
Abstract
Historical geological events and climatic changes have played important roles in shaping population differentiation and distribution within species. In this study, we analysed the distribution, expansion and colonization patterns, and genetic differentiation among Psittacanthus rhynchanthus populations in Mesoamerica. Specifically, we determine the effects of major historical events and geographic barriers on population divergence using nuclear and chloroplast DNA sequence data and the impact of Pleistocene glacial cycles on the distribution dynamics of P. rhynchanthus using ecological niche modelling (ENM). Our results showed that P. rhynchanthus populations split into two lineages, one distributed on the Yucatán Peninsula and the second along the Pacific and Atlantic slopes and Honduras, approximately 1 million years ago. The two lineages were fragmented at the last interglacial according to ENM predictions and experienced postglacial range expansion during the Last Glacial Maximum. Analysis of climate differentiation and niche models showed that both lineages have different climatic preferences, where the climatic characteristics of the Yucatán populations are not fully represented in the rest of the distribution range of P. rhynchanthus. Additionally, our study suggests that Pleistocene expansion of suitable habitat, environmental segregation (ecological barrier between regions) and, perhaps host shifts, have acted as the isolation mechanisms between the two lineages. Our results provide new insight as to understanding the distribution and phylogeographic patterns and the possible mechanisms underlying intraspecific evolutionary relationships of plants in the Mesoamerican tropical lowlands.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant genetic differentiation of widespread species in temperate North America is usually driven by climate changes during the Pleistocene (Hewitt 2000). According to several plant phylogeographic surveys in the region, a south–north decrease in genetic diversity and low intraspecific genetic differentiation has been found accompanied by a rapid postglacial expansion northwards (e.g. Jaramillo-Correa et al. 2009; Vargas-Rodríguez et al. 2015; Colin and Eguiarte 2016). In contrast, the effects of the Pleistocene glacial cycles were seemly less drastic in Mesoamerica (Caballero et al. 2010; Caballero-Rodríguez et al. 2018; Mastretta-Yanes et al. 2018), though some plant species exhibit marked structuring geographic patterns matching the presence of biogeographical barriers (e.g. Gutiérrez-Rodríguez et al. 2011; Ornelas and Rodríguez-Gómez 2015), environmental complexity of the region (e.g. Cavers et al. 2003; Ornelas et al. 2010, 2019a; Ortiz-Rodriguez et al. 2018a), and often accompanied by morphological differentiation (e.g. Cavender-Bares et al. 2011; Ornelas and González 2014; Ruiz-Sanchez and Ornelas 2014). Given the age of biogeographic barriers in Mesoamerica (Isthmus of Panama, 23–10 Ma; Talamanca Cordillera, 3.9 Ma; Nicaraguan Depression, 10–4 Ma; Motagua–Polochic–Jocotán fault system, 8–3 Ma; Isthmus of Tehuantepec, 5–2 Ma, e.g. Daza et al. 2010; Ornelas et al. 2013; Winston et al. 2017), lineage divergence or genetic discontinuities are expected to be observed across these barriers from the Miocene to the Pliocene in species with a low dispersal potential.
Within the Mesoamerican region (Sánchez-González et al. 2013; Morrone 2014; Montaño-Arias et al. 2018), phylogeographic patterns seem to be correlated with its complex geography and the ecological preferences of plant species. For those distributed in the highlands or semiarid zones of Mexico and Central America, a marked geographic structure of genetic variation is observed (Moreno-Letelier and Piñero 2009; Gutiérrez-Rodríguez et al. 2011; Ornelas and González 2014; Ramírez-Barahona and Eguiarte 2014; Ruiz-Sanchez and Specht 2014; Ornelas and Rodríguez-Gómez 2015; Ornelas et al. 2018a), whereas plant species that inhabit the tropical lowlands show low geographic structuring of genetic variation and signs of recent population expansion (e.g. Cavers et al. 2003; Chávez-Pesqueira and Núñez-Farfán 2016; Ornelas et al. 2016; Licona-Vera et al. 2018). Therefore, the demographic and phylogeographic patterns of Mesoamerican taxa that conform to either an in situ survival model or an expansion–contraction model suggest that the constituents of Mesoamerican biota could have responded idiosyncratically to past climate changes and colonization of the region.
Psittacanthus (Loranthaceae) is a widespread genus of hemiparasitic mistletoes across the Americas. It includes ca. 119 species, of which most are found in South America and about 23 are endemic to Mexico and Central America (Kuijt 2009). The distribution, expansion and colonization patterns, and genetic differentiation among populations of mistletoe species are influenced by interactive factors including the ecology of interactions with their hosts and their pollen and seed vectors (Lira-Noriega et al. 2015; Díaz Infante et al. 2016; Yule et al. 2016; Ramírez-Barahona et al. 2017), and the topography, microclimate and vegetation along with the formation of biogeographic barriers and uplift of mountain ranges in the region (Ornelas et al. 2016, 2018a, 2019b; Pérez-Crespo et al. 2017; Baena-Díaz et al. 2018; Licona-Vera et al. 2018). Psittacanthus rhynchanthus (Benth.) Kuijt (Fig. 1) is a forest-adapted species distributed on the Pacific (from Jalisco to Chiapas) and Atlantic (Puebla, Veracruz and Tabasco) slopes in Mexico, and continuing onto the Yucatán Peninsula, Central America, and northern Colombia, Venezuela and Trinidad (Kuijt 2009). It tends to prefer lower elevations (sea level to c. 2000 m above sea level), often in coastal situations, in tropical evergreen forest, tropical semi-deciduous forest and seasonally dry tropical forest (SDTF), and less often in thorn forest, oak forest and cloud forest (Kuijt 2009). The distribution of P. rhynchanthus in both slopes of Mexico is a common pattern among other plant taxa with affinities to the tropical lowlands (Rzedowski 1963; Luna-Vega 2008).

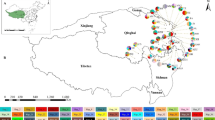
Psittacanthus rhynchanthus in tropical lowlands at Pinoltepec, Veracruz, Mexico (a, photograph by María Teresa Mejía-Saules), Patulul, Guatemala (b, photograph by Pablo Carrillo), Celestún, Yucatán (c, photograph by Etelvina Gándara), and Petatlán, Guerrero (d, photograph by Eduardo Ruiz-Sanchez). e Collection sites of P. rhynchanthus on a relief map showing the three biogeographic barriers discussed in this study: Isthmus of Tehuantepec (IT), Motagua–Polochic–Jocotán (MPJ) fault system and the Nicaraguan Depression (ND), and the climatic filter barrier (dashed line). Coordinates of collection sites provided in Online Resource 2
The distribution range and habitat preferences of P. rhynchanthus are well suited for investigation of the role of climatic factors and historical geographical barriers in shaping population genetic differentiation across tropical lowland forests of northern Mesoamerica. Previous phylogeographic studies in Mexico have shown that the Sierra Madre Occidental, Sierra Madre Oriental, and the Sierra Madre del Sur had acted as physical barriers for dispersal of lowland plant and vertebrate species (e.g. Guevara-Chumacero et al. 2010; McCormack et al. 2011; Ruiz-Sanchez and Ornelas 2014; Suárez-Atilano et al. 2014), or that the drier surrounding valleys of these mountain ranges isolate populations and restrict dispersal (e.g. González et al. 2011; Ornelas et al. 2016, 2018a; Pérez-Crespo et al. 2017, but see Arteaga et al. 2011; Chávez-Pesqueira and Núñez-Farfán 2016). In the highlands of Mesoamerica, the distributions of plant species and the partitioning of their genetic diversity were significantly influenced by the climatic fluctuations during the Pleistocene (e.g. Ramírez-Barahona and Eguiarte 2013; Mastretta-Yanes et al. 2015). Genetic divergence data contrasted with past distribution modelling indicate that populations of plant species experienced range or altitudinal shifts or remained in situ during the Last Glacial Maximum (LGM) within their current fragmented distribution (e.g. Ornelas and González 2014; Ramírez-Barahona and Eguiarte 2014; Ornelas et al. 2016, 2019b). For Psittacanthus species distributed in the highlands of Mexico (P. schiedeanus and P. calyculatus), ecological niche modelling (ENM) indicates that their distribution ranges (conditions of suitable habitat) were relatively unstable during the last 140,000 years, with expansion from the last interglacial (LIG; 120,000–140,000 years ago) to LGM (ca. 20,000 years ago) and contraction from LGM to present-day climate (Ornelas et al. 2016; Pérez-Crespo et al. 2017). For species distributed in xeric environments (P. auriculatus and P. sonorae), ENM suggests a different pattern, with contraction and fragmentation from LIG to LGM and expansion from Mid-Holocene (MH; ca. 6000 years ago) to present in P. auriculatus (Ornelas et al. 2018a), and no major changes since the LIG in P. sonorae (Ornelas et al. 2019b). Lastly, range shifts from southern Guatemala into the Yucatán Peninsula were suggested by ENM in P. mayanus, with expansion from LIG to LGM, contraction from LGM to MH and colonization of the northernmost portion of the Yucatán Peninsula, and expansion from MH to present (Licona-Vera et al. 2018). However, plant species in the lowlands might have different range dynamics in response to glacial cycles.
Here, we determine the effects of major historical events and geographic barriers and the impact of past climatic oscillations on population divergence, demographic history and distribution dynamics of P. rhynchanthus. Specifically, we examine nuclear and chloroplast DNA sequence variation of P. rhynchanthus to explore whether: (1) structuring occurs among its populations and whether (2) genetic differentiation and structuring are concordant with the Miocene–Pliocene formation of mountain ranges or (3) the distribution of suitable habitat during the Pleistocene. The effects of past climatic fluctuations on the distribution of suitable habitat for P. rhynchanthus were investigated using ENM to determine whether the extended or contracted ranges of the tropical deciduous forest vegetation in the lowlands of northern Mesoamerica played a role on structuring geographic patterns of the mistletoe’s genetic variation. We predict a south-to-north contraction/expansion in the reconstructed distribution of suitable habitat for P. rhynchanthus, as expected for taxa in the tropical lowlands (Haffer 1969; Prance 1973; Hewitt 1996, 2000; but see Colinvaux et al. 1996, 2001; Pennington et al. 2004). Neotropical species populations have responded to Pleistocene climatic change, from long-lasting glacial cycles to brief interglacials and back again (e.g. Haffer 1969; Prance 1973; Pennington et al. 2004; Licona-Vera et al. 2018). In response to glacial cycles, several populations of lowland forests became isolated and differentiated from one another by the expansion of non-forest vegetation adapted to more xeric conditions during periods of geographic isolation and Pleistocene cool-dry climatic cycles (e.g. Haffer 1969; Prance 1973). The isolated forests were again connected during humid climatic cycles when the intervening non-forest vegetation became forested allowing contracted populations (refugia) to extend their ranges (see Haffer 1969). Under this scenario, the reconstructed distribution of suitable habitat for the mistletoe would be of a contracted distribution southwards (range contraction model) or fragmented into southerly refugia (range fragmentation model) during glacial periods and an expanded and continuous distribution northwards along the Pacific and Atlantic coasts of Mexico during interglacials (see Table 1 for further details). In addition, populations on the Yucatán Peninsula separated from those along the Mexican slopes are a Miocene–Pliocene division (old isolation model), and further genetic differentiation linked to changes in the distribution of suitable habitat during the Pleistocene in which the xeric conditions in the peninsula acted as an effective barrier preventing contact with populations from the Petén region (ecological barrier; Savage 1966; Lavin et al. 1991; Licona-Vera et al. 2018; Table 1, Online Resource 1).
