
Abstract
Campylobacter phage vB_CjeM_Los1 was recently isolated from a slaughterhouse in the Republic of Ireland using the host Campylobacter jejuni subsp. jejuni PT14, and full-genome sequencing and annotation were performed. The genome was found to be 134,073 bp in length and to contain 169 predicted open reading frames. Transmission electron microscopy images of vB_CjeM_Los1 revealed that it belongs to the family Myoviridae, with tail fibres observed in both extended and folded conformations, as seen in T4. The genome size and morphology of vB_CjeM_Los1 suggest that it belongs to the genus Cp8virus, and seven other Campylobacter phages with similar size characteristics have also been fully sequenced. In this work, comparative studies were performed in relation to genomic rearrangements and conservation within each of the eight genomes. None of the eight genomes were found to have undergone internal rearrangements, and their sequences retained more than 98% identity with one another despite the widespread geographical distribution of each phage. Whole-genome phylogenetics were also performed, and clades were shown to be representative of the differing number of tRNAs present in each phage. This may be an indication of lineages within the genus, despite their striking homology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Campylobacter jejuni is a Gram-negative microaerophilic human pathogen that is commonly found in the intestinal tracts of poultry. It is the number one bacterial cause of gastroenteritis in Europe, and in 2004, campylobacteriosis became a notifiable disease in Ireland [1]. Infection with C. jejuni (90% of campylobacteriosis cases) typically occurs through the improper handling of broiler meat; however, ingestion of lettuce and takeaway foods have also been identified as risk factors for contracting the disease [2]. It is estimated that 9.3 million cases of Campylobacter infection occur in Europe every year, with annual costs of approximately €2.4 billion [3]. Due to the self-limiting nature of the disease, many cases may go unreported, and therefore the actual incidence rates may be much higher. Since the implementation of mandatory reporting for Campylobacter infections in 2004, the Irish incidence rates have seen an overall increase [4].
In 2008, the European Food Safety Authority (EFSA) carried out an EU-wide survey on broiler carcasses and found an 83.1% presence of Campylobacter in 394 Irish broilers analysed, which was above the European average of 71.2% [5]. Current methods of Campylobacter reduction in broiler houses include strict biosecurity with the presence of fly screens to eliminate contamination from external sources, thorough cleaning of the broiler house in between flocks and reduction of the slaughter age [3]. Antibiotics used in broiler houses to reduce Campylobacter colonisation have included fluoroquinolones, which were primarily licenced for use in poultry in Spain in 1986 and, subsequently, worldwide. However, the reported emergence of Campylobacter resistance to such antibiotics has become a concern, as ciprofloxacin is one of the most commonly prescribed antibiotics in the case of serious campylobacteriosis in humans [6].
In recent years, bacteriophages have been proposed as a promising biocontrol measure for Campylobacter, and hence, significant research effort has been focussed on its phages. Virulent phages offer many advantages over antibiotics when attempting to eliminate bacteria from a surface, or even from a human or animal host. They are auto-dosing, in that phages, when administered as a single dose, will propagate in the presence of their bacterial host and generate new virus particles to continue to reduce the number of bacteria. Phages are generally specific for one bacterial species or even particular strains and so do not have the capability to diminish the microflora of a host, potentially leading to the establishment of opportunistic infections [7]. They are non-toxic, unlike some antibiotics, and they also have the capability to penetrate through bacterial biofilms, such as the ones produced by members of the genus Campylobacter [8, 9]. It is estimated that phages are the most abundant biological systems on the planet, and the number of phage particles may exceed 1031 [10]. Phages are typically found in their host environment, with reports of Campylobacter phage isolation from poultry and duck intestinal samples, abattoirs, poultry faeces and retail chicken [11,12,13].
Information collected on lytic Campylobacter phages from the National Collection of Type Cultures (NCTC, UK) has previously allowed these to be designated into groups based on characteristics determined by transmission electron microscopy (TEM) and pulsed field gel electrophoresis (PFGE) [14]. Further group characteristics have come to light in recent years, such as isolation frequency and bacterial defects associated with phage resistance (Table 1). These phages all belong to the family Myoviridae and contain genomic dsDNA. More recently, Javed et al. proposed an updated classification system, including the former group II phages in the genus Cp220likevirus, and the former group III phages in the genus Cp8unalikevirus, both within the subfamily Eucampyvirinae [15]. The International Committee on Taxonomy of Viruses’ (ICTV) 2016 Virus Taxonomy Release officially renamed these genera “Cp220virus” and “Cp8virus”, respectively, using Campylobacter virus CP81 as the Cp8virus type species [16].
Many in vitro and in vivo trials have been conducted to evaluate the efficacy of phages in the reduction of viable C. jejuni. It is estimated that reducing the numbers of Campylobacter cells in the intestines of broiler birds at slaughter by 3 log10 units could result in a 90% decrease in disease risk in humans. A 1 log10 unit reduction of Campylobacter cell numbers on broiler carcasses post-slaughter could result in a 50-90% risk reduction [3]. A study undertaken by Loc-Carrillo et al. determined that oral administration of Cp8viruses CP8 and CP34 in broilers could reduce faecal Campylobacter counts by up to 5 log10 CFU per g of cecal contents, and Campylobacter-contaminated chicken skin showed a reduction of approximately 1.2 log10 units when challenged with phage NCTC 12673 in comparison to a phage-free control [20, 23]. Here, we detail the bioinformatics analysis of the first fully sequenced Campylobacter phage isolated in the Republic of Ireland with comparative genomics of other fully sequenced Campylobacter phage of similar morphological descriptions and genome size.
Materials and methods
Phage isolation
Campylobacter jejuni subsp. jejuni PT14 (hereafter C. jejuni PT14) was used in this study due to its extensive characterisation and previous success in Campylobacter phage isolation [24]. C. jejuni PT14 was cultured on Blood-Free Campylobacter Selectivity Agar Base (Sigma Aldrich, UK) and incubated at 42 °C microaerophilically (Campygen Gas Generating Systems, Oxoid) for 24-48 h. The bacteria were then harvested into NZCYM broth (Sigma Aldrich, UK) prior to further use. Poultry faecal samples, acquired from Shannonvale Foods Ltd., Cork, were diluted 1:10 with SM buffer (100 mM NaCl, 8 mM MgSO4∙7H2O, 50 mM Tris-HCl, pH 7.5) and placed on a shaking platform overnight at room temperature. The samples were then centrifuged at 4500 g for 30 min, and the supernatant was filtered through 0.22-μm filter units. An enrichment step was included to isolate well-propagating phages. To approximately 10 ml of NZCYM broth, 3-4 ml of sample supernatant, 1 ml of C. jejuni PT14 suspension and 50 mM CaCl2 were added. These were incubated microaerophilically at 42 °C for 48 h on a shaking platform and then centrifuged and filtered as above. NZCYM overlays (0.4% agarose) were prepared and kept molten at 45 °C. Decimal dilutions of the enriched samples were prepared in SM buffer. Four hundred μl of C. jejuni PT14 suspension and 50 mM CaCl2 was added to each overlay, followed by 100 μl of each decimal dilution. The overlays were poured onto Anaerobe Basal Agar (Oxoid), allowed to set, and incubated microaerophilically for 24-48 h at 42 °C before inspection for plaque formation.
