
Summary
Tamarix chinensis Lour. is a perennial shrub that is highly adapted to flooded and salty conditions with a wide distribution. In this paper, a phylogeographic history of T. chinensis including 20 populations from China was inferred using two chloroplast DNA (cpDNA) segments (trnL-trnF, and rps16) and one nuclear ribosomal DNA (nrDNA) region. A total of 11 ribotypes and 16 chlorotypes were identified. The total nucleotide diversity (HT) of the nrITS and cpDNA were 0.803 and 0.635 respectively, showing a moderate level of genetic variation. The haplotype trees of both nrITS and cpDNA exhibited an H1-centred radiation differentiation pattern. In addition, both cpDNA and nrITS data showed no significant population differentiation within T. chinensis. AMOVA results revealed that almost all genetic variations existed within the populations. Furthermore, we found relatively stronger population differentiation based on nrITS rather than that of cpDNA. nrITS indicated a significant positive correlation between the genetic differentiation coefficient and geographic distance. These results implied that seed dispersal is more efficient than pollen dispersal in T. chinensis. The molecular data and ecological niche modelling also indicated that T. chinensis retreated into refugia on a large-scale during the Last Glacial Maximum (LGM) period and then there was sudden population expansion and recolonisation of suitable habitats after the glacial period.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Profound climatic oscillations during the Quaternary glacial period and subsequent periodic fluctuations resulted in repeated acute environmental changes, which also substantially shaped species distribution, evolution, and extinction (Comes & Kadereit 1998; Hewitt 2000, 2004). Many plant species sought refuge during the Pleistocene Epoch (Petit et al. 2003). The distributions of these glacial refugia, the potential recolonisation routes, and the subsequent evolution and speciation of plants during glacial and post-glacial epochs has been extensively studied for Europe, East Asia, North America and the Qinghai-Tibetan plateau (Horsak et al. 2019; Muellner-Riehl 2019; Park & Donoghue 2019; Song et al. 2018; Xia et al. 2018).
China’s warm-temperate zone generally refers to the area between 32°30' – 42°30' and 103°30' – 124°10', which comprises an area of approximately 7 × 105 km2 (Shangguan et al. 2009). The typical vegetation in this region is deciduous broadleaved forest (Gao et al. 2001). This warm temperate zone had not been strongly influenced by massive Quaternary glaciers since the Tertiary Period. So unlike the large-scale extinction of broadleaved species in Europe and North America, the majority of species among the vegetation were preserved in this region. Although no massive ice sheet directly covered China’s warm-temperate zone during the Quaternary glacial period, the climate in this region became cooler and drier, the evolution and distribution of species in this region were still affected by climate fluctuations (Park & Donoghue 2019). The glacial refugia and potential migration pathways of species in Europe and North America have been widely studied (Tiffney & Manchester 2001; García-Vázquez et al. 2017; Horsak et al. 2019; Schoenswetter & Schneeweiss 2019). Regarding the East Asian flora, the drastic climate change caused by the uplift of the Qinghai-Tibet Plateau was the main reason for divergence in many species, resulting in the emergence of endemic species and biodiversity among alpine plants in East Asia (Sun et al. 2015; Li et al. 2013; Wan et al. 2016). In recent years, a significant amount of research has been conducted on the phylogeography of plants in the temperate zones of Central Asia, such as the origination of temperate forests. The existence of glacial refugia in north and south Qinling has been documented, for example for Ostryopsis davidiana Decne. (Tian et al. 2009) and Dysosma versipellis (Hance) M.Cheng & T.S.Ying (Qiu et al. 2011). Certain dominant species of the temperate coniferous-broadleaf mixed forests of northeast China, such as the Korean pine (Pinus koraiensis Siebold & Zucc.) (Aizawa et al. 2012), Mongolian oak (Quercus mongolica Fisch. ex Turcz.) (Zeng et al. 2011), and Manchurian ash (Fraxinus mandshurica Rupr.) (Hu et al. 2008), may have sought refuge in situ in the Changbai Mountain region during glacial periods and only underwent limited post-glacial spatial expansion. However, other studies have indicated that temperate deciduous forests migrated southwards during the Last Glacial Maximum (LGM) (Yu et al. 2000; Ni et al. 2006).
Tamarix chinensis Lour. is native to China and widely distributed in China’s warm-temperate zone, occurring in several other northern, eastern and north-western provinces (such as Liaoning, Jiangsu, Shandong, Hebei, Anhui, Henan, and some regions of Shaanxi and Gansu etc.). Tamarix chinensis is a perennial shrub that is highly adapted to flooded and salty conditions. Moreover, this plant has become an invasive species in America due to its fast growth and tolerance. The genetic structure of T. chinensis in the USA (Gaskin & Schaal 2002) and the genetic diversity in the Yellow River Delta (Jiang et al. 2012) have been investigated. Liang et al. (2018a, b) reported that T. chinensis and T. austromongolica Nakai populations which are widely distributed in the Yellow River, showed low population genetic differentiation, and neither had obvious geographic structure. Their divergence time coincided well with the formation of the modern Yellow River and expanded approximately from the upper to lower reaches of the Yellow River during the Quaternary period. Although preliminary studies have been conducted on the genetic structure and genealogical geography of T. chinensis, the results were not entirely consistent, which may be due to the differences in sampling range, and how this species responded to Pleistocene climate change was not clear.