Materials and methods
The species
Psittacanthus rhynchanthus, recognized by the bud apexes conspicuously curved and acutely beaked and the long reddish stamen hairs borne behind the smooth pollen sacs (Kuijt 1987; Fig. 1a–d), commonly grows on Bursera simaruba (L.) Sarg. (Burseraceae) and less frequently on Spondias purpurea L. (Anacardiaceae) trees in the Mesoamerican region; in Venezuela, Platymiscium platystachyum Griseb. (Fabaceae) is the main host of this mistletoe (Kuijt 2009). Its coral hermaphroditic flowers are visited and pollinated by hummingbirds (León-Camargo and Rangel-Ch. 2015), and several species of birds consume and disperse their one-seeded fleshy fruits. Psittacanthus rhynchanthus is part of a species complex characterized by the curved apexes of scarlet flower buds, which includes P. americanus on the eastern Caribbean islands and P. wurdackii endemic to Venezuela (Kuijt 2009). Aside from the strictly insular occurrence, P. americanus is differentiated from P. rhynchanthus by its flowers with the inner petal surfaces glabrous and terete stems (Kuijt 2009). The narrow-leaved P. wurdackii has been regarded as an extreme form of P. rhynchanthus, with narrow-falcate leaves and nearly glabrous internal floral morphology (Kuijt 2009). Given the close relationship between P. americanus and P. rhynchanthus, Kuijt (2009) stated that the source plants for migration into the Caribbean from Mesoamerica were P. rhynchanthus (Kuijt 2009).
Sample collection and DNA extraction
We sampled 146 individuals in 22 localities, representative of the species range in Mexico, Guatemala and Honduras (Fig. 1e; Online Resource 2). Sampling localities (hereafter referred to as populations) were targeted based on Kuijt (2009) and Mexican herbaria, including 5 populations located along the Pacific in western Mexico, 7 populations along the Atlantic slope of Mexico, 4 populations from Chiapas, 4 populations from the Yucatán Peninsula, 2 populations from Guatemala, and one population from Honduras. One voucher specimen was collected per population in most cases and deposited at XAL (Instituto de Ecología, A.C., INECOL). The sampling strategy was aimed at avoiding collection of several individuals per host tree.
Leaf tissue samples were preserved in silica gel desiccant until DNA extractions were performed. Total genomic DNA was extracted with the CTAB method (Doyle and Doyle 1987) or using the DNeasy Plant Mini kit (Qiagen, Valencia, CA, USA) following the manufacturer protocol.
PCR and sequencing
The internal transcribed spacer (ITS) region of the nuclear ribosomal DNA (nrDNA) and trnL-trnF and atpB-rbcL intergenic spacer regions from the chloroplast genome (cpDNA) was amplified by PCR and sequenced for 146 individuals. These markers have been screened for variability in Psittacanthus at the species and population levels (e.g. Ornelas et al. 2019b). The genetic structure of the cpDNA sequences may provide a picture of the impact of seed movement on gene flow, because cpDNA is often maternally inherited in most angiosperms, whereas the genetic structure of the nrDNA sequences should reflect both pollen and seed movement (Ornelas et al. 2016).
For ITS amplifications, we used the primers designed for the genus (Ornelas et al. 2016), whereas for the trnL-trnF, atpB and rbcL we used the primers described by Taberlet et al. (1991) and Chiang et al. (1998). Protocols for DNA extraction, for amplification by PCR assays and for sequencing the PCR products with some minor modifications as described elsewhere (Ornelas et al. 2016, 2018a). PCR products were sequenced bidirectionally using the BigDye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA, and analysed on a 310 automated DNA sequencer (Applied Biosystems) at the INECOL’s sequencing facility, or at University of Washington High Throughput Genomics Unit, Seattle, Washington. Edited sequences were aligned manually with BIO-EDIT version 7.2.5 (Hall 1999) and SE-AL version 2.0a111 (http://tree.bio.ed.ac.uk/software/seal). The resulting sequences were submitted to GenBank (accession nos. ITS: MN381235–MN38186, trnL-trnF: MN272734–MN272842, atpB-rbcL: MN272843–MN272951). We additionally downloaded DNA sequences from the GenBank of representatives of Psittacanthus palmeri and P. sonorae from Ornelas et al. (2019b) to be used as out-group.
Divergence time estimation and haplotype relationships
We used *BEAST (Heled and Drummond 2010), as implemented in BEAST version 1.6.1 (Drummond and Rambaut 2007), to estimate a species tree and divergence time between groups of P. rhynchanthus from both the nrDNA data and the concatenated cpDNA sequence data (Online Resource 2). Nucleotide substitution models with jModeltest (Darriba et al. 2012) were incorporated as HKY for ITS and JC for trnL-trnF and atpB-rbcL. The simulation was run with P. rhynchanthus samples assigned as PAC + ATL + HON or YUC, and samples of P. palmeri and P. sonorae as out-group according to Ortiz-Rodriguez et al. (2018b). Analysis was performed following settings and substitution rates as in Ornelas et al. (2019b). We also analysed the nrDNA and concatenated cpDNA sequence data to infer the timing for the origin of P. rhynchanthus using a constant population size coalescent model with BEAST version 1.8. The optimum model setting was determined by the results of jModeltest. We assigned a normal calibration prior distribution, and the tree root node was calibrated using a mean age of 6.23 Ma (SD ± 1.06, range 8.31–4.1 Ma), which corresponds to the estimated mean age for the genus Psittacanthus (Ornelas et al. 2016; Pérez-Crespo et al. 2017; Licona-Vera et al. 2018; Ortiz-Rodriguez et al. 2018b). We ran 30 million generations for the cpDNA data set of the MCMC chain, with a sampling frequency of 3000 generations. Convergence of the posterior distributions of the parameters was evaluated by monitoring the effective sample sizes (ESS > 200) and trace plots in Tracer (http://tree.bio.ed.ac.uk/software/tracer/). A maximum credibility tree, which represents the maximum posterior topology, was calculated in TreeAnnotator (Drummond and Rambaut 2007) after discarding the first 10% of trees as burn-in. The aligned data set and the phylogenetic tree are available in TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S24997).
Relationships among P. rhynchanthus haplotypes are presented as statistical parsimony networks generated using the program TCS version 1.2.1 (Clement et al. 2000), with gaps treated as single evolutionary events and a 95% connection probability limit. Loops were resolved following the criteria given by Pfenninger and Posada (2002). We report herein results for the combined trnL-trnF/atpB-rbcL data set.
The most likely number of genetically differentiated clusters was estimated using BAPS version 5.3 (Corander et al. 2008) with settings as in Ornelas et al. (2019b). For each marker, we surveyed the probability of a different number of genetic clusters (K = 2 to K = 15) under the independent loci model and two independent runs with 10 replicates for each K, accepting the partition with the K value that had a higher likelihood and posterior probability.
Population indices and geographic structure of populations
Haplotype diversity indices for each population (hS, vS) and at the species level (hT, vT), and coefficients of population differentiation (GST, NST) were estimated using PERMUT version 1.0 (Pons and Petit 1996). Phylogeographic structure at the species range was further tested contrasting the NST and GST values with 10,000 permutations and the U-statistic (Pons and Petit 1996). Molecular diversity indices (h, gene diversity; π, nucleotide diversity) and pairwise comparisons of FST values between regions were calculated using Arlequin version 3.1.1 (Excoffier et al. 2005). Populations with 1 or 2 samples were lumped with closest population (2 samples from population 10 into population 9 and 1 sample from population 13 into population 14). To account for differences in the number of sampled individuals between genetic groups, individual-based and population-based rarefaction curves were generated to assess the proportion of haplotype richness sampled for each group recovered (Gotelli and Colwell 2001; see also Ornelas et al. 2019b). A trend towards an asymptotic relationship infers haplotype saturation. In contrast, a steep slope suggests that a large fraction of the available haplotype diversity remains unsampled (Gotelli and Colwell 2001). This same method allows the number of haplotypes between populations to be compared when the number of samples is different. If the 95% confidence interval of the haplotype richness estimates from two populations overlaps, it can be concluded that there is no significant difference between them (Gotelli and Colwell 2001). Individual-based curves were generated following the procedure of Chao and Jost (2012), as implemented in the R ‘iNEXT’ package (Hsieh et al. 2016), using 1000 randomizations and extrapolating to 150 individuals that represented a little more than the total number of individuals sampled in this study. Population-based curves were generated in EstimateS version 9.0 (Colwell 2013) using 10,000 replications and extrapolating to 30 populations that represent more than the number of localities sampled in this study.
Analysis of molecular variance (AMOVA; Excoffier et al. 1992), as implemented in Arlequin, was carried out to evaluate partitioning of the total genetic variation among groups and sampling locations. Four AMOVA models were performed according to the haplotype network analysis with populations treated as: (a) a single group to determine the amount of variation partitioned among and within populations, and populations on the Yucatán Peninsula (YUC), Pacific slope (PAC), Atlantic slope (ATL) and the Honduras population (HON) resembling biogeographic subregions within Mesoamerica (Fig. 1e; Online Resource 2) grouped into two or three lineages: (b) YUC and the other populations, (c) YUC + HON and PAC + ATL, (d) PAC + HON, ATL and YUC, or (e) YUC + HON, PAC and ATL. AMOVAs were performed using the Jukes and Cantor model for the combined trnL-trnF/atpB-rbcL sequences and 10,000 permutations to determine the statistical significance of each AMOVA model.
We also conducted a spatial analysis of molecular variance (SAMOVA), as implemented in SAMOVA version 1.0 (Dupanloup et al. 2002), to identify groups of locations that are geographically and genetically differentiated from each other. The SAMOVA was performed maximizing the proportion of total genetic variance due to differences among groups of locations (FCT), as suggested by Dupanloup et al. (2002), with 10,000 simulated annealing simulations and geographic groups from 2 to 10 groups (Online Resource 2).
Historical demography
We inferred signatures of demographic expansion by calculating Fu’s Fs (Fu 1997), Tajima’s D (Tajima 1989) and Ramos-Onsins and Rozas’ R2 (Ramos-Onsins and Rozas 2002) statistics of neutrality and conducting mismatch distribution (Harpending 1994) analysis with Arlequin and ‘pegas’ package (Paradis 2010) using R version 3.4.1 (R Development Core Team 2013; https://www.r-project.org/). Fu’s Fs is the best for large sample sizes while R2 is the most suited statistics when sample size is small (Ramos-Onsins and Rozas 2002). Mismatch distribution analysis was carried out using the sudden population expansion model of Schneider and Excoffier (1999) with 20,000 bootstrap replicates. The differences between observed mismatch distributions and expected distribution, under a sudden population expansion model, were tested by examining the sum of squared deviations (SSD) test and the Harpending’s raggedness index (Hri) according to Rogers and Harpending (1992).