Phage propagation, lytic spectrum analysis and adsorption tests
Visible plaques were removed from the overlay and placed into SM buffer, which was subsequently filter-sterilised and serially diluted. Four hundred μl of C. jejuni PT14 suspension and 50 mM CaCl2 were added to molten NZCYM overlays, followed by 100 μl of each decimal dilution. The overlays were poured onto Anaerobe Basal Agar, allowed to set, and incubated microaerophilically for 24-48 h at 42 °C. This procedure was performed in triplicate to purify the phage. To propagate the phage, multiple overlays were prepared containing 400 μl of C. jejuni PT14 suspension, 50 mM CaCl2 and a phage titre sufficient to produce confluent lysis after incubation. To harvest the phage (hereafter named Los1), 4 ml of SM buffer was poured onto each overlay and the plates were placed on a shaking platform for 2 h. The SM buffer was then removed from each plate and filtered with 0.22-μm filter units, and the titre (plaque forming units [PFU]/ml) was determined by plaque assay. The lytic spectrum of the phage was investigated by performing plaque assays of the phage on a lawn of C. jejuni and C. coli isolates. To monitor phage adsorption, C. jejuni PT14 and phage Los1 were combined at a multiplicity of infection (MOI) of 0.0025 in NZCYM broth prewarmed to 42 °C. Aliquots were removed initially (in triplicate) and filtered. Aliquots were then taken for membrane filtration every 5 min for 20 min. Plaque assays were performed as described previously to enumerate unadsorbed phage. The experiment was performed in triplicate.
Transmission electron microscopy (TEM)
For TEM analysis, Los1 was propagated to a titre of 108 PFU/ml as described above and treated overnight with 15% polyethylene glycol (PEG) 8000 at 4 °C. This was then centrifuged at 5000g for 1 h, the supernatant was discarded, and the pellet was resuspended in 5 ml of SM buffer. The residual PEG was removed by adding an equal volume of chloroform and centrifuging for 5 min at 10,000 g. The aqueous phase was removed and used for negative staining of phages with 1% (w/v) uranyl acetate and with 1% w/v) ammonium molybdate on ultra-thin carbon films. The specimen was subsequently picked up with 400 mesh grids and used for transmission electron microscopy (Tecnai 10, FEI Thermo Fisher Scientific, The Netherlands) at an acceleration voltage of 80 kV.
Phage DNA sequencing and bioinformatics analysis of Los1
Phenol extractions were initially used to extract phage DNA from high-titre phage suspensions. However, the DNA yield and purity were too low to be deemed acceptable for sequencing. This correlates with the findings of Aruntyunov et al., who observed that the majority of Campylobacter phage NCTC1673 DNA remained in the phenol phase during extractions due to protein-bound DNA [25]. As an alternative approach, the Wizard® DNA Clean-Up System (Promega) was used to extract the phage DNA from a Los1 suspension treated overnight with 10 U of DNase and 10 μg of RNase A at 37 °C. Four ml of resin was mixed with the phage suspension, the mixture was passaged through the binding column, and the filtrate was discarded. The column was washed twice with 80% isopropyl alcohol (IPA) and centrifuged at 20,000g for 5 min to remove residual IPA. Nuclease-free water was heated to 90 °C and added to the column, which was then centrifuged at 20,000g to elute the phage DNA. Whole-genome sequencing of the phage DNA was performed using the Illumina platform, and reads were assembled using SPAdes v. 3.5.0 [26]. Open Reading Frames (ORFs) were predicted using GLIMMER v.3.02 [27] and Prodigal v.1.20 [28]. A putative function was assigned to each ORF based on BLASTP analysis at NCBI (http://www.ncbi.nlm.nih.gov/) and Pfam matches (EMBL-EBI) [29] with an e-value cutoff of 1.0. The programme Snapgene Viewer (GSL Biotech; available at www.snapgene.com) allowed for construction and visualisation of the genome map. TMHMM Server v.2.0 (http://www.cbs.dtu.dk/services/TMHMM/) was used to predict transmembrane regions, and putative signal peptide cleavage sites were determined using the SignalP 4.1 server [30]. The presence of inteins was investigated by aligning each ORF against the intein database, InBase (http://tools.neb.com/~vincze/blast/index.php?blastdb=inbase, no longer supported by NEB Biolabs) with a maximum e-value of 1.0. To align recurring motifs in the phage genome and identify ribosomal binding sites, 100-bp upstream regions of each ORF were analysed by MEME Suite v. 4.11.2, using MEME Motif Discovery [31]. tRNAscan SE v.1.21 (Eddy lab) [32] was used to identify phage tRNA, searching with tRNAscan and EufindtRNA with strict and relaxed (INT cutoff = -32.1) parameters, respectively. Codon usage frequencies for Los1 and a C. jejuni type strain (subsp. jejuni NCTC 11168) were generated using the codon usage finder Kazusa (available at http://www.kazusa.or.jp/codon/), and these were compared in frequency/1000, taking into consideration the tRNAs present in both genomes. Using BLASTP matches for the large terminase protein subunit, sequences were aligned using MUSCLE, and a maximum-likelihood phylogenetic tree was constructed with Mega7 [33] in an attempt to predict the phage packaging mechanism for Los1. Bootstrap analysis was performed with 100 replicates.
Comparative genomics of Campylobacter phages
To date, seven Campylobacter phages of similar genomic size to Los1 have been sequenced. Table 2 contains information about these phages and Los1, including the number of ORFs and tRNAs predicted in each phage, the geographical origin of the phage, and the sample type from which each phage was isolated. Using the sequences of all eight genomes listed in Table 2, global alignments were generated using progressiveMAUVE [34] to compute sequence identity and to visualise any genomic rearrangements. BLAST Ring Image Generator (BRIG) [35] was employed to generate a circular image displaying the BLASTP comparison of ORFs from the abovementioned Campylobacter phages. All-against-all dot plots were constructed with whole-genome fasta files in Gepard [36]. Mulan (MUltple sequence Local AligNment and visualization tool) is a program that utilises a TBA (Threaded Blockset Aligner) algorithm for whole genomes. The eight Campylobacter phage genomes, were submitted to Mulan, and following multi-sequence alignments, a neighbour-joining phylogenetic tree was constructed [37].