In this study, two chloroplast DNA (cpDNA) regions and one nuclear ribosomal DNA (nrDNA) region were used to examine the phylogeographical pattern of Tamarix chinensis. Additionally, we used Ecological Niche Modeling (ENM) to supplement the results of the molecular approaches. By reconstructing potential geographic distribution of species during different historical periods, ENM can provide innovative insights into questions in ecology and evolution (Alvarado-Serrano & Knowles 2014).
We addressed the following questions: (i) what was the diversity and the genetic structure of Tamarix chinensis populations in China? (ii) how did the species respond to the climatic oscillations during the Pleistocene?
Material and Methods
Population Sampling
8 – 10 leaf samples were collected from 20 natural populations of Tamarix chinensis in China. In each population, individuals were spaced at least 10 m apart from each other. GPS records and voucher specimens were also collected. Leaf samples were dried and stored in silica gel immediately after field sampling. To avoid interference from human activity as far as possible, natural distribution was prioritised. Individuals were removed after laboratory identification using morphology or molecular phylogeny if they did not belong to T. chinensis. Further, some individuals failed in PCR or sequencing. A total of 135 individuals were analysed in this study (Table 1).
DNA extraction, PCR amplification, and sequencing
Total genomic DNA was extracted from approximately 20 – 30 mg of dry leaf tissue using HP Plant DNA extract kits (Omega, GA, USA), according to the manufacturer's protocol. The concentration and purity of the extracted DNA were quantified using a NanoDrop 2000 (ThermoScientific, DE, USA). After preliminary screening of 12 fragments, we chose a nuclear internal transcribed spacer (ITS) region and two chloroplast DNA (cpDNA) regions: trnL-trnF and rps16 for the full survey because they contained the most polymorphic sites. The sequences of these primers are listed in Table S1. PCR amplification was performed in a total volume of 25 μL, with 2.5 μL10×PCR buffer (Mg2+concentration of 25 mM), 2 μL of each dNTP (10 mM), 1.5 U of Taq Polymerase (Takara), 10 – 30 ng genomic DNA, and 2 μL of each primer (5 mM). An S1000TM thermo cycler (Bio-Rad, CA, USA) was used with the following settings for ITS: 95°C for 3 min; 30 cycles of 95°C for 30 s, 58°C for 30 s, 72°C for 1 min; and 72°C for 10 min. The cpDNA regions were amplified using the same conditions with the exception of annealing at 52°C for 1 min and a final elongation at 72°C for 1 min. After product purification with an alcohol/EDTA/NaAc protocol, PCR products were then sequenced on an ABI 3730 automated sequencer (Honortech Co., Ltd, Beijing, China). All sequence data has been deposited in GenBank.
Molecular data analysis
DNA sequences were aligned using ClustalX version 1.83 (Thompson et al. 1997). The presence of “double peaks” at polymorphic sites in the chromatogram was manually checked. The number of indels and levels of polymorphism for each gene region were determined using DnaSP version 5.0 (Librado & Rozas 2009). Haplotype diversity (h) and nucleotide diversity (π) were calculated for each population (hS and πS) and at the species level (hT and πT) using DnaSP version 5.0 (Librado & Rozas 2009). A network of the haplotypes was constructed using TCS 1.21 (Clement et al. 2000), with the default parsimony connection limit of 95%, and each indel was treated as a single mutation event.
Total haplotype diversity (HT), within-population diversity (HS), GST (coefficient of genetic variation over all populations), and NST (coefficient of genetic variation influenced by both haplotype frequencies and genetic distances between haplotypes) were calculated by the program PERMUT (Pons & Petit 1996).
To infer the most likely number of population genetic clusters (K) in the cpDNA and nrDNA dataset, we performed Bayesian analysis of population structure as implemented in STRUCTURE version 2.2 (Evanno et al. 2005). K ranged from 1 – 10, with 10 replicates performed for each K and using a burn-in period of 2 × 105 and 5 × 104 Monte Carlo and Markov chains. The "no-admixture model" and independent allele frequencies were chosen for this analysis. The most likely number of clusters was identified using the maximal value of L (K) returned by STRUCTURE.
Genetic divergence among populations was inferred from Nei's estimator of population substructure (GST) as well as from ΦST obtained from non-hierarchical analyses of molecular variance (AMOVAs) in ARLEQUIN version 3.5 (Excoffier & Lischer 2010). Hierarchical AMOVA was also used to quantify the partitioning of cpDNA and nrDNA variance between regional groups of populations (ΦCT) and between populations within such groups (ΦSC). Significance levels of Φ statistics were based on 10000 permutations.