Bayesian skyline plots (BSP; Drummond et al. 2005) of changes in effective population size (Ne) through time were carried out in BEAST because departures from neutrality are often caused by changes in Ne. We used the cpDNA (trnL-F, atpB-rbcL) data set and chose a HKY substitution model selected with jModeltest, with settings and substitution rates as in Ornelas et al. (2019b). Two independent runs of 30 million generations each were conducted, and trees and parameters were sampled every 1000 iterations, with a burn-in of 10%. Results of each run were visualized using Tracer to ensure that stationarity and convergence had been reached and that the ESS was higher than 200.
Palaeodistribution modelling and environmental variation
We used ENM (Elith et al. 2011) to examine the potential range shifts of suitable habitat occupied by P. rhynchanthus in response to climatic oscillations during the last 140 thousand years. Hypothetical models and the expected effects from the LIG (c. 140–120 ka) to the present for groups of populations of the mistletoe are shown in Online Resource 1.
We constructed ENM models with MaxEnt version 3.3.3k (Phillips et al. 2006) and 19 bioclimatic variables from WorldClim (Hijmans et al. 2005; Booth et al. 2014) at a c. 1-km2 (2.5 arc-min) spatial resolution. We obtained data for past climate layers for two LGM (2.5 arc-min) past conditions based on the Community Climate System Model (CCSM) and Model for the Interdisciplinary Research on Climate (MIROC) global models (Hijmans et al. 2005; Braconnot et al. 2007), as well as the LIG (at 30 arc-seconds). Locations of P. rhynchanthus were obtained from field collections of the authors and from herbarium records online (https://www.gbif.org/species/7288106) in the Global Biodiversity Information Facility (GBIF.org (1st March 2016) GBIF Occurrence Download, https://doi.org/10.15468/dl.ey2xst). Data from the online database were checked to exclude misidentification and duplicate records. A total of 73 spatially unique locations were used for the analysis. Data layers were manipulated in R.
We first constructed ENM models for the mistletoe under current climate conditions and employed all 19 climatic variables and 20 cross-validation replicates, from which 80% of the distribution coordinates were used for training and 20% for testing along with comparisons of the variable importance (%) and jackknife plots. In each iteration, the contribution of every single variable to the general distribution was determined by jackknife statistical technique, which allowed the variables with the greatest influence on the probability of persistence of P. rhynchanthus and spatial distribution in the region to be identified. To exclude highly correlated variables (correlation values ≥ 0.8), pairwise correlations were examined among the 19 variables within the distribution of P. rhynchanthus with PAST version 3.06 (SAS Institute Inc. Cary, NC, USA; https://folk.uio.no/ohammer/past/) and removed the variable with lower explanatory power based on their relative contributions to the MaxEnt model. After removing the highly correlated variables, six variables (BIO4 = Temperature Seasonality, BIO5 = Max Temperature of Warmest Month, BIO6 = Min Temperature of Coldest Month, BIO10 = Mean Temperature of Warmest Quarter, BIO13 = Precipitation of Wettest Month, BIO15 = Precipitation Seasonality; BIO18 = Precipitation of Warmest Quarter, BIO19 = Precipitation of Coldest Quarter) were used to construct the final models with 20 cross-validation replicates without extrapolation and considering the average output grids as the final predictive models.
Given that ENMs do not address the historical aspects relating to species distribution (hypothetical ‘M’ region or accessible areas to the species via dispersal over relevant periods of time; Barve et al. 2011), we used a geographical clipping based on the biogeographic provinces proposed by Morrone (2005, 2014) and a map of ecoregions (http://maps.tnc.org/gis_data.html). This geographical clipping represents accessible areas to P. rhynchanthus, representing elevation range limits and historical barriers and potential boundaries on the landscape to dispersal (Barve et al. 2011). Verified occurrence records for P. rhynchanthus were projected onto a map of Mexico and used the shapefiles after geographical clipping to select the hypothetical ‘M’ region representing areas similar to those that included the points of occurrence of P. rhynchanthus. Climate layers were clipped with the ‘M’ region for use in MaxEnt analysis.
To incorporate the evolutionary information available for P. rhynchanthus during the ecological niche modelling (see Smith et al. 2019), we split P. rhynchanthus into two lineages (YUC and PAC + ATL + HON; see “Results” section) and modelled each separately, which improved niche estimates as compared to those using lumped data for all known populations of the species (AUC = 0.732 ± 0.127; TSS = 0.312 ± 0.17). Resulting lineage distribution models under current climate conditions were projected onto the LGM (at 2.5 arc-min) and LIG (at 30 arc-seconds) conditions following the ‘bilinear’ method through the ‘resample’ package in R. Past environmental layers of the LIG (Otto-Bliesner et al. 2006) and LGM (Otto-Bliesner et al. 2007) were also drawn from WorldClim, choosing for the LGM the CCSM and MIROC models from the Paleoclimate Modelling Intercomparison Project Phase II database (Braconnot et al. 2007). The CCSM and MIROC climate models simulate different climate conditions, with cooler sea-surface temperature conditions assumed in CCSM than in MIROC, resulting in higher annual precipitation in CCSM than in MIROC (Otto-Bliesner et al. 2007). Previous evaluations of the performance of CCSM and MIROC concluded that MIROC performs well for areas near the equator, while CCSM performs better at high latitudes (e.g. Masson-Delmotte et al. 2006), suggesting that the predictions of the LGM distribution that are based on CCSM may be more reliable.
We evaluated the performance of the models by calculating the true skill statistic (TSS), a threshold-dependent measure of model performance that evaluates the accuracy of predictive maps generated by presence-only data (Allouche et al. 2006; Liu et al. 2013). TSS is the sum of sensitivity and specificity minus one. (Sensitivity is the proportion of presences correctly predicted, and specificity is the proportion of correctly predicted absences.) The TSS was calculated for each replicate using the 10th percentile training presence logistic threshold (T10LT), and the resulting TSS values averaged among replicates. TSS varies from − 1 to + 1, where negative values and values near zero indicate that distributions are no better than random, whereas values ranging between 0.4 and 0.8 are considered as acceptable models (Fielding and Bell 1997; Landis and Koch 1977). The performance of the models was also evaluated by calculating the area under the receiver operating characteristic curve (AUC; Swets 1988), where 1 is the maximum prediction and 0.5 suggests a random prediction.
Lastly, we carried out a principal components analysis (PCA) in PAST on the 19 bioclimatic variables data from WorldClim to examine P. rhynchanthus population divergence related to habitat and ecological variation. Differences in PC scores between locations separated by geography (PAC + ATL + HON and YUC) were tested with one-way ANOVAs.
Bayesian Phylogeographic and Ecological Clustering (BPEC)
The Bayesian Phylogeographic and Ecological Clustering (BPEC) method, as implemented in the ‘BPEC’ package (Manolopoulou et al. 2011; Manolopoulou and Emerson 2012) in R, was followed to identify clusters genetically and geographically distinct and the ancestral locations for P. rhynchanthus. The method accounts for haplotype connection ambiguities due to the presence of loops and estimates posterior probabilities under a coalescent-based migration-mutation model (Manolopoulou et al. 2020). The haplotype tree model used in BPEC assigns posterior probabilities of haplotypes to the most ancestral, which then are associated with sampling locations to infer the most ancestral location. Unlike standard parsimony, BPEC fits a prior over all possible trees to identify trees with high posterior probability in a fully model-based framework, thus accommodating for uncertainty in haplotype relationships, which is one of the main criticisms of TCS (Knowles 2008). We analysed all cpDNA haplotypes, their distributions and climatic preferences based on coordinates of occurrence data and the values of the first two PCs from a PCA using the 19 bioclimatic variables from WorldClim for each of the coordinates. After several preliminary trials, we run the final analysis with 2 specified as the prior for maximum number of migrations and relaxation of the parsimony criterion not allowed to reach convergence. MCMC chains were run for 10 million steps, with 10,000 posterior samples being saved for analysis.
Results
Divergence time estimation and haplotype relationships
The aligned sequences of ITS, trnL-F and atpB-rbcL for haplotype networks were 552, 268 and 438 base pairs (bp) long, respectively (Fig. 2a). The combined trnL-F/atpB-rbcL data set (706 bp, 109 sequences) contained 11 variable and 7 parsimony informative sites. For ITS, we had amplification problems and decided to use the data for those successfully sequenced individuals (57 sequences) only to illustrate variation in the network and for species tree and divergence time estimation.

Statistical parsimony networks of ITS and single trnL-F and atpB-rbcL data (a) and statistical parsimony network of combined trnL-F/atpB-rbcL data (b) of thirteen Psittacanthus rhynchanthus haplotypes overlaid on a map of North America (c). Haplotype designations in the networks correspond to those in Online Resource 4, and the size of the circles is proportional to the frequency of each haplotype. Haplotypes are coded with a different colour according to geographic regions: Pacific slope (dark blue), Atlantic slope (light blue), Yucatán Peninsula (yellow), and Honduras (light purple), and a number codes each of the ribotypes or haplotypes. The numbers inside the haplotypes in the network indicate the number of individuals that share that haplotype. Loops (dashed lines) were resolved following the criteria given by Pfenninger and Posada (2002). The numbers by the haplotypes in the map correspond to localities fully described in Fig. 1 and Online Resources 2 and 4. Pie charts represent haplotypes found in each sampling locality. The size of sections of the pie charts corresponds to the proportion of individuals with a given haplotype. In the inset (d), a Bayesian analysis of population genetic structure (BAPS) is presented based on the trnL-F/atpB-rbcL sequences. Colours indicate different genetic clusters (K = 2), the yellow cluster include individuals from the Yucatán Peninsula, and the blue cluster includes individuals from the Pacific and Atlantic slopes and Honduras. Numbers indicate different populations (localities) as fully described in Online Resource 2
The *BEAST tree of multilocus data (ITS + cpDNA) for differentiation between the P. rhynchanthus groups (Online Resource 3) indicated strong support for the monophyly of P. rhynchanthus, with divergence estimated at 0.525 Ma (95% HPD 1.17–0.1 Ma, PP = 0.96) for the split between populations from the Yucatán Peninsula and populations from the Atlantic and Pacific slopes and Honduras. The BEAST analysis placed the split between P. rhynchanthus and the other Psittacanthus species at 1.25 Ma (95% HPD: 2.64–0.15 Ma, PP = 1) using the nrDNA and concatenated cpDNA (Online Resource 3).
Thirteen haplotypes were identified for the trnL-F/atpB-rbcL concatenated data (Online Resource 4). The network revealed two common haplotypes, H1 and H12 (Fig. 2b). Haplotype H1, the most frequent haplotype (56 samples, 51% of the individuals) and core of network, was retrieved from most populations except those from the Yucatán Peninsula and Honduras (Fig. 2c). Haplotype H12, the second most common (21 samples, 19.3% of the individuals) and five steps separated from H1, was exclusively located in peninsular populations along with H10–H13 haplotypes. Other haplotypes were exclusively located in Honduras (H8 and H9) or Veracruz (H3, H5 and H6), except one individual from La Peña, Michoacán (H3), and one individual from San Salvador, Guatemala (H3) (Fig. 2c; Online Resource 4).