Results and discussion
Phage isolation, lytic spectrum and transmission electron microscopy
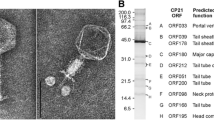
Phage Los1 was isolated from a fresh poultry faecal sample taken from a holding crate at a slaughterhouse in Cork, Ireland. After plaque assay analysis of the enriched sample, clearings were visible, and hence, purification and propagation of the phage were performed, which typically yielded titres of ~ 109 PFU/ml. TEM images revealed that phage Los1 belongs to the family Myoviridae (head diameter: 94.7 ± 3.4 nm, tail length: 102.5 ± 3.1 nm, tail width: 19.3 ± 0.8 nm [n = 12]). The tail fibres (with ca. 1/3 of the tail length) can be seen in a number of conformations, including phage particles with fibres folded up on the tail in an upwards direction (Fig. 1c) and also attached in a downwards ‘rosette-like’ position from the tail end (Fig. 1a). These differing tail fibre conformations have also been observed for other C. jejuni (group III) phages and also T4 [42, 43]. For the long tail fibres of the myovirus T4 [43], it has been postulated that maintaining tail fibres against the virion body can allow the phage to diffuse more rapidly in liquid media in search for a host. Delicate tail fibres may also be folded in free phage for protection against low pH and unfavourable temperatures; however, this can compromise the adsorption rate and successful DNA injection into the bacterial host [42, 43]. Tail fibres can undergo conformational changes and extend when their respective receptor proteins are detected, or when conditions allow phage progeny to remain viable [42]. The adsorption of phage Los1 to C. jejuni PT14 in broth typically results in approximately 50% unbound phage after a 20-min incubation at an MOI of 0.0025; the large proportion of unbound phage is possibly due to retracted tail fibres. The host range of Los1 was briefly investigated using plaque assays against 26 C. jejuni strains and six C. coli strains and was found to have no infectivity against any of the C. coli strains tested and to form plaques on 27% of the C. jejuni isolates (Supplemental Material, Table S1.1). This limitation of infectivity to C. jejuni strains is in accordance with the lytic spectra observed for other Campylobacter phages with similar head diameter [41, 44, 45].

Transmission electron micrographs of phage Los1 stained with 1% (w/v) uranyl acetate (a-d) or with 1% (w/v) ammonium molybdate (image e). The arrow in image a indicates distal globular structures of tail fibers forming a rosette-like structure beneath the tail. Open triangles in image c indicate tail fibers attached in upward positions on the tail surface. The black capsid and contracted tail seen in image d indicates receptor binding at globular vesicles with subsequent DNA liberation. In image e, tail fibres are shown detached from the tail due to the alternative staining method
Bioinformatic analysis Los1 genome sequence
General genomic features
Sequence analysis of phage Los1 revealed a 127-bp terminal repeat, indicating a circularly permuted genome, and removal of such yielded a single-copy genome of 134,073 bp with a GC content of 26.2%, 4.3% lower than that of its propagating host, C. jejuni PT14 [24]. This higher AT content, commonly seen in phage relative to their host, is thought to be transcriptionally advantageous, as polymerases may succeed in melting phage DNA with more ease than host DNA [46]. In combination with the TEM image (Fig. 1) depicting a myovirus with an icosahedral head of approximately 95 nm in diameter, the genome size allows a tentative categorisation of phage Los1 into the genus Cp8virus. Coding sequences account for 92.7% of this genome, with 169 ORFs predicted (gene density, 1.26), many of which overlap. The vast majority of these (approx. 87%) are encoded on the reverse strand. Phage Los1 predominantly uses AUG as a start codon for protein synthesis; however, GUG and CUG are also used, albeit at lower frequencies (0.59% and 3.7%, respectively). BLASTP and Pfam analysis allowed a putative function to be assigned to 71 of the predicted ORFs. The highest-scoring homologs, along with their scores, can be seen in Supplementary Data 2. Eleven hef-like homing endonucleases were also identified, each containing regions of homology to each other.
As noted in Campylobacter phage Cp81 [39], the genome of Los1 is not modular, with a seemingly arbitrary arrangement of ORFs. No obvious organisation could be observed, and many protein subunits are present along the genome at some distance from one another, such as the terminase (Los1_001, Los1_027) and the DNA primase (Los1_054, Los1_160). This may be, in part, due to the presence of homing endonucleases.
tRNAs and Los1 ORF codon usage
Four tRNAs are present in the genome of Los1, clustered between Los1_040 and los1_041. These tRNAs (met-CAT, asn-GTT, arg-TCT and tyr-GTA) are also present in the host [24], so their retention in the genome would not appear to confer a transcriptional advantage for phage genes. When codon usage in Los1 was compared to the codon usage within a C. jejuni genome in the Kasuza database (seen in Supplemental Material 1, Fig. S1.1), the codons for which there are tRNAs in Los1 (aside from tRNAmet) were used at a slightly higher frequency in Los1. This, however, is the case for many other codons for which there are no tRNAs present. The requirement of Los1 tRNAs for transcription of highly expressed genes was investigated, and while AGA was the predominantly used codon for arginine in all ORFs, AAC and TAC were used at low frequencies, with the exception of both putative topoisomerase subunits (Los1_129, Los1_131), baseplate subunits (Los1_146, Los1_156), the ssDNA binding protein (Los1_167), the tail fibre protein (Los1_162) and the putative RNAse H (Los1_164). Retention of Los1 tRNA may be beneficial in the transcription of these particular ORFs.
Replisome of Los1, DNA modification and transcription
Phage Los1 encodes for many putative proteins involved in DNA replication, repair and nucleotide modification. As in the highly characterised replisome of T4 [47], Los1 contains genes for a DNA polymerase (Los1_048), primase and helicase proteins, which may come together to form the primosome (Los1_036, Los1_037, Los1_052, Los1_150), a sliding clamp and clamp loader subunits (Los1_158, Los1_119, Los1_156, respectively) and a ssDNA binding protein (Los1_167). Two ORFs encode the phage topoisomerase, Los1_129 and Los1_131, which may have originated as one larger ORF but are now separated by a hef-like gene. Many other genes are predicted to have functions in DNA synthesis, such as a thymidylate synthetase (Los1_003), ribonucleotide reductase (subunits Los1_017, Los1_108, Los1_109) and thymidine kinase (Los1_052), and DNA repair, for example, the putative DNA repair and recombination protein (Los1_038). Phage DNA methylation functions to protect phage DNA from host restriction systems [48], and genes for three methylases were predicted in Los1 (Los1_042, Los1_043, Los1_049). Los1_042 putatively encodes an adenine-specific methylase, which is significant due to the low GC content of the genome, indicating that base methylations are widespread in the Los1 genome. The DNA of Campylobacter phages are notoriously difficult to digest using restriction enzymes [13], even when known restriction sites are located in the genome, which is unsurprising considering the DNA protection conferred by base modifications.
