumar PL, Saxena KB, Kulkarni NK, Muniyappa V, Waliyar F (2004) Sterility mosaic disease: the ‘green plague’ of pigeonpea. Plant Dis 88:436–445
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Kannaiyan J, Nene YL, Reddy MV, Ryan JG, Raju TN (1984) Prevalence of pigeonpea disease and associated crop losses in Asia, Africa and the Americas. Trop Pest Manag 30:62–71
Kulkarni NK, Kumar PL, Muniyappa V, Jones AT, Reddy DVR (2002) Transmission of Pigeon pea sterility mosaic virus by the eriophyid mite, Aceria cajani (Acari: Arthropoda). Plant Dis 86:1297–1302
Kumar PL, Duncan GC, Roberts IM, Jones AT, Reddy DVR (2002) Cytopathology of Pigeonpea sterility mosaic virus in pigeonpea and Nicotiana benthamiana: similarities with those of eriophyid mite-borne agents of undefined aetiology. Ann Appl Biol 140:87–96
Kumar PL, Jones AT, Reddy DVR (2003) A novel mite-transmitted virus with a divided RNA genome closely associated with pigeonpea sterility mosaic disease. Phytopathology 93:71–81
Kumar PL, Jones AT, Waliyar F (2004) Biology, etiology and management of pigeonpea sterility mosaic disease. Annu Rev Plant Pathol 3:1–24
Kumar PL, Latha TKS, Kulkarni NK, Raghavendra N, Saxena KB, Waliyar F, Rangaswamy KT, Muniyappa V, Doriswamy S, Jones AT (2005) Broad-Based resistance to Pigeonpea sterility mosaic disease in wild relatives of pigeonpea (Cajanus: Phaseolae). Ann Appl Biol 146:371–379
Laney AG, Gergerich RC, Tzanetakis IE (2010) Redbud yellow ringspot disease: 30 years of research. Phytopathology 100:S201
Laney AG, Gergerich RC, Keller KE, Martin RR, Tzanetakis IE (2011a) In: GenBank database. Accession no. JF795479–JF795482
Laney AG, Keller KE, Martin RR, Tzanetakis IE (2011) A discovery 70 years in the making: characterization of the Rose rosette virus. J Gen Virol 92:1727–1732
Marck C (1988) DNA Strider: a “C” programme for the fast analysis of DNA and protein sequences on the Apple Macintosh family computers. Nucl Acids Res 16:1829–1836
Martelli GP, Castellano MA, Lafortezza R (1993) An ultrastructural study of fig mosaic. Phytopathol Medit 32:33–43
Matsuda S, Vert JP, Saigo H, Ueda N, Toh H, Akutsu T (2005) A novel representation of protein sequences for prediction of subcellular location using support vector machines. Protein Sci 14:2804–2813
McGavin WJ, Mitchell C, Cock PJA, Wright KM, MacFarlane SA (2012) Raspberry leaf blotch virus, a putative new member of the genus Emaravirus, encodes a novel genomic RNA. J Gen Virol 93:430–437
Mielke N, Muehlbach HP (2007) A novel, multipartite, negative-strand RNA virus is associated with the ringspot disease of European mountain ash (Sorbus aucuparia L.). J Gen Virol 88:1337–1346
Mielke-Ehret N, Muehlbach HP (2012) Emaravirus: a novel genus of multipartite, negative strand RNA plant viruses. Viruses 4:1515–1536
Muehlbach HP, Mielke-Ehret N (2011) Genus Emaravirus. In: King AMQ, Adams MJ, Carstens EB, Lefkowitz E (eds) Virus taxonomy. Ninth report of the international committee on the taxonomy of viruses. Elsevier-Academic Press, Amsterdam, pp 767–769
Nene YL, Kannaiyan J, Haware MP, Reddy MV (1981) Pigeonpea diseases: resistance-screening techniques. Information Bulletin No. 9. International Crops Research Institute for the Semi-Arid Tropics, Patancheru, Andhra Pradesh, India, pp 1–14
Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 85:2444–2448
Singh AK, Agrawal KC, Rathi YPS (1999) Sterility mosaic of pigeonpea: a challenge of 20th Century. Indian J Virol 15:85–92
Skare JM, Wijkamp I, Rezende JAM, Kitajima EW, Park JW, Desvoyes B, Rush CM, Michels G, Scholthof KBG, Scholthof HB (2006) A new eriophyid mite-borne membrane-enveloped virus-like complex isolated from plants. Virology 347:343–353
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tatineni S, McMechan AJ, Wosula EN, Wegulo SN, Graybosch RA, French R, Heinb GL (2014) An eriophyid mite-transmitted plant virus contains eight genomic RNA segments with unusual heterogeneity in the nucleocapsid protein. J Virol 88:11834–11845
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 24:4876–4882
Yu C, Karlin DG, Lu Y, Wright K, Chen J, MacFarlane S (2013) Experimental and bioinformatic evidence that Raspberry leaf blotch emaravirus p4 is a movement protein of the 30K superfamily. J Gen Virol 94:2117–2128
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Acknowledgments
The authors are grateful to Prof. G. P Martelli for his critical reading of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
In order from RNA1 to RNA6, accession numbers are: HF912243-HF912246, HG939489, HG939490.
Rights and permissions
About this article

Cite this article
Elbeaino, T., Digiaro, M., Uppala, M. et al. Deep sequencing of dsRNAs recovered from mosaic-diseased pigeonpea reveals the presence of a novel emaravirus: pigeonpea sterility mosaic virus 2. Arch Virol 160, 2019–2029 (2015). https://doi.org/10.1007/s00705-015-2479-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2479-y