BAPS analyses with trnL-F/atpB-rbcL sequences inferred existence of two genetic clusters (K = 2) (log marginal likelihood = − 193.439, PP = 1.0). In agreement with the shallow genetic divergence and haplotype distribution shown in Fig. 2, these two genetic clusters group as: the yellow cluster includes individuals from the YUC populations, and the blue cluster includes individuals from populations in the Pacific and Atlantic slopes and Honduras (Fig. 2d). Therefore, we used the two-population structure (YUC, PAC + ATL + HON), as described by the haplotype network analyses, for downstream analyses.
Population indices and geographic structure of populations
Differentiation among populations based on trnL-F/atpB-rbcL (GST = 0.600, SE = 0.0913) indicated that P. rhynchanthus is genetically subdivided. Genetic diversity across all populations (hT = 0.729, SE = 0.0846; vT = 0.731, SE = 0.1637) was higher than the average within-population value (hS = 0.291, SE = 0.0735; vS = 0.226, SE = 0.0679). PERMUT analysis showed that NST (0.691, SE = 0.0834) and GST values were statistically different (P < 0.05), indicating phylogeographical structuring. Pairwise comparisons of FST values were low and significant when PAC and ATL + HON populations were compared (FST = 0.231, P < 0.001), whereas those for YUC/PAC and YUC/ATL + HON were high and significant (FST = 0.921 and 0.762, respectively, P < 0.001).
When populations are treated as a single group, the AMOVA showed that 13.5% of the genetic variation for trnL-F/atpB-rbcL was explained by differences within populations and 84.5% by differences between populations (Table 2). The three groupings tested in AMOVA revealed a high degree of population structuring, with highest FCT value (FCT = 0.80) obtained when populations are grouped as two lineages: populations from the Yucatán Peninsula and populations from the Pacific and Atlantic slopes and the Honduras population (Table 2). A significant but smaller proportion of the variation was attributed to differences between groups when sampling sites were grouped differently (Table 2).
SAMOVA results revealed significant FCT values for groups between K = 2 and K = 10, with high FCT value for K = 2 (Table 2). When K = 2, the spatial genetic analysis identified one group formed by populations of the Yucatán Peninsula and Honduras treated as one group and the other one encompassing the populations of the Pacific and Atlantic slopes and no single-population group was formed. This configuration was generally consistent with the clustering depicted in BAPS (Fig. 2d) and the best AMOVA model (Table 2). Although FCT = 0.899 was higher at K = 3 than at K = 2 (0.892), an additional increase in the number of K led to higher FCT values than K = 3 but dissolution of group structure and single-population groups were formed.
Genetic diversity (h) and nucleotide diversity (π) were higher for the PAC + ATL + HON population (Table 3). Rarefaction curves based on populations provide evidence that differences in haplotype richness between geographic regions (YUC, PAC + ATL + HON) are not significant according to the 95% confidence intervals (Online Resource 5).
Historical demography
For trnL-F/atpB-rbcL, Fu’s Fs and Tajima’s D had negative but small non-significant (P > 0.05) values for the YUC and PAC + ATL + HON group, indicating that populations are neutrally evolving and though values are negative we failed to reject the null hypothesis of a constant population size (Table 3). In contrast, SSD and Hri showed low and non-significant values indicating a good fit, so the expansion model was not rejected, except for the SSD of the PAC + ATL + HON group (Table 3). R2 statistic showed positive, small and highly significant values for both groups, indicating that these groups presented past demographic expansion (Table 3). The Bayesian skyline plots suggest that the effective population size was stable over time in P. rhynchanthus and in the groups of populations, except a marginal decrease after the LGM in the PAC + ATL + HON group (Online Resource 6).
Palaeodistribution modelling and environmental variation
For the P. rhynchanthus lineage models of YUC and PAC + ATL + HON, the AUC values indicated that model prediction performed well (0.881 ± 0.073 and 0.841 ± 0.118, respectively). Our TSS results using the 10 percentile training presence logistic threshold (10PTPLT) resulted in a good proportion of correctly classified training observations (mean ± SE, YUC= 0.587 ± 0.33, PAC + ATL + HON = 0.552 ± 0.337). We applied the 10PTPLT (mean ± SE, YUC = 0.283 ± 0.020, PAC + ATL + HON = 0.280 ± 0.031) to the final models, where grid cells with model values below this given threshold were eliminated. Therefore, we converted our output grids accordingly to define the presence/absence maps for the two P. rhynchanthus models. The predicted potential distribution of P. rhynchanthus under current conditions was similar to its actual distribution. Interestingly, the potential distribution of suitable habitat for the YUC lineage has changed much since the LIG (Fig. 3a–d). The predicted distribution at the LIG (Fig. 3a) differed greatly from LGM predictions, with southward range expansion predicted from LIG to LGM according to the LGM-CCSM model (Fig. 3b) or severe contractions into small areas in the tip of the peninsula according to the LGM-MIROC model (Fig. 3c), to southward range expansion predicted from LGM to the present (Fig. 3d). For the PAC + ATL + HON lineage, the potential range under current conditions (Fig. 3e) also differed to its actual potential distribution (Fig. 3h). The predicted distribution at the LIG (Fig. 3e) differed greatly from LGM predictions, with range expansion into the Atlantic slope, the Balsas Basin and the western Pacific slope according to both LGM-CCSM and LGM-MIROC models (Fig. 3f–g), to a widespread but more fragmented distribution to the present (Fig. 3h). Overall, the models show there are large and different range expansion patterns, from LIG to the present.

Results from the MaxEnt analyses showing ecological niche models for the YUC (left) and PAC + ATL + HON (right) Psittacanthus rhynchanthus groups: a, e at last interglacial (LIG, 140–120 ka BP), b, f Last Glacial Maximum (LGM, CCSM, 21 ka BP), c, g Last Glacial Maximum (LGM, MIROC, 21 ka BP), and d, h at present (0 ka), respectively
The PCA indicated three niche axes that together explain 80% of the environmental variation in P. rhynchanthus (Fig. 4a, Online Resource 7). The first niche axis (41.9% of variation) is positively associated with temperature and precipitation variables, and the second niche axis (26.3%) is positively associated with temperature variables and negatively associated with annual precipitation (BIO12). The third (11.8%) is positively associated with precipitation seasonality variables (Online Resource 7). Univariate ANOVAs of PC scores reveal significant differences between the YUC and PAC + ATL + HON populations (Online Resource 7).

a Principal components analysis (PCA) on the 19 bioclimatic variables with the first principal component (PC1) largely a measure of precipitation conditions, and the second principal component (PC2) mainly determined by temperature measures. Symbols correspond to Psittacanthus rhynchanthus YUC (yellow) and PAC + ATL + HON (blue) lineages. Density plots of PC1 and PC2 scores of P. rhynchanthus lineages. b Analyses of Bayesian phylogeographical and ecological clustering (BPEC) using cpDNA. Each coloured contour plot indicates a different phylogeographic cluster identified in Psittacanthus rhynchanthus, and the shaded similar colours indicate uncertainty about the respective clusters. Sample locations are given as circles, with the three inferred ancestral sites Chetumal (Quintana Roo), Escárcega (Campeche) and El Porvenir (Honduras) also marked with an asterisk symbol
Bayesian Phylogeographic and Ecological Clustering (BPEC)
Chloroplast haplotypes were assigned by BPEC to two phylogeographic clusters (YUC and ATL + PAC + HON) with high posterior probabilities (> 0.98) (Fig. 4b), with most likely ancestral locations being Chetumal (Quintana Roo), Escárcega (Campeche) and El Porvenir (Honduras) (Fig. 4b). Haplotypes from locations of the Yucatán Peninsula were assigned to one cluster, whereas the haplotypes from locations of the Atlantic and Pacific slopes of Mexico and Honduras were assigned to another cluster (Fig. 4b). Low posterior cluster assignment probabilities (< 0.4) were obtained for the haplotypes in the Atlantic slope of Mexico to be assigned to a distinct cluster, with uncertainty about the other clusters (Fig. 4b); thus, despite the private haplotypes for this region, they are considered together with those from the Pacific slope and Honduras as a single cluster.
Discussion
Genetic diversity and structure of the Psittacanthus rhynchanthus populations
Despite similar life-history traits among Psittacanthus species (hermaphroditic flowers, predominately outcrossed, vector-mediated seed dispersal), there is evidence of biological attributes appearing to be associated with differences in genetic structure and diversity (e.g. Loveless and Hamrick 1984; Carvalho et al. 2019). For instance, low levels of genetic variation and genetic structure may be reflecting historical effects of population expansion with secondary contact and gene flow among populations, which periodically reset genetic differentiation within lineages (Ramírez-Barahona et al. 2017; Baena-Díaz et al. 2018). Genetic differentiation among P. rhynchanthus populations (GST = 0.600) was slightly higher as compared with other Psittacanthus species with more restricted distributions (P. auriculatus = 0.561; P. mayanus = 0.461; P. sonorae = 0.416; Licona-Vera et al. 2018; Ornelas et al. 2018a, 2019b), and lower as compared with other widespread species (P. calyculatus = 0.869; P. schiedeanus = 0.692; Ornelas et al. 2016; Pérez-Crespo et al. 2017). Total haplotype diversity at the species level (hT = 0.729) was nearly three times higher than haplotype diversity for each P. rhynchanthus populations (hS = 0.291), a pattern similar and values intermediate to those observed in other Psittacanthus species studied to date (P. schiedeanus, hT = 0.553, hS = 0.150, Ornelas et al. 2016; P. calyculatus, hT = 0.811, hS = 0.150, Pérez-Crespo et al. 2017; P. auriculatus, hT = 0.793, hS = 0.348, Ornelas et al. 2018a; P. mayanus, hT = 0.245, hS = 0.132, Licona-Vera et al. 2018; P. sonorae, hT = 0.553, hS = 0.323, Ornelas et al. 2019b). Total haplotype diversity in P. rhynchanthus was higher than the mean for cpDNA as reported by Petit et al. (2005) in 170 species of angiosperms (hT = 0.670). Rarefaction curves indicate that the number of haplotypes discovered on each group was near the maximum number expected. If in fact more haplotypes had been discovered with a more thorough sampling (on a per population basis), one would expect to see a further weakening of the currently observed genetic groups and a higher probability of uncovering haplotypes that are shared among regions.