Los1 does not appear to encode its own RNA polymerase; however, a conserved motif (e value 1.2e-23) was found upstream of 12 ORFs containing the consensus sequence of the C. jejuni-10 promoter region, indicated by pink arrows in Fig. 2. Los1_068 was identified as the sigma factor for a late transcription gene with sequence similarity to the same gene in t4; however, no obvious t4-like late promoter was identified. One hundred forty-three ORFS were also preceded by a motif matching a consensus ribosomal binding site (RBS) sequence in C. jejuni further downstream from the putative -10 sequence (Fig. S1.2) [49].

Genome map of phage Los1 with legend (inset). ORFs are coloured in relation to putative function as determined by BLASTP and Pfam matches (see Supplemental Material 2). Putative promoters are indicated by pink arrows (colour figure online)
Phage DNA packaging mechanism
Los1 contains one intein, located at the N-terminal end of the large terminase subunit (Los1_027), which may facilitate the assembly of both terminase subunits. Phylogenetic analysis of large terminase subunits from phages with known packaging mechanisms resulted in the incorporation of Los1_027 into the clade of terminases belonging to phages with t4-like headful DNA packaging.
ORFs involved in progeny release
Los1_127 was found to contain a soluble transglycosylase (SLT) domain, which may function to degrade host peptidoglycan. SLT domains have been found in phage endolysins with N-acetyl-β-muramidase and carboxypeptidase/endopeptidase activity, and they appear to be limited to phages with specificity for proteobacteria [50]. The nucleotide sequence of Los1_127 also seems to be highly conserved among Los1 and the Campylobacter phages listed in Table 2 (BLASTN e value, 0 for all alignments). Los1_127 also contains a putative signal peptide at the N-terminus of the protein, with a predicted cleavage site between positions 21 and 22 (D-score 0.644), indicating that this protein may function as a signal arrest release (SAR) endolysin [51, 52]. Experimental studies are necessary to confirm this possibility. No Los1 protein was found to be homologous to a previously determined holin in BLASTP searches; however, if Los1_127 is a SAR endolysin, it is likely that any holin present in the genome would be a pinholin, acting to allow the passage of ions across the host membrane to allow for membrane depolarisation rather than allowing passage of the entire endolysin to the periplasm [53]. A likely pinholin candidate is Los1_053, a small protein that is 62 residues in length and contains two predicted transmembrane domains and shows homology to a Bacillus simplex Na +/K + antiporter (e value, 2.2). This ORF is also highly conserved among the Campylobacter phages listed in Table 2. Phage holins are typically encoded adjacent to the endolysin gene; however, as stated above, the genome of Los1 lacks modularity. A large gene encoding a putative peptidase (Los1_140) was identified, 889 amino acids in length, making it the second-largest protein in Los1 (second to Los1_162, encoding the large tail fibre subunit). This sequence was not predicted to contain any transmembrane, signal or binding domains, and the only functional domain that could be identified was a D-alanyl-D-alanine carboxypeptidase enzymatic domain (e-value, 1.6 × 10−8) between residues 459 and 558. These domains have been predicted in many Gram-positive and Gram-negative phage endolysins in comparative studies, but some have been experimentally proven to function as L-alanine-D-glutamate endopeptidases [50]. As of yet it is unclear whether or not this protein has a function in degradation of peptide bonds in the host murein.
Los1 DNA binding protein
Regarding the difficulties experienced when extracting DNA from Los1 particles, it was expected that one ORF from the genome would encode a DNA-binding protein as was demonstrated experimentally for Gp001 of Campylobacter phage NCTC 12673 [25]. In that study, it was found that Gp001 encoded a protein that, when complexed with the DNA of NCTC 12673, even at low pH, allowed it to evade degradation by bacterial nucleases and remain stable in the acidic environment that may be encountered in the animal gut. Los1_118 was found to share 100% sequence identity with Gp001 (BLASTN), and it can thus be assumed that this same DNA-binding protein hindered Los1 DNA extraction attempts.
Los1 YopX proteins
Two putative YopX (Yersinia outer protein X) family proteins were found during the annotation of Los1, Los1_087 and Los1_088. While found in other phages [54, 55], their role in phage genomes is largely unknown. YOPs are secreted proteins that contribute to the pathogenicity of Yersinia pestis and the evasion of the host’s innate immune system, and specifically, YOPX has been shown to play a role in the adhesiveness of bacteria to eukaryotic cells and mediation of serum resistance [56]. Both YOPX proteins in Los1, when subjected to BLASTP analysis, showed varying degrees of sequence similarity to hypothetical proteins in C. jejuni and C. coli. If these proteins confer the same advantages to invasive Campylobacter as in Y. pestis, it could be hypothesized that their presence in Los1 may promote the invasion and retention of phage-infected Campylobacter into host epithelial cells, and out of the harsh environment of the intestinal tract, where free phages may be less stable (Fig. 3).