Genetic divergence between populations (FST) both ITS and cpDNA were obtained with DnaSP. Mantel test was implemented between matrix of FST and geographic distance (km) in R software using the Vegan package.
To infer demographic processes, the null hypothesis of the sudden expansion model in ARLEQUIN was tested through comparing the observed and expected distributions of pairwise sequence differences (i.e. mismatch distributions). The sum of squared deviations (SSD) and raggedness index (RAG) were used to test the goodness-of-fit of the observed mismatch distribution to the model expectation. In addition, tests of selective neutrality, namely Tajima’s D (Tajima 1989) and Fu’s Fs (Fu 1997) values, were used to infer potential population growth and expansion. If the expansion model was not rejected in the mismatch analysis and neutrality test, the relationship s = 2 ut (Rogers & Harpending 1992) was used to estimate the age of expansion (t), where l is the substitution rate for DNA sequences.
Past and current distribution inferences
Ecological niche modelling was carried out in MAXENT version 3.3.3 (Phillips et al. 2006) to predict suitable past (i.e. LGM, LIG[Last Interglacial]) and present climate envelopes for Tamarix chinensis. Information on the geographic distribution of T. chinensis was based on the 45 populations included in this study, in which 25 points were obtained from the Chinese Virtual Herbarium (http://www.cvh.org.cn/cms_cvh) and the remaining 20 points were from the sampling sites. Current bioclimatic variables, LIG and LGM data were downloaded from the WorldClim database (http://www.worldclim.org/). LGM data were simulated using the Community Climate System Model (Collins et al. 2006), and 19 environmental parameters were used to model the potential distribution of the species. To test the performance of each model, 20% of the data in each run was randomly selected by Maxent and compared with the model output generated with the remaining data. The area under the receiver operating characteristic curves (area under curve [AUC]) was used to compare model performance (Phillips et al. 2006)).
Results
Genetic diversity and population structure
The nrITS region was sequenced in Tamarix chinensis (105 individuals, 20 populations). nrITS sequences were deposited in the GenBank database under accession numbers: MH475502 – MH475606. The region was aligned with a consensus length of 616bp, which contained 10 nucleotide substitutions. Based on these polymorphisms, 11 haplo-262 types (H1 – 11) were identified among all the samples 263 surveyed (Table 1). Among those detected, the most widespread were H1 (in 17 of 20 populations) and H5 (in 14 of 20 populations). The haplotype and nucleotide diversities of T. chinensis were HT = 0.814 and πT = 2.17×10-3. H9 and H11 were unique haplotypes, occurring in P7 (Jinzhong, Shanxi) and P6 (Linfen, Shanxi), respectively. The highest nucleotide and haplotype diversities were found in Qianggang (Jiangsu) (Table 1).
Two cpDNA segments from each of 134 Tamarix chinensis individuals (20 populations) were sequenced. The sequences were deposited in the GenBank database under accession numbers: trnL-trnF: MH634851 – MH634985; rps16: MH634716 – MH634850. The length of aligned sequences of trnF- trnL and rps16 were 1542 bp, which contained 14 nucleotide substitutions, detecting a total of 16 chloroplast haplotypes (H1 – H16). Among those detected, the most widespread were H1 (in all populations). The haplotype and nucleotide diversities of T. chinensis were HT = 0.586 and πT =0.50×10-3. Ten unique chlorotypes were found in the species, with the highest diversity in P5 (Dingbian, Shaanxi) and P6 (Linfen, Shaanxi), respectively. The highest nucleotide and 285 haplotype diversities also were found in these two 286 populations (Table 1).
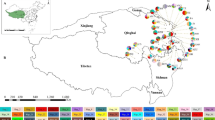
The haplotype network constructed based on the nrITS and cpDNA sequences exhibited a star-like topology, with haplotypes radiating from the central haplotype H1. This indicates that H1 is the ancestral haplotype, and the radiating haplotypes were derived through differentiation, suggesting that population expansion may have occurred previously. When the nrITS and cpDNA haplotypes were projected onto a map of geographic locations of the populations, we found that H1 originated from the northwest region (Figs 1 and 2).

A Statistical parsimony network of haplotypes H1 – H11 of nrITS (R = ribotype); B geographic distribution and genealogical relationships of nrITS haplotypes recovered from Tamarix chinensis populations in China.

A Statistical parsimony network of haplotypes H1 – H16 of cpDNA (C = chlorotype); B geographic distribution and genealogical relationships of cpDNA, haplotypes recovered from Tamarix chinensis populations in China.
Genetic differentiation among populations
Cluster analysis was performed on all populations using STRUCTURE, which resulted in no meaningful K values in either nrITS nor cpDNA (not shown). Based on the characteristics of haplotypes and geographic regions, the 20 populations can be divided into two groups artificially: the northwest region group (including BY, LZ, JB, DG, and DB) and the east region group (the remaining 15 populations). The results of genetic variation analysis based on the nrITS and cpDNA haplotype sequences of Tamarix chinensis revealed that the total genetic diversity of all populations (nrITS HT