Gene diversity (h) levels for P. rhynchanthus lineages (h = 0.296–0.537) ranged intermediate between lineages of other cpDNA sequence-based studies in Psittacanthus (P. schiedeanus: 0.14–0.23; Ornelas et al. 2016; P. calyculatus: 0.415–0.789; Pérez-Crespo et al. 2017; P. auriculatus: 0.125–0.398; Ornelas et al. 2018a; P. mayanus: 0.361–0.679; Licona-Vera et al. 2018; P. sonorae: 0.148–0.595; Ornelas et al. 2019b). Nucleotide diversities (π = 0.0008–0.0030) in P. rhynchanthus were low as compared to those reported for other lineages within widespread Psittacanthus species (P. schiedeanus = 0.0000–0.0198, Ornelas et al. 2016; P. calyculatus = 0.0102–0.0565, Pérez-Crespo et al. 2017), but higher than those for range-restricted species (P. auriculatus = 0.0001–0.0005, Ornelas et al. 2018a; P. mayanus = 0.0002–0.0023, Licona-Vera et al. 2018; P. sonorae = 0.0003–0.0008, Ornelas et al. 2019b). The moderate h and π values in P. rhynchanthus, between those of widespread and range-restricted Psittacanthus species, may be attributed to: (1) bird-ingested, long-distance seed dispersal that results in the homogeneous distribution of cpDNA haplotypes, (2) similar ecological environment across its distribution that could limit genetic differences, or to (3) a uniform distribution (e.g. among host species) that promotes migration and genetic homogeneity.
This phylogeographic study is first to show genetic differentiation between populations on the Yucatán Peninsula (YUC) and those along the Pacific and Atlantic slopes (PAC + ATL + HON). The haplotype network, *BEAST species tree, FST pairwise comparisons, AMOVAs, and BAPS support the existence of two clusters (YUC, PAC + ATL + HON), population differentiation, and fairly high levels of genetic structuring, especially when considering the cpDNA data. The slightly more sensitive BPEC analysis revealed the same two geographically structured clusters, suggesting that similarity in climatic conditions facilitates the genetic exchange within each region. On the other hand, this analysis retrieved populations of the Yucatán Peninsula and Honduras as the ancestral locations, suggesting that P. rhynchanthus colonized the northern range of its distribution from ancestral populations located in Central America and probably into the Caribbean region from the Yucatán Peninsula. The scenario that the colonization of the present-day range of P. rhynchanthus happened from the south and that the source plants for migration into Mesoamerica and into the Caribbean from Mesoamerica was P. rhynchanthus (Kuijt 2009) requires further phylogenetic testing for the sister relationship between P. rhynchanthus and P. americanus on the eastern Caribbean islands and P. wurdackii endemic to Venezuela (Kuijt 2009).
As indicated in Table 2, for trnL-trnF/atpB-rbcL, the very high FST values between populations at all levels supports isolation and genetic drift in P. rhynchanthus. A recent review on the genetic diversity of plants (Carvalho et al. 2019), a higher mean FST was recorded in widely distributed plant species, in comparison with the species with a more restricted range. This is because widely distributed plants have greater phenotypic plasticity and fewer adaptive restrictions (Falk and Holsinger 1991). When phylogeny was taken into account, the FST values were influenced by habitat and the combination of habitat with distribution (Carvalho et al. 2019). Assuming low dispersal capabilities in this mistletoe, the isolation between populations on the Yucatán Peninsula and those along the Mexican slopes (ecological barrier), and the Pleistocene changes in the distribution of suitable habitat that accentuated isolation, appear to explain genetic differentiation between regions. Life-history traits of some mistletoes such as seed-mediated dispersal, broad host range, high adult survivorship and migratory seed dispersers (Ornelas et al. 2016; Licona-Vera et al. 2018) might explain its genetic homogeneity within regions. Several bird species consume the fruits of P. rhynchanthus and probably disperse the seeds over long distances if consumed by migratory bird species. Therefore, it is possible that the low genetic structure and genetic differentiation within regions have been eroded by large-scale seed recruitment and high historical rates of gene flow (Ornelas et al. 2016; Licona-Vera et al. 2018). In P. rhynchanthus, haplotypes of the combined trnL-trnF/atpB-rbcL (H10–H13) were found to be exclusive to the Yucatán Peninsula, which favours the hypothesis of restricted bird seed-mediated gene flow between regions.
Demographic history
Unimodal mismatch distribution values (SSD and Hri) coupled with the Ramos-Onsins and Rozas statistic (R2), which is very powerful in detecting population expansions in small samples (Ramos-Onsins and Rozas 2002), clearly suggests a scenario of ancestral population expansion in P. rhynchanthus. Population expansion, also supported by the star-like pattern radiating from ancestral haplotypes and the low-to-moderate values of nucleotide and haplotype diversity, was not caused by changes in effective population size according to the BSP analysis. Consistent with the lack of significant changes in effective population sizes over time in the BSP analysis, the negative but no-significant Fu’s Fs and Tajima’s D for the cpDNA data indicate a weak population expansion signature, or that a low mutation rate of cpDNA could explain these contradictory signatures of demographic changes, which would be expected under equilibrium due to effective population size and sample size differences. Thus, it is possible that PAC + ATL + HON and YUC populations have experienced recent demographic expansion from an ancestral population with a small effective population size.
From the LIG to present conditions, ENM models of P. rhynchanthus suggest for the YUC lineage a recent southern expansion into the Petén province and a northern expansion into the Balsas Depression and along both the Pacific and Atlantic slopes for the PAC + ATL + HON lineage. These patterns are consistent with the hypothesis that isolation by the semi-deciduous tropical rain forest along the Petén province and Chiapas (ecological barrier) may have restricted northward gene flow from locations of the Pacific and Atlantic slopes in Mexico into the SDTF of the Yucatán province (e.g. Savage 1966; Lavin et al. 1991; Licona-Vera et al. 2018). Together, our results are consistent with both a model of isolation and reduced gene flow by both physical and environmental barriers, and expansion/contraction of species ranges responding to climatic changes during the Pleistocene (Hewitt 2000). Given the few phylogeographic studies in the region, the phylogeography of P. rhynchanthus including the ENM results is particularly important to understand the evolution of biota in the tropical lowlands of Mexico.
The two LGM climate models (CCSM and MIROC) differed regarding the potential distribution of suitable habitat for P. rhynchanthus, particularly for the YUC lineage (Fig. 3). Despite differences between those models, prediction of the CCSM model is consistent with the theory of Quaternary dry forest refugia as originally posed for the Neotropics, in which tropical forests became contracted and fragmented in their distribution and isolated forest patches were separated by an expanding savannah-like vegetation at the LGM (e.g. Haffer 1969; Carnaval and Moritz 2008). Although palaeoecological data are limited for northern Mesoamerica (Ramírez-Barahona and Eguiarte 2013), current evidence suggests that precipitation was reduced in the wet season at the LGM, resulting in either arid conditions extending over much of Mexico, Central America and the Caribbean (e.g. Anselmetti et al. 2006), or that the precipitation decrease was not significant in Mesoamerica (Hodell et al. 2008; Bush et al. 2009; Mueller et al. 2010), indicative of more stable forest areas at the tropical lowlands. As compared with the LIG model, both LGM models predict expanded suitable habitat for P. rhynchanthus. According to palynological data for the Yucatán Peninsula (Leyden 1984), it is possible that the widely distributed xeric vegetation facilitated the expansion of P. rhynchanthus to coastal Yucatán at the LGM, and then, it was contracted by expanding more mesic vegetation towards the present conditions. Thus, the uncovered distribution for P. rhynchanthus on the Yucatán Peninsula supports the existence of patchy climates and long-term stable areas in the region that served as source of genetic variability during population expansion, as suggested for other tropical regions during the LGM (e.g. Carnaval et al. 2009).
The existence of unique haplotypes on the Yucatán Peninsula and Veracruz suggests that P. rhynchanthus persisted in the northern portion of the Yucatán Peninsula (YUC lineage) at the LGM and that the southern part of the Yucatán Peninsula into the Petén region was colonized more recently. In contrast, the PAC + ATL + HON lineage expanded its distribution northwards according to ENM models. This suggests that the ancestral populations of P. rhynchanthus colonized the Balsas Depression and Atlantic slope and expanded northwards along the Pacific slope at the LGM and then contracted that distribution to present conditions in areas currently covered by SDTF. Given the host dependence of mistletoes, it is possible that the observed genetic structuring and range dynamics of P. rhynchanthus through time were influenced by host–mistletoes interactions across geography (but see Ornelas et al. 2019b). However, detailed data on prevalence or specificity of these mistletoes to its main host, Bursera simaruba, are not available to assess whether the observed mistletoe range dynamics across space and time follows changes in the distribution of its host by contrasting models that include data on host distributions with those that use only climate data for the mistletoe (Ornelas et al. 2018b).
Phylogeographic history and origin of Psittacanthus rhynchanthus
Studies on the evolution of plants in the SDTF, widespread along the coasts of Mexico, are scarce. Becerra (2005) used a time-calibrated phylogeny to reconstruct the geographic centre, time of origin and diversification rate of Bursera at different times, and relate those to the origin and expansion of the SDTF in Mexico. Her results suggest that the oldest lineages of Bursera diversified mostly in Western Mexico, as the Sierra Madre Occidental and later the Trans-Mexican Volcanic Belt were formed, whereas the more recent lineages diverged in the south-central part of the country. According to her results, Bursera probably originated between 30 and 20 million years ago (Mya) and began a relatively rapid diversification, when conditions (dry, warm, and seasonal climate) were likely favourable for the establishment of the SDTF as well (Becerra 2005). These results suggest that the SDTF was first established in the west of Mexico and from there expanded to central and south Mexico, and from the south of Mexico into Central America more recently, 2.5 Mya according to fossil evidence (Becerra 2005). The evolutionary history of the Mexican Malpighiaceae pool species and its occupancy of the SDTF in Mexico were also tested with phylogenetic methods. In this system, a ‘stepping-stone’ dispersal scenario seems to explain the majority of migration events from South America to Mexico, with mainly Central America as a bridge between South America and Mexico (Willis et al. 2014). Also, the significant increase in migration rates and species diversification beginning in the Miocene, in conjunction with the development of land corridors through Central America, played a key role in the initial establishment of the Mexican Malpighiaceae species pool and in situ adaptation to dry forest environments and precipitation seasonality until they became restricted to Mexico during the mid-Miocene, around 13.7 Mya on average (Willis et al. 2014). These results suggest that the SDTF, geographically restricted during the Late Oligocene, expanded greatly until the mid-Miocene (Willis et al. 2014).
Bursera simaruba L.Sarg., a species complex of Neotropical trees, is the most common host over much of P. rhynchanthus’s range (Kuijt 2009). A conspicuous element in tropical forests below 1400 m, the distribution of B. simaruba generally matches the distribution of P. rhynchanthus, including both coasts of Mexico to northern South America, Florida, and the West Indies (Rosell et al. 2010). Divergence dates within the Psittacanthus ‘Bursera group’ (P. nudus, P. palmeri, P. sonorae) appear to be related to those of its main Bursera host species. The split between B. simaruba, the only known Bursera species parasitized by P. rhynchanthus (Kuijt 2009), and its closest relative B. itzae Lundell occurred ~ 8.4 Ma (De-Nova et al. 2012), whereas Psittacanthus species showed a Late Miocene–Pliocene (7.6–3.4 Ma) initial divergence to P. rhynchanthus (Ornelas et al. 2016; Ortiz-Rodriguez et al. 2018b). Based on the Bursera phylogenetic tree (De-Nova et al. 2012), recent lineages of Psittacanthus (P. mayanus Standl. & Steyerm., P. rhynchanthus) invaded ancestors of the Simaruba clade in the Middle Miocene, after shifting from the SDTF to the tropical rain forest (De-Nova et al. 2012). Here, our BEAST analysis placed the origin of the P. rhynchanthus clade at 2.64–0.15 Ma (Mid-Pleistocene) and a more recent divergence (1.17–0.1 Ma) for the split between the YUC and PAC + ATL + HON lineages according to the *BEAST species tree analysis. Although caution should be taken with molecular dating, these estimates do not coincide with the timing of intense uplift of the Trans-Mexican Volcanic Belt and climate change from the Miocene to the Pleistocene, which may have produced the conditions for Bursera diversification, and expansion of the SDTF and colonization of mistletoes (Becerra 2005; De-Nova et al. 2012), but congruent with previous estimates for the origins and diversification within other Psittacanthus species (e.g. Licona-Vera et al. 2018; Ornelas et al. 2019b).