Maximum-likelihood phylogenetic tree generated using phage large terminase subunits. The intein sequence located in the Los1 terminase subunit was removed to give a more accurate prediction of function. Bootstrapping values above 50 are displayed at the nodes. Coloured labels correspond to differing phage DNA packaging mechanisms: P22-like headful (orange), 3’-extended COS ends (pink), T1-like headful (grey), T4-like headful (green), λ-like 5’-extended COS ends (light blue), T7-like direct terminal repeats (blue), mu-like headful (black), P2-like 5’-extended COS ends (purple), T1-like headful (grey) (colour figure online)
Comparative genomics of Campylobacter group III phages
To date, eight fully assembled Campylobacter group III phage genomes, all of similar size, have been sequenced, including vB_CjeM_los1 (Table 2). These were globally aligned using progressiveMAUVE, but two of the eight genomes aligned in the reverse compliment direction and required inversion. Subsequently, when all sequences were aligned, it was revealed that all were circular permutations of another (Fig. 4), which was also noted in phage CP81 with Bal-31 exonuclease assays [39]. When the genome sequences were then corrected to begin at their respective regions homologous to Los1_001 and aligned, locally collinear block (LCB) weights and lengths indicated that all eight sequences differed by just 1.43%, excluding an LCB (green LCB within a hef-like ORF in Fig. 5) seen in just CPX and NCTC 12673. Considering their significant similarity to campylobacter virus CP81 (the type member of the genus Cp8virus), the place of the seven other listed Campylobacter phage genomes in the genus Cp8virus can be established. Also noted is the conservation of arrangement from genome to genome. hef-like genes, which are free-standing selfish genetic elements with no discernible role in phage progeny production [57], were identified in each of the phage genomes, and the number of these genes in each member of the genus Cp8virus can be seen in Table 2. These endonucleases cleave phage DNA without affecting phage viability and allow the homing endonuclease gene to insert itself into hef-free cognate sites in other phage genomes [58]. These sites can be within genes, and in all of the eight genomes, the topoisomerase gene has apparently been divided into two subunits by the insertion of a hef-like sequence. There is evidence of three other hef-related gene-splitting events in all genomes, in accordance with the documented findings of a genome analysis of phage CP81 [39]. There are three tail tube proteins in each of the Cp8viruses, which might have originated as one larger protein (grey ORFs in Fig. 5). Two genes lie adjacent to one another (possibly split by an ancient nonsense mutation), while a third part can be found a significant distance away, next to a hef-like gene, which may be responsible for the separation. Many Cp8virus proteins share a degree of sequence identity with T4, leading to the assumption that an evolutionary relationship exists. However, unlike T4, with its highly organised genome [59], Cp8viruses do not display this ordered arrangement of early, middle and late genes, possibly caused in part by hef homologues. In T4, the genes encoding the major head protein and capsid protein are side by side (gp23 and gp24, respectively), but in the Cp8virus genomes, they are distantly located on opposite strands (orange-coloured ORFs in Fig. 5). Both ORFs are in the vicinity of hef-like genes. A similar case can be observed in all genomes with the portal vertex protein and the large terminase subunit (pink-coloured ORFs in Fig. 5). Using Los1 as an example, these proteins are encoded 24 ORFs apart but may have evolved from the same T4 DNA packaging protein, as they align with two regions of T4’s gp17. Hef-like homing endonucleases are also present in the vicinity of these two Cp8virus ORFs, indicating a role in the gene split. As can be seen in Fig. 5, in six of the phage genomes, the ribonucleotide-diphosphate reductase subunit is flanked by two hef-like genes, one encoded upstream on the plus strand and one downstream on the minus strand. In phages CP30A and PC5, an additional hef-like gene appears to have inserted itself on the minus strand between the upstream hef-like sequence and the ribonucleotide-diphosphate reductase subunit. While slightly varying numbers of these selfish elements are present within each of the eight Campylobacter phages, no instance of genomic rearrangement from genome to genome caused by these homing endonucleases could be identified. Duplication of regions flanking the hef-like sequences was also not observed.

progressiveMAUVE alignment of Campylobacter phage genomes showing circular permutations

progressiveMAUVE alignment of Campylobacter phage genomes in GenBank format. White boxes along the length of each genome represent individual ORFs, and the position above or below the line corresponds to whether the ORF is encoded on the plus (above) or minus (below) strand of DNA. Conserved genome segments are indicated by locally collinear blocks (LCBs) for which weights have been computed (data not shown). hef-like genes are indicated with green boxes, topoisomerase subunits are coloured in blue, ORFs coloured yellow indicate the putative ribonucleotide diphosphate reductase subunit, tail tube protein sequences are shaded grey, pink ORFs indicate the large terminase subunit, and the portal vertexf protein and major head and capsid proteins are coloured orange. Diagonal red lines in the genomes of Cp8 and CpX indicate a region in each genome with homology to a hef-like homing endonuclease but containing a nonsense mutation (colour figure online)
Details of BLASTP comparisons of all Los1 ORFs with predicted ORFs of other Cp8viruses are shown in Fig. 6. What is obvious is the high degree of conservation within the majority of genes, despite the worldwide distribution of these phages (Table 2). Proteins predicted to have involvement in phage structure, DNA replication, and lysis have retained sequence similarity to one another. Genomic regions displaying the highest levels of variation within the genomes include clusters of small genes for which no function could be ascertained, as well as hef homologues. In certain cases, single Los1 ORFs are shown to be separated into two homologous ORFs in other phage, and vice versa. For instance, Los1_162 and Los1_163 (tail fibre subunits) are encoded in one large ORF in Cp30a (ORF 49). This may be attributed to random nonsense mutations and also might account, in part, for the differing number of ORFs predicted in each of the phages. It can also be considered that ORF prediction software has advanced since the first report of Cp8virus genome sequences in 2011[38, 39]. Some protein sequences in the GenBank file of NCTC 12673 were not available, and these correspond to the gaps seen in the relevant ring in Fig. 6. BLASTN, however, confirmed that non-hypothetical proteins of Los1 (for example, Los1_001) did indeed have homologous counterparts in NCTC 12673. Also, the genome of phage Cp81 was opened at a region that split an ORF encoding one of the tail fibre subunits (seen as a gap in the CP81 ring in Fig. 6). Opening of the genome at an alternative location led to the assembly of this subunit for further analysis (Fig. 7).

BRIG output showing BLASTP ORF comparisons of Cp8virus GenBank files, using Los1 as the reference genome. Upper and lower identity thresholds were set to 70% (above which, ORFs are represented with solid colours) and 50% (above which ORFs show shaded colouring), respectively. The innermost circle displays GC content. Coloured rings denote phage genomes; CP8, blue; CP30A, dark blue; CP81, purple; CPX, fuschia; PC14, orange; NCTC12673, green; PC5, dark green. Regions of variability highlighted using * indicate hef homologues, and the numbers bordering the image correspond to the following ORFs in Los1: 1, putative exonuclease (Los1_044); 2, putative methyltransferase (Los1_049); 3, variable region of ORFs with no discernible function; 4, putative baseplate hub and tail lysozyme (Los1_146); 5, putative baseplate wedge subunit (Los1_156); 6, tail fibre subunits (Los1_162, Los1_163) (colour figure online)

Gepard dot plots of phage genome sequences. A. Eight Campylobacter phage sequences. Diagonal, continuous black lines demonstrate the similarity of the genomes to one another without rearrangements or major deletions. Minor deletions can be visualised in the warping of these lines. The background noise can be attributed to repetitive sites in the genomes, which is unsurprising, considering the high AT content of each, and also ORFs such as hef-like genes, which may appear as repeated elements, considering their homology to one another. B. Sequences from Los1, Campylobacter phage Cp220 (from the genus Cp220virus), Enterobacter phage T4, and Bacillus phage SPP1. The absence of diagonal lines in segments containing two different phage genome sequences indicates a lack of similarity between those corresponding genome sequences
The tail fibre subunits were compiled from the Campylobacter phages listed above and aligned with ORF 049 in phage CP30A using BLASTP. While the sequences themselves retain a high percentage of identity to segments of ORF 049 in CP30A, the locations at which the protein is divided up into subunits vary dramatically (Fig. 8). These varying ORF subunits within the tail fibre protein result from previously identified mutations [41] and may result in shorter tail fibres.