Conclusions
Our results revealed the overall phylogeographical patterns in the lowland forest-adapted P. rhynchanthus mistletoe, with strong genetic differentiation among populations and phylogeographic structuring separating populations from the Yucatán Peninsula and populations from the Pacific and Atlantic slopes of Mexico. Although some P. rhynchanthus populations are genetically differentiated (Veracruz and Honduras), the widespread haplotypes and low genetic differentiation on the Yucatán Peninsula (H12) and Pacific and Atlantic slopes (H1) suggest effective plastid gene flow. Results of mismatch distribution analysis and ENMs are suggestive of recent demographic expansion for populations of P. rhynchanthus within these regions, most likely during the LGM, without changes in effective population size. In contrast, population differentiation of P. rhynchanthus, likely before the LIG, and climate differentiation between regions are consistent with a fragmentation (old isolation) model, suggesting that gene flow has been restricted despite the expanded distribution of suitable habitat during the Pleistocene glacial cycles.
References
Allouche O, Tsoar A, Kadmon R (2006) Assessing the accuracy of species distribution models: prevalence, kappa, and the true skill statistic (TSS). J Appl Ecol 43:1223–1232. https://doi.org/10.1111/j.1365-2664.2006.01214.x
Anselmetti F, Ariztegui D, Hodell DA, Hillesheim MB, Brenner M, Gilli A, McKenzie JA, Mueller AD (2006) Late Quaternary climate-induced lake level variations in Lake Petén Itzá, Guatemala, inferred from seismic stratigraphic analysis. Palaeogeogr Palaeoclim Palaeoecol 230:52–69. https://doi.org/10.1016/j.palaeo.2005.06.037
Arteaga MC, McCormack JE, Eguiarte LE, Medellín RA (2011) Genetic admixture in multidimensional environmental space: asymmetrical niche similarity promotes gene flow in armadillos (Dasypus novemcinctus). Evolution 65:2470–2480. https://doi.org/10.1111/j.1558-5646.2011.01329.x
Baena-Díaz F, Ramírez-Barahona S, Ornelas JF (2018) Hybridization and differential introgression associated with environmental shifts in a mistletoe species complex. Sci Rep 8:5591. https://doi.org/10.1038/s41598-018-23707-6
Barve N, Barve V, Jiménez-Valverde A, Lira-Noriega A, Maher SP, Peterson AT, Soberón J, Villalobos F (2011) The crucial role of the accessible area in ecological niche modeling and species distribution modeling. Ecol Model 222:1810–1819. https://doi.org/10.1016/j.ecolmodel.2011.02.011
Becerra JX (2005) Timing the origin and expansion of the Mexican tropical dry forest. Proc Natl Acad Sci USA 102:10919–10923. https://doi.org/10.1073/pnas.0409127102
Booth TH, Nix HA, Busby JR, Hutchinson MF (2014) BIOCLIM: the first species distribution modelling package, its early applications and relevance to most current MAXENT studies. Diversity Distrib 20:1–9. https://doi.org/10.1111/ddi.12144
Braconnot P, Otto-Bliesner B, Harrison S, Joussaume S, Peterchmitt JY, Abe-Ouchi A, Crucifix M, Driesschaert E, Fichefet Th, Hewitt CD, Kageyama M, Kitoh A, Loutre M-F, Marti O, Merkel U, Ramstein G, Valdes P, Weber L, Yu Y, Zhao Y (2007) Results of PMIP2 coupled simulations of the Mid-Holocene and Last Glacial Maximum—part 2: feedbacks with emphasis on the location of the ITCZ and mid- and high latitudes heat budget. Clim Past 3:279–296. https://doi.org/10.5194/cp-3-279-2007
Bush MB, Correa-Metrio AY, Hodell DA, Brenner M, Anselmetti FS, Ariztegui D, Mueller AD, Curtis JH, Grzesik DA, Burton C, Gilli A (2009) Re-evaluation of climate change in lowland Central America during the last glacial maximum using new sediment cores from Lake Petén- Itzá, Guatemala. In: Vimeuz F, Sylvestre F, Khodri M (eds) Past climate variability in South America and Surrounding Regions, from the Last Glacial Maximum to the Holocene, vol. 14. Springer, Houten, pp 113–128
Caballero M, Lozano-García S, Vázquez-Selem L, Ortega B (2010) Evidencias de cambio climático y ambiental en registros glaciales y en cuencas lacustres del centro de México durante el último máximo glacial. Bol Soc Geol Mex 62:359–377
Caballero-Rodríguez D, Correa-Metrio A, Lozano-García S, Sosa-Nájera S, Ortega B, Sanchez-Dzib Y, Aguirre-Navarro K, Sandoval-Montaño A (2018) Late-Quaternary spatiotemporal dynamics of vegetation in Central Mexico. Rev Palaeobot Palynol 250:44–52. https://doi.org/10.1016/j.revpalbo.2017.12.004
Carnaval AC, Moritz C (2008) Historical climate modelling predicts patterns of current biodiversity in the Brazilian Atlantic forest. J Biogeogr 35:1187–1201. https://doi.org/10.1111/j.1365-2699.2007.01870.x
Carnaval AC, Hickerson MJ, Haddad CFB, Rodrigues MT, Moritz C (2009) Stability predicts genetic diversity in the Brazilian Atlantic Forest hotspot. Science 23:785–789. https://doi.org/10.1126/science.1166955
Carvalho YGS, Vitorino LC, de Souza UJB, Bessa LA (2019) Recent trends in research on the genetic diversity of plants: implications for conservation. Diversity 11:62. https://doi.org/10.3390/d11040062
Cavender-Bares J, Gonzalez-Rodriguez A, Pahlich A, Koehler K, Deacon N (2011) Phylogeography and climatic niche evolution in live oaks (Quercus series Virentes) from the tropics to the temperate zone. J Biogeogr 38:962–981. https://doi.org/10.1111/j.1365-2699.2010.02451.x
Cavers S, Navarro C, Lowe AJ (2003) Chloroplast DNA phylogeography reveals colonization history of a Neotropical tree, Cedrela odorata L., in Mesoamerica. Molec Ecol 12:1451–1460. https://doi.org/10.1046/j.1365-294X.2003.01810.x
Chao A, Jost L (2012) Coverage-based rarefaction and extrapolation: standardizing samples by completeness rather than size. Ecology 93:2533–2547. https://doi.org/10.1890/11-1952.1
Chávez-Pesqueira M, Núñez-Farfán J (2016) Genetic diversity and structure of wild populations of Carica papaya in Northern Mesoamerica inferred by nuclear microsatellites and chloroplast markers. Ann Bot (Oxford) 118:1293–1306. https://doi.org/10.1093/aob/mcw183
Chiang TY, Schaal BA, Peng CI (1998) Universal primers for amplification and sequencing a noncoding spacer between the atpB and rbcL genes of chloroplast DNA. Bot Bull Acad Sinica 39:245–250
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Molec Ecol 9:1657–1659. https://doi.org/10.1046/j.1365-294x.2000.01020.x
Colin R, Eguiarte LE (2016) Phylogeographic analyses and genetic structure illustrate the complex evolutionary history of Phragmites australis in Mexico. Amer J Bot 103:876–887. https://doi.org/10.3732/ajb.1500399
Colinvaux PA, de Oliveira PE, Moreno JE, Miller MC, Bush MB (1996) A long pollen record from lowland Amazonia: forest and cooling in glacial times. Science 274:85–87. https://doi.org/10.1126/science.274.5284.85
Colinvaux PA, Irion G, Rasanen ME, Bush MB, de Mello J (2001) A paradigm to be discarded: geological and paleoecological data falsify the Haffer & Prance refuge hypothesis of Amazonian speciation. Amazoniana 16:609–646
Colwell RK (2013) EstimateS: statistical estimation of species richness and shared species from samples. Version 9 and earlier. User’s Guide and application. Available at: http://purl.oclc.org/estimates. Accessed 23 Aug 2019
Corander J, Sirén J, Arjas E (2008) Bayesian spatial modeling of genetic population structure. Comput Stat 23:111–129. https://doi.org/10.1007/s00180-007-0072-x
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Meth 9:772. https://doi.org/10.1038/nmeth.2109
Daza JM, Castoe TA, Parkinson CL (2010) Using regional comparative phylogeographic data from snake lineages to infer historical processes in Middle America. Ecography 33:343–354. https://doi.org/10.1111/j.1600-0587.2010.06281.x
De-Nova A, Medina R, Montero JC, Weeks A, Rosell JA, Olson ME, Eguiarte LE, Magallón S (2012) Insights into the historical construction of species-rich Mesoamerican seasonally dry tropical forests: the diversification of Bursera (Burseraceae, Sapindales). New Phytol 193:276–287. https://doi.org/10.1111/j.1469-8137.2011.03909.x
Díaz Infante S, Lara C, Arizmendi MC, Eguiarte LE, Ornelas JF (2016) Reproductive ecology and isolation of Psittacanthus calyculatus and P. auriculatus mistletoes (Loranthaceae). PeerJ 4:e2491. https://doi.org/10.7717/peerj.2491
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure from small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. https://doi.org/10.1186/1471-2148-7-214
Drummond AJ, Rambaut A, Shapiro B, Pybus OG (2005) Bayesian coalescent inference of past population dynamics from molecular sequences. Molec Biol Evol 22:1185–1192. https://doi.org/10.1093/molbev/msi103
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Molec Ecol 11:2571–2581. https://doi.org/10.1046/j.1365-294X.2002.01650.x
Elith J, Phillips SJ, Hastie T, Dudík M, Chee YE, Yates CJ (2011) A statistical explanation of MaxEnt for ecologists. Diversity Distrib 17:43–57. https://doi.org/10.1111/j.1472-4642.2010.00725.x
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinf Online 1:47–50
Falk DA, Holsinger KE (1991) Genetics and conservation of rare plants. Oxford University Press, New York
Fielding AH, Bell JF (1997) A review of methods for the assessment of prediction errors in conservation presence/absence models. Environm Conservation 24:38–49
Fu YX (1997) Statistical neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
González C, Ornelas JF, Gutiérrez-Rodríguez C (2011) Selection and geographic isolation influence hummingbird speciation: genetic, acoustic and morphological divergence in the wedge-tailed sabrewing (Campylopterus curvipennis). BMC Evol Biol 11:38. https://doi.org/10.1186/1471-2148-11-38
Gotelli NJ, Colwell RK (2001) Quantifying biodiversity: procedures and pitfalls in the measurement and comparison of species richness. Ecol Lett 4:379–391. https://doi.org/10.1046/j.1461-0248.2001.00230.x
Guevara-Chumacero LM, López-Wilchis R, Pedroche FF, Juste J, Ibáñez C, Barriga-Sosa IDLA (2010) Molecular phylogeography of Pteronotus davyi (Chiroptera: Mormoopidae) in Mexico. J Mammol 91:220–232. https://doi.org/10.1644/08-MAMM-A-212R3.1
Gutiérrez-Rodríguez C, Ornelas JF, Rodríguez-Gómez F (2011) Chloroplast DNA phylogeography of a distylous shrub (Palicourea padifolia, Rubiaceae) reveals past fragmentation and demographic expansion in Mexican cloud forests. Molec Phylogen Evol 61:603–615. https://doi.org/10.1016/j.ympev.2011.08.023
Haffer J (1969) Speciation in Amazonian forest birds. Science 165:131–136. https://doi.org/10.1126/science.165.3889.131
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98. https://doi.org/10.12691/ajidm-4-3-3
Harpending RC (1994) Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human Biol 66:591–600
Heled J, Drummond AJ (2010) Bayesian inference of species trees from multilocus data. Molec Biol Evol 27:570–580. https://doi.org/10.1093/molbev/msp274
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276. https://doi.org/10.1111/j.1095-8312.1996.tb01434.x
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913. https://doi.org/10.1038/35016000
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. https://doi.org/10.1002/joc.1276
Hodell DA, Anselmetti FS, Ariztegui D, Brenner M, Curtis JH, Gilli A, Grzesik DA, Guilderson TJ, Müller AD, Bush MB, Correa-Metrio A, Escobar J, Kutterolf S (2008) An 85-ka record of climate change in lowland Central America. Quatern Sci Rev 27:1152–1165. https://doi.org/10.1016/j.quascirev.2008.02.008
Hsieh TC, Ma KH, Chao A (2016) iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers). Meth Ecol Evol 7:1451–1456. https://doi.org/10.1111/2041-210X.12613
Jaramillo-Correa JP, Beaulieu J, Khasa DP, Bousquet J (2009) Inferring the past from the present phylogeographic structure of North American forest trees: seeing the forest for the genes. Canad J Pl Res 39:286–307. https://doi.org/10.1139/X08-181
Knowles LL (2008) Why does a method that fails continue to be used? Evolution 62:2713–2717. https://doi.org/10.1111/j.1558-5646.2008.00481.x
Kuijt J (1987) Novelties in Mesoamerican mistletoes (Loranthaceae and Viscaceae). Ann Missouri Bot Gard 74:511–532
Kuijt J (2009) Monograph of Psittacanthus (Loranthaceae). Syst Bot Monogr 86:1–362
Landis JR, Koch GG (1977) The measurement of observer agreement for categorical data. Biometrics 33:159–174
Lavin M, Mathews S, Hughes C (1991) Chloroplast DNA variation in Gliricidia sepium (Leguminosae): intraspecific phylogeny and tokogeny. Amer J Bot 78:1576–1585. https://doi.org/10.1002/j.1537-2197.1991.tb11437.x
León-Camargo D, Rangel-Ch JO (2015) Interacción colibrí-flor en tres remanentes de bosque tropical seco (BST) del Municipio de Chimichagua (Cesar Colombia). Caldasia 37:107–123
Leyden BW (1984) Guatemalan forest synthesis after Pleistocene aridity. Proc Natl Acad Sci USA 81:4856–4859. https://doi.org/10.1073/pnas.81.15.4856
Licona-Vera Y, Ortiz-Rodriguez AE, Vásquez-Aguilar AA, Ornelas JF (2018) Lay mistletoes on the Yucatan Peninsula: post-glacial expansion and genetic differentiation of Psittacanthus mayanus (Loranthaceae). Bot J Linn Soc 186:334–360. https://doi.org/10.1093/botlinnean/box098
Lira-Noriega A, Toro-Núñez O, Oaks JR, Mort ME (2015) The roles of history and ecology in chloroplast phylogeographic patterns of the bird-dispersed plant parasite Phoradendron californicum (Viscaceae) in the Sonoran Desert. Amer J Bot 102:149–164. https://doi.org/10.3732/ajb.1400277
Liu C, White M, Newell G (2013) Selecting thresholds for the prediction of species occurrence with presence-only data. J Biogeogr 40:778–789. https://doi.org/10.1111/jbi.12058
Loveless MD, Hamrick JL (1984) Ecological determinants of genetic structure in plant populations. Annual Rev Ecol Syst 15:65–95. https://doi.org/10.1146/annurev.es.15.110184.000433
Luna-Vega I (2008) Aplicaciones de la biogeografía histórica a la distribución de las plantas mexicanas. Revista Mex Biodivers 79:217–241
Manolopoulou I, Emerson BC (2012) Phylogeographic ancestral inference using the coalescent model on haplotype trees. J Comp Biol 19:745–755. https://doi.org/10.1089/cmb.2012.0038
Manolopoulou I, Legarreta L, Emerson BC, Brooks S, Tavaré S (2011) A Bayesian approach to phylogeographic clustering. Interface Focus 1:909–921. https://doi.org/10.1098/rsfs.2011.0054
Manolopoulou I, Hille A, Emerson B (2020) BPEC: an R package for Bayesian phylogeographic and ecological clustering. J Stats Softw (accepted). arXiv:1604.01617
Masson-Delmotte V, Kageyama M, Braconnot P, Charbit S, Krinner G, Ritz C, Guilyardi E, Jouzel J, Abe-Ouchi A, Crucifix M, Gladstone RM, Hewitt CD, Kitoh A, LeGrande AN, Marti O, Merkel U, Motoi T, Ohgaito R, Otto-Bliesner B, Peltier WR, Ross I, Valdes PJ, Vettoretti G, Weber SL, Wolk F, Yu Y (2006) Past and future polar amplification of climate change: climate model intercomparisons and ice-core constraints. Clim Dynam 26:513–529. https://doi.org/10.1007/s00382-005-0081-9
Mastretta-Yanes A, Moreno-Letelier A, Piñero D, Jorgensen TH, Emerson BC (2015) Biodiversity in the Mexican highlands and the interaction of geology, geography and climate within the Trans- Mexican Volcanic Belt. J Biogeogr 42:1586–1600. https://doi.org/10.1111/jbi.12546
Mastretta-Yanes A, Xue AT, Moreno-Letelier A, Jorgensen TH, Alvarez N, Piñero D, Emerson BC (2018) Long-term in situ persistence of biodiversity in tropical sky islands revealed by landscape genomics. Molec Ecol 27:432–448. https://doi.org/10.1111/mec.14461
McCormack JE, Heled J, Delaney KS, Peterson AT, Knowles LL (2011) Calibrating divergence times on species trees versus gene trees: implications for speciation history of Aphelocoma jays. Evolution 65:184–202. https://doi.org/10.1111/j.1558-5646.2010.01097.x
Montaño-Arias G, Luna-Vega I, Morrone JJ, Espinosa D (2018) Biogeographical identity of the Mesoamerican dominion wwith emphasis on seasonally dry tropical forests. Phytotaxa 376:277–290. https://doi.org/10.11646/phytotaxa.376.6.3
Moreno-Letelier A, Piñero D (2009) Phylogeographic structure of Pinus strobiformis Engelm. across the Chihuahuan Desert filter-barrier. J Biogeogr 36:121–131. https://doi.org/10.1111/j.1365-2699.2008.02001.x
Morrone JJ (2005) Hacia una síntesis biogeográfica de México. Revista Mex Biodivers 76:207–252
Morrone JJ (2014) Biogeographical regionalisation of the Neotropical region. Zootaxa 3782:1–110. https://doi.org/10.11646/zootaxa.3782.1.1
Mueller AD, Anselmetii FS, Ariztequi D, Brenner M, Hodell DA, Curtis JH, Escobar J, Gilli A, Grzesik DA, Guilderson TP, Kutterolf S, Plötze (2010) Late Quaternary palaeoenvironment of northern Guatemala: evidence from deep drill cores and seismic statigraphy of Lake Petén Itzá. Sedimentology 57:1220–1245. https://doi.org/10.1111/j.1365-3091.2009.01144.x
Ornelas JF, González C (2014) Interglacial genetic diversification of Moussonia deppeana (Gesneriaceae), a hummingbird-pollinated, cloud forest shrub in northern Mesoamerica. Molec Ecol 23:4119–4136. https://doi.org/10.1111/mec.12841
Ornelas JF, Rodríguez-Gómez F (2015) Influence of Pleistocene glacial/interglacial cycles of the genetic structure of the mistletoe cactus Rhipsalis baccifera (Cactaceae) in Mesoamerica. J Heredity 106:196–210. https://doi.org/10.1093/jhered/esu113
Ornelas JF, Ruiz-Sanchez E, Sosa V (2010) Phylogeography of Podocarpus matudae (Podocarpaceae): pre-Quaternary relicts in northern Mesoamerican cloud forests. J Biogeogr 37:2384–2396. https://doi.org/10.1111/j.1365-2699.2010.02372.x
Ornelas JF, Sosa V, Soltis DE, Daza JM, González C, Soltis PS, Gutiérrez-Rodríguez C, Espinosa de los Monteros A, Castoe TA, Bell C, Ruiz-Sanchez E (2013) Comparative phylogeographic analyses illustrate the complex evolutionary history of threatened cloud forests of northern Mesoamerica. PLoS ONE 8:e56283. https://doi.org/10.1371/journal.pone.0056283
Ornelas JF, Gándara E, Vásquez-Aguilar AA, Ramírez-Barahona S, Ortiz-Rodriguez AE, González C, Mejía Saules MT, Ruiz-Sanchez E (2016) A mistletoe tale: postglacial invasion of Psittacanthus schiedeanus (Loranthaceae) to Mesoamerican cloud forests revealed by molecular data and species distribution modeling. BMC Evol Biol 16:78. https://doi.org/10.1186/s12862-016-0648-6
Ornelas JF, Licona-Vera Y, Vásquez-Aguilar AA (2018a) Genetic differentiation and fragmentation in response to climate change of the narrow endemic Psittacanthus auriculatus. Trop Conservation Sci 11:1–15. https://doi.org/10.1177/1940082918755513
Ornelas JF, Licona-Vera Y, Ortiz-Rodriguez AE (2018b) Contrasting responses of generalized/specialized mistletoe-host interactions under climate change. Écoscience 25:223–234. https://doi.org/10.1080/11956860.2018.1439297
Ornelas JF, Ortiz-Rodriguez AE, Ruiz-Sanchez E, Sosa V, Pérez-Farrera MA (2019a) Ups and downs: genetic differentiation among populations of the Podocarpus (Podocarpaceae) species in Mesoamerica. Molec Phylogen Evol 138:17–30. https://doi.org/10.1016/j.ympev.2019.05.025
Ornelas JF, García JM, Ortiz-Rodriguez AE, Licona-Vera Y, Gándara E, Molina-Freaner F, Vásquez-Aguilar AA (2019b) Tracking host trees: phylogeography of endemic Psittacanthus sonorae (Loranthaceae) mistletoe in the Sonoran Desert. J Heredity 110:229–246. https://doi.org/10.1093/jhered/esy065
Ortiz-Rodriguez A, Ornelas JF, Ruiz-Sanchez E (2018a) A jungle tale: molecular phylogeny and divergence time estimates of the Desmopsis-Stenanona clade (Annonaceae) in Mesoamerica. Molec Phylogen Evol 122:80–94. https://doi.org/10.1016/j.ympev.2018.01.021
Ortiz-Rodriguez AE, Guerrero EY, Ornelas JF (2018b) Phylogenetic position of Neotropical Bursera-specialist mistletoes: the evolution of deciduousness and succulent leaves in Psittacanthus (Loranthaceae). Bot Sci 96:443–461. https://doi.org/10.17129/botsci.1961