Tail fibre subunits of Campylobacter phages as encoded in their respective genomes. The scale refers to the number of nucleotides. As can be seen from the image, many of the tail fibres are encoded by two subunits of varying size. The protein is intact in CP30A and NCTC12673 and is encoded in three parts in phage CPX
In previous studies, the receptor-binding protein (RBP) was found to be localised to the C-terminus of the tail fibre subunit in phage NCTC12673 [60]. BLASTP analysis of this sequence revealed homologous sequences not only in Cp8viruses but also in four members of the genus Cp220virus, all with sequence identity above 91%. The putative RBPs in Cp220viruses are also located at the C-terminus of their respective tail fibre proteins. These sequence homologs were aligned using MUSCLE (Fig. S1.3), and phylogenetic analysis was performed. While some bootstrap values are below 50%, this tree (Fig. S1.4) shows that neither Cp8viruses nor Cp220viruses cluster together as separate clades. Host resistance to Cp220viruses results in motility defects in Campylobacter strains, indicating that flagellin is the phage receptor, while Cp8viruses appear to use the capsular polysaccharide for host binding. The presence of putative RBPs in Cp220viruses homologous to those seen in Cp8viruses is surprising; however, Sørensen et al. noted that, in transmission electron microscopy images of Campylobacter type II and type III phages (since renamed Cp8viruses and Cp220viruses, respectively), the tail fibres of type II phage contained distal globular structures, possibly indicating an additional tail fibre subunit [42].
Conservation of ORFs is evident among the Cp8viruses, and regarding tRNAs, all of the listed genomes contain tRNAs for met-CAT, asn-GTT and tyr-GTA. Los1 and CP30A contain and additional tRNAARG, and CP81, CPX and CP8 also have a tRNALEU. When whole-genome alignments were performed using the program Mulan, a phylogenetic tree was also generated based on the neighbour-joining method (Fig. 9). It may be noteworthy that, phylogenetically, Cp8viruses have been grouped in a manner that represents their tRNA content, and tRNA analysis may be an indication of evolutionary relationship.

Whole-genome neighbour-joining phylogenetic tree of Cp8viruses generated using Mulan. Tree distances are in number of substitutions per 1 kb. Cp8viruses are coloured according to the number of tRNAs in their genome: 5 tRNAs, red; 3 tRNAs, green; and 4 tRNAs, blue (colour figure online)
Conclusions
With growing concerns regarding food safety and antibiotic resistance, bacteriophages are being looked to as an alternative for reduction of bacterial pathogens in food production. There have been frequent new reports of phage isolation and characterisation generating data to allow phages to be grouped taxonomically into families and genera. There are eight Campylobacter group III phages fully sequenced to date that show homology to the newly isolated phage Los1 (Table 2). In the ICTV proposal leading to the creation of the genus Cp8virus [61], the reasoning for such groupings was that Campylobacter phages NCTC 12673 and CPX shared 95% sequence identity to phage CP81. As highlighted in Fig. 4, all eight Campylobacter phages share over 98% sequence identity, cementing their placement in the genus. The level of conservation is even more impressive when considering that the phages were isolated in locations across Europe and the USA. The greatest level of diversity between the genomes can be seen outside of known major functional ORFs and within regions encoding hef-like proteins and smaller hypothetical proteins. In relation to proteins such as the putative endolysin and holin, their identity and retention in all genomes would further indicate their functional importance.
As regards the opening of Cp8virus genome sequences annotated so far, there does not appear to be a consensus start position, but for future genus members, it may be ideal to ensure that the sequences, firstly, are all read in the same direction (with the majority of ORFs encoded on the negative DNA strand) and, secondly, begin with the ORF of the terminase subunit homologous to Los_001, if available. This would make further comparative studies more straightforward.
More studies, both in vitro and in silico are necessary to correctly assign function to current hypothetical proteins of these phages. While approximately 42% of Los1 ORFs have been elucidated, a large proportion still remain unknown and may give further insight into the replication methods and survival mechanisms of Cp8viruses.
References
Health Protection Surveillance Centre (2014) Epidemiology of campylobacter in Ireland, 2013. http://www.hpsc.ie/A-Z/Gastroenteric/Campylobacter/Publications/AnnualReportsonCampylobacteriosis/. Accessed 12 Feb 2017
Danis K, Di Renzi M, O’Neill W, Smyth B, McKeown P, Foley B, Tohani V, Devine M (2009) Risk factors for sporadic Campylobacter infection: an all-Ireland case–control study. Euro Surveillance 14(7)
European Food Safety Authority (2011) Scientific opinion on Campylobacter in broiler meat production: control options and performance objectives and/or targets at different stages of the food chain. EFSA J. https://doi.org/10.2903/j.efsa.2011.2105
Health Protection Surveillance Centre (2004-2015) Epidemiology of campylobacter in Ireland. http://www.hpsc.ie/A-Z/Gastroenteric/Campylobacter/Publications/AnnualReportsonCampylobacteriosis/. Accessed 12 Feb 2017
European Food Safety Authority (2010) Analysis of the baseline survey on the prevalence of Campylobacter in broiler batches and of Campylobacter and Salmonella on broiler carcasses in the EU, 2008—Part A: Campylobacter and Salmonella pr. EFSA J. https://doi.org/10.2903/j.efsa.2010.1503
Engberg J, Aarestrup FM, Taylor DE, Gerner-Smidt P, Nachamkin I (2001) Quinolone and macrolide resistance in Campylobacter jejuni and C. coli: resistance mechanisms and trends in human isolates. Emerg Infect Dis 7(1):24–34. https://doi.org/10.3201/eid0701.700024