Otto-Bliesner BL, Marshall SJ, Overpeck JT, Miller GH, Hu A (2006) Simulating Arctic climate warmth and icefield retreat in the Last Interglaciation. Science 311:1751–1753. https://doi.org/10.1126/science.1120808
Otto-Bliesner BL, Hewitt CD, Marchitto TM, Brady E, Abe-Ouchi A, Crucifix M, Murakami S, Weber SL (2007) Last Glacial Maximum ocean thermohaline circulation: PMIP2 model intercomparisons and data constraints. Geophys Res Lett 34:L12706. https://doi.org/10.1029/2007GL029475
Paradis E (2010) pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26:419–420. https://doi.org/10.1093/bioinformatics/btp696
Pennington RT, Lavin M, Prado DE, Pendry CA, Pell SK, Butterworth CA (2004) Historical climate change and speciation: neotropical seasonally dry forest plants show patterns of both Tertiary and Quaternary diversification. Phil Trans R Soc Lond B 359:515–537. https://doi.org/10.1098/rstb.2003.1435
Pérez-Crespo MJ, Ornelas JF, González-Rodríguez A, Ruiz-Sanchez E, Vásquez-Aguilar AA, Ramírez-Barahona S (2017) Phylogeography and population differentiation in the Psittacanthus calyculatus (Loranthaceae) mistletoe: a complex scenario of climate-volcanism interaction along the Trans-Mexican Volcanic Belt. J Biogeogr 44:2501–2514. https://doi.org/10.1111/jbi.13070
Petit RJ, Duminil J, Fineschi S, Hampe A, Salvini D, Vendramin GG (2005) Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Molec Ecol 14:689–701. https://doi.org/10.1111/j.1365-294X.2004.02410.x
Pfenninger M, Posada D (2002) Phylogeographic history of the land snail Candidula unifasciata (Helicellinae, Stylommatophora): fragmentation, corridor migration, and secondary contact. Evolution 56:1776–1788. https://doi.org/10.1111/j.0014-3820.2002.tb00191.x
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190:231–259. https://doi.org/10.1016/j.ecolmodel.2005.03.026
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144:1237–1245
Prance GT (1973) Phytogeographic support for the theory of Pleistocene forest refuges in the Amazon Basin, based upon evidence from distribution pattern in Caryocaraceae, Chrysobalanaceae, Dichapetalaceae and Lecythidaceae. Acta Amazon 3:5–28
Ramírez-Barahona S, Eguiarte LE (2013) The role of glacial cycles in promoting genetic diversity in the Neotropics: the case of cloud forests during the Last Glacial Maximum. Ecol Evol 3:725–738. https://doi.org/10.1002/ece3.483
Ramírez-Barahona S, Eguiarte LE (2014) Changes in the distribution of cloud forests during the last glacial period predict the patterns of genetic diversity and demographic history of the tree fern Alsophila firma (Cyatheaceae). J Biogeogr 41:2396–2407. https://doi.org/10.1111/jbi.12396
Ramírez-Barahona S, González C, González-Rodríguez A, Ornelas JF (2017) The influence of climatic niche preferences on the population genetic structure of a mistletoe species complex. New Phytol 214:1751–1761. https://doi.org/10.1111/nph.14471
Ramos-Onsins R, Rozas R (2002) Statistical properties of new neutrality tests against population growth. Molec Biol Evol 19:2092–2100
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise differences. Molec Biol Evol 9:552–569. https://doi.org/10.1093/oxfordjournals.molbev.a040727
Rosell JA, Olson ME, Weeks A, De-Nova JA, Medina Lemos R, Pérez Camacho J, Feria TP, Gómez-Bermejo R, Montero JC, Eguiarte LE (2010) Diversification in species complexes: tests of species origin and delimitation in the Bursera simaruba clade of tropical trees (Burseraceae). Molec Phylogen Evol 57:798–811. https://doi.org/10.1016/j.ympev.2010.08.004
Ruiz-Sanchez E, Ornelas JF (2014) Phylogeography of Liquidambar styraciflua (Altingiaceae) in Mesoamerica: survivors of a Neogene widespread temperate forest (or cloud forest) in North America? Ecol Evol 4:311–328. https://doi.org/10.1002/ece3.938
Ruiz-Sanchez E, Specht CD (2014) Ecological speciation in Nolina parviflora (Asparagaceae): lacking spatial connectivity along of the Trans-Mexican Volcanic Belt. PLoS ONE 9:e98754. https://doi.org/10.1371/journal.pone.0098754
Rzedowski J (1963) El extremo boreal del bosque tropical siempre verde en Norteamérica continental. Vegetation 11:173–198. https://doi.org/10.1007/BF00298831
Sánchez-González LA, Navarro-Sigüenza AG, Ornelas JF, Morrone JJ (2013) What’s in a name? Mesoamerica. Revista Mex Biodivers 84:1305–1308. https://doi.org/10.7550/rmb.34171
Savage JM (1966) The origins and history of the Central American herpetofauna. Copeia 1966:719–766
Schneider S, Excoffier L (1999) Estimation of demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics 152:1079–1089
Smith AB, Godsoe W, Rodríguez-Sánchez F, Wang HH, Warren D (2019) Niche estimation above and below the species level. Trends Ecol Evol 34:261–273. https://doi.org/10.1016/j.tree.2018.10.012
Suárez-Atilano M, Burbrink F, Vázquez-Domínguez E (2014) Phylogeographical structure within Boa constrictor imperator across the lowlands and mountains of Central America and Mexico. J Biogeogr 41:2371–2384. https://doi.org/10.1111/jbi.12372
Swets JA (1988) Measuring the accuracy of diagnostic systems. Science 240:1285–1293. https://doi.org/10.1126/science.3287615
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Pl Molec Biol 17:1105–1109. https://doi.org/10.1007/BF00037152
Tajima F (1989) Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Vargas-Rodríguez YL, Platt WJ, Urbatsch LE, Foltz DW (2015) Large scale patterns of genetic variation and differentiation in sugar maple from tropical Central America to temperate North America. BMC Evol Biol 15:257. https://doi.org/10.1186/s12862-015-0518-7
Willis CG, Franzone BF, Zhenxiang X, Davis CC (2014) The establishment of Central American migratory corridors and the biogeographic origins of seasonally dry tropical forests in Mexico. Frontiers Genet 5:1–14. https://doi.org/10.3389/fgene.2014.00433
Winston ME, Kronauer DJC, Moreau CS (2017) Early and dynamic colonization of Central America drives speciation in Neotropical army ants. Molec Ecol 26:859–870. https://doi.org/10.1111/mec.13846
Yule KM, Koop JAH, Alexandre NM, Johnston LR, Whiteman NK (2016) Population structure of a vector-borne plant parasite. Molec Ecol 25:3332–3343. https://doi.org/10.1111/mec.13693
Acknowledgements
We thank Cristina Bárcenas, Pablo Carrillo, Luis Cervantes✝, Etelvina Gándara, Felicitas Lagunes, José Luis Martínez, María Teresa Mejía-Saules, Eduardo Ruiz-Sanchez and Victoria Sosa who helped in obtaining samples and/or generated sequences for this work; and Ricarda Riina and two anonymous reviewers for useful comments on previous versions of the manuscript. Permission to conduct our fieldwork was granted by the Mexican government (Instituto Nacional de Ecología, Secretaría del Medio Ambiente y Recursos Naturales, SGPA/DGGFS/712/1299/12), and collecting permits from Guatemala (Universidad del Valle, UVAL) and Honduras (Centro Universitario Regional del Litoral Atlántico, CURLA) herbaria. This work was supported by competitive grants (grant numbers 61710, 155686, A1-S-26134) from the Consejo Nacional de Ciencia y Tecnología (CONACyT; http://www.conacyt.mx) and research funds (20030/10563) from the Departamento de Biología Evolutiva, Instituto de Ecología, AC (INECOL) awarded to Juan Francisco Ornelas. Doctoral scholarships from CONACyT were granted to A.E.O.R. (262563), Y.L.V. (262561) and E.A.L.H. (584159), and a research assistant scholarship from CONACyT was granted to M.H.S.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
The authors comply will all rules of the journal following the COPE guidelines; all authors have contributed and approved the final manuscript.
Additional information
Handling Editor: Ricarda Riina.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Information on Electronic Supplementary Material
Information on Electronic Supplementary Material
Online Resource 1. Potential effects of Pleistocene glacial/interglacial cycles on the distribution of suitable habitat for populations of the mistletoe, Psittacanthus rhynchanthus for the last interglacial (LIG, 140–120 ka BP), Last Glacial Maximum (LGM, 21 ka BP) and the present (0 ka).
Online Resource 2. Geographic information and sample sizes of the 22 Psittacanthus rhynchanthus localities sampled in this study.
Online Resource 3. Time divergence estimates and phylogenetic analysis for P. rhynchanthus individuals based on nrDNA and combined cpDNA (trnL-F/atpB-rbcL) sequence data using a Bayesian approach.
Online Resource 4. Numbers of genetically analysed samples (n) for each molecular marker (ITS and trnL-F/atpB-rbcL), and numbers of distinct ribotypes (R) and haplotypes (H) found in Psittacanthus rhynchanthus individuals sampled.
Online Resource 5. Rarefaction analyses for (a) individual- and (b) population-based haplotype richness (95% confidence intervals, CI) accumulation of cpDNA haplotypes based on random sampling of the Psittacanthus rhynchanthus data set.
Online Resource 6. Bayesian skyline plots showing historical demographic trends of Psittacanthus rhynchanthus showing changes in effective population size (Ne; mean ± 95% central posterior density) over time.
Online Resource 7. Factor loadings from the principal components analysis of Psittacanthus rhynchanthus in Mesoamerica on temperature and precipitation variables from WorldClim.
Rights and permissions
About this article

Cite this article
Ortiz-Rodriguez, A.E., Licona-Vera, Y., Vásquez-Aguilar, A.A. et al. Genetic differentiation among Psittacanthus rhynchanthus (Loranthaceae) populations: novel phylogeographic patterns in the Mesoamerican tropical lowlands. Plant Syst Evol 306, 10 (2020). https://doi.org/10.1007/s00606-020-01638-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00606-020-01638-y