Wittebole X, De Rooke S, Opal SM (2013) A historical overview of bacteriophage therapy as an alternative to antibiotics for the treatment of bacterial pathogens. Virulence 5(1):226–235. https://doi.org/10.4161/viru.25991
Loc-Carrillo C, Abedon ST (2011) Pros and cons of phage therapy. Bacteriophage 1(2):111–114. https://doi.org/10.4161/bact.1.2.14590
Siringan P, Connerton PL, Payne RJH, Connerton IF (2011) Bacteriophage-mediated dispersal of Campylobacter jejuni biofilms. Appl Environ Microbiol 77(10):3320–3326. https://doi.org/10.1128/AEM.02704-10
Chibani-Chennoufi S, Bruttin A, Dillmann ML, Brüssow H (2004) Phage-host interaction: an ecological perspective. J Bacteriol 186(12):3677–3686. https://doi.org/10.1128/JB.186.12.3677-3686.2004
Hansen VM, Rosenquist H, Baggesen DL, Brown S, Christensen BB (2007) Characterization of Campylobacter phages including analysis of host range by selected Campylobacter Penner serotypes. BMC Microbiol. https://doi.org/10.1186/1471-2180-7-90
Grajewski BA, Kusek JW, Gelfand HM (1985) Development of a bacteriophage typing system for Campylobacter jejuni and Campylobacter coli. J Clin Microbiol 22(1):13–18
Atterbury RJ, Connerton PL, Dodd CER, Rees CED, Connerton IF (2003) Isolation and characterization of Campylobacter bacteriophages from retail poultry. Appl Environ Microbiol 69(8):4511–4518. https://doi.org/10.1128/AEM.69.8.4511-4518.2003
Sails AD, Wareing DRA, Bolton FJ, Fox AJ, Curry A (1998) Characterisation of 16 Campylobacter jejuni and C. coli typing bacteriophages. Med Microbiol 47(1):123–128. https://doi.org/10.1099/00222615-47-2-123
Javed MA, Ackermann H, Azeredo J, Kropinski A (2013) A suggested classification for two groups of Campylobacter Myoviruses. Arch Virol. https://doi.org/10.1007/s00705-013-1788-2
Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, Kropinski A, Krupovic M, Kuhn JH, Mushegian A, Nibert M, Sabandzovic S, Sanfacon H, Siddel SG, Simmonds P, Varsani A, Zerbini FM, Gorbalenya AE, Davidson AJ (2016) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Adv Virol. https://doi.org/10.1007/s00705-016-2977-6
Connerton PL, Timms AR, Connerton IF (2011) Campylobacter bacteriophages and bacteriophage therapy. J Appl Microbiol 111(1):255–265. https://doi.org/10.1111/j.1365-2672.2011.05012.x
El-Shibiny A, Connerton PL, Connerton IF (2007) Campylobacter succession in broiler chickens. Vet Microbiol 125(3–4):323–332. https://doi.org/10.1016/j.vetmic.2007.05.023
Hwang S, Yun J, Kim KP, Heu S, Lee S, Ryu S (2009) Isolation and characterization of bacteriophages specific for Campylobacter jejuni. Microbiol Immunol 53(10):559–566. https://doi.org/10.1111/j.1348-0421.2009.00163.x
Loc Carrillo CM, Atterbury RJ, El-Shibiny A, Connerton PL, Dillon E, Scott A, Connerton IF (2005) Bacteriophage therapy to reduce Campylobacter jejuni colonization of broiler chickens. Appl Environ Microbiol 71(11):6554–6563. https://doi.org/10.1128/AEM.71.11.6554-6563.2005
Loc Carrillo CM, Connerton PL, Pearson T, Connerton IF (2007) Free-range layer chickens as a source of Campylobacter bacteriophage. Antonie Van Leeuwenhoek 92(3):275–284. https://doi.org/10.1007/s10482-007-9156-4
Coward C, Grant AJ, Swift C, Philp J, Towler R, Heydarian M, Frost JA, Maskell DJ (2006) Phase-variable surface structures are required for infection of Campylobacter jejuni by bacteriophages. Appl Environ Microbiol 72(7):4638–4647. https://doi.org/10.1128/AEM.00184-06
Goode D, Allen VM, Barrow PA (2003) Reduction of experimental Salmonella and Campylobacter contamination of chicken skin by application of lytic bacteriophages. Appl Environ Microbiol 69(8):5032–5036. https://doi.org/10.1128/AEM.69.8.5032-5036.2003
Brathwaite KJ, Siringan P, Moreton J, Wilson R, Connerton IF (2013) Complete genome sequence of universal bacteriophage host strain Campylobacter jejuni subsp. jejuni PT14. Genome Announc 1(6):e00969–13. https://doi.org/10.1128/genomea.00969-13
Arutyunov D, Szymanski CM, Calendar R (2015) A novel DNA-binding protein from Campylobacter jejuni bacteriophage NCTC12673. FEMS Microbiol Lett 362(21). https://doi.org/10.1093/femsle/fnv160
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–477. https://doi.org/10.1089/cmb.2012.0021
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL (1999) Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27(23):4636–4641
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform 11(1):119. https://doi.org/10.1186/1471-2105-11-119
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44(D1):D279–D285. https://doi.org/10.1093/nar/gkv1344
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. https://doi.org/10.1038/nmeth.1701
Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. In: Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. AAAI Press, Menlo Park, California, pp 28–36
Lowe TM, Eddy SR (1997) tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147. https://doi.org/10.1371/journal.pone.0011147
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA (2011) BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. https://doi.org/10.1186/1471-2164-12-4
Krumsiek J, Arnold R, Rattei T (2007) Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23(8):1026–1028. https://doi.org/10.1093/bioinformatics/btm039
Ovcharenko I, Loots GG, Giardine BM, Hou M, Ma J, Hardison RC, Stubbs L, Miller W (2005) Mulan: Multiple-sequence local alignment and visualization for studying function and evolution. Genome Res 15(1):184–194. https://doi.org/10.1101/gr.3007205
Kropinski AM, Arutyunov D, Foss M, Cunningham A, Ding W, Singh A, Pavlov AR, Henry M, Evoy S, Kelly J, Szymanski CM (2011) Genome and proteome of Campylobacter jejuni bacteriophage NCTC 12673. Appl Environ Microbiol 77(23):8265–8271. https://doi.org/10.1128/AEM.05562-11
Hammerl JA, Jäckel C, Reetz J, Beck S, Alter T, Lurz R, Barretto C, Brüssow H, Hertwig S (2011) Campylobacter jejuni group III phage CP81 contains many T4-Like genes without belonging to the T4-type phage group: Implications for the evolution of T4 phages. J Virol 85(17):8597–8605. https://doi.org/10.1128/JVI.00395-11
Loc Carrillo C, Atterbury RJ, El-Shibiny A, Connerton PL, Dillon E, Scott A, Connerton IF (2005) Bacteriophage therapy to reduce Campylobacter jejuni colonization of broiler chickens. Appl Environ Microbiol 71(11):6554–6563. https://doi.org/10.1128/AEM.71.11.6554-6563.2005
Janež N, Kokošin A, Zaletel E, Vranac T, Kovač J, Vučković D, Možina SS, Šerbec VC, Zhang Q, Accetto T, Podgornik A, Peterka M (2014) Identification and characterisation of new Campylobacter group III phages of animal origin. FEMS Microbiol Lett 359(1):64–71
Sørensen MCH, Gencay YE, Birk T, Baldvinsson SB, Jäckel C, Hammerl JA, Vegge CS, Neve H, Brøndsted L (2015) Primary isolation strain determines both phage type and receptors recognised by Campylobacter jejuni bacteriophages. PLoS One 10(1):e0116287. https://doi.org/10.1371/journal.pone.0116287
Conley MP, Wood WB (1975) Bacteriophage T4 whiskers: a rudimentary environment-sensing device. Proc Natl Acad Sci USA 72(9):3701–3705
Javed MA, van Alphen LB, Sacher J, Ding W, Kelly J, Nargang C, Smith DF, Cummings RD, Szymanski CM (2015) A receptor-binding protein of Campylobacter jejuni bacteriophage NCTC 12673 recognizes flagellin glycosylated with acetamidino-modified pseudaminic acid. Mol Microbiol 95(1):101–115. https://doi.org/10.1111/mmi.12849
Firlieyanti AS, Connerton PL, Connerton IF (2016) Campylobacters and their bacteriophages from chicken liver: The prospect for phage biocontrol. Int J Food Microbiol 237(1):121–127. https://doi.org/10.1016/j.ijfoodmicro.2016.08.026
Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Rüger W (2003) Bacteriophage T4 genome. Microbiol Mol Biol Rev 67(1):86–156. https://doi.org/10.1128/MMBR.67.1.86-156.2003
Noble E, Spiering MM, Benkovic SJ (2015) Coordinated DNA replication by the bacteriophage T4 replisome. Viruses. 7(6):3186–3200. https://doi.org/10.3390/v7062766
Labrie SJ, Samson JE, Moineau S (2010) Bacteriophage resistance mechanisms. Nat Rev Microbiol 8(5):317–327. https://doi.org/10.1038/nrmicro2315
Wösten MMSM, Boeve M, Koot MGA, van Nuenen AC, van der Zeijst BAM (1998) Identification of Campylobacter jejuni promoter sequences. J Bacteriol 180(3):594–599
Oliveira H, Melo LDR, Santos SB, Nobrega FL, Ferreira EC, Cerca N, Azeredo J, Kluskens LD (2013) Molecular aspects and comparative genomics of bacteriophage endolysins. J Virol 87(8):4558–4570. https://doi.org/10.1128/JVI.03277-12
São-José C, Parreira R, Vieira G, Santos MA. 2000. The N-terminal region of the Oenococcus oeni bacteriophage fOg44 lysin behaves as a bona fide signal peptide in Escherichia coli and as a cis-inhibitory element, preventing lytic activity on oenococca. Journal of Bacteriology. 182 (20), 5823 - 5831
Kakikawa M, Yokoi KJ, Kimoto H, Nakano M, Kawasaki K, Taketo A, Kodaira K (2002) Molecular analysis of the lysis protein Lys encoded by Lactobacillus plantarum phage phig1e. Gene 299(1–2):227–234
Park T, Struck DK, Dankenbring CA, Young R (2007) The Pinholin of lambdoid phage 21: control of lysis by membrane depolarization. J Bacteriol 189(24):9135–9139. https://doi.org/10.1128/JB.00847-07
Yasmin A, Kenny JG, Shankar J, Darby AC, Hall N, Edwards C, Horsburgh MJ (2010) Comparative genomics and transduction potential of Enterococcus faecalis temperate bacteriophages. J Bacteriol 192(4):1122–1130. https://doi.org/10.1128/JB.01293-09
Merrill BD, Grose JH, Breakwell DP, Burnett SH (2014) Characterization of Paenibacillus larvae bacteriophages and their genomic relationships to firmicute bacteriophages. BMC Genomics. https://doi.org/10.1186/1471-2164-15-745
Kolodziejek AM, Schnider DR, Rohde HN, Wojtowicz AJ, Bohach GA, Minnich SA, Hovde CJ (2010) Outer membrane protein X (Ail) contributes to Yersinia pestis virulence in pneumonic plague and its activity is dependent on the lipopolysaccharide core length. Infect Immun 78(12):5233–5243. https://doi.org/10.1128/IAI.00783-10
Friedrich NC, Torrents E, Gibb EA, Sahlin M, Sjöberg BM, Edgell DR (2007) Insertion of a homing endonuclease creates a genes-in-pieces ribonucleotide reductase that retains function. PNAS 104(15):6176–6181. https://doi.org/10.1073/pnas.0609915104
Sandegren L, Nord D, Sjöberg BM (2005) SegH and Hef: two novel homing endonucleases whose genes replace the mobC and mobE genes in several T4-related phages. Nucleic Acids Res 33(19):6203–6213. https://doi.org/10.1093/nar/gki932
Wood WB, Revel HR (1976) The genome of bacteriophage T4. Bacteriol Rev 40(4):847–868
Javed MA, Poshtiban S, Arutyunov D, Evoy S, Szymanski CM (2013) Bacteriophage receptor binding protein based assays for the simultaneous detection of Campylobacter jejuni and Campylobacter coli. PLoS One 8(7):e69770. https://doi.org/10.1371/journal.pone.0069770
Kropinski AM, Lavigne R, Ackermann HW, Szymanski C, Javed MA, Connerton I, Timms A, Azeredo J, Carvalho CM, Hertwig S, Hammerl JA (2013) Proposal: create two genera (Cp220likevirus and Cp8unalikevirus) within a new subfamily (Eucampyvirinae) in the family Myoviridae. In: International Committee on the Taxonomy of Viruses. http://www.ictvonline.org/proposals-14/2013.004a-kB.A.v4.Eucampyvirinae.pdf
Acknowledgements
Lisa O’Sullivan was funded by a Teagasc Walsh Fellowship (Project ref: 2013003) and by Teagasc (Project ref: 6323).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Handling Editor: T. K. Frey.
This paper is dedicated to the memory of Alan Lucid, who passed away suddenly on the 31st of August 2017 during the manuscript preparation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
O’Sullivan, L., Lucid, A., Neve, H. et al. Comparative genomics of Cp8viruses with special reference to Campylobacter phage vB_CjeM_los1, isolated from a slaughterhouse in Ireland. Arch Virol 163, 2139–2154 (2018). https://doi.org/10.1007/s00705-018-3845-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-3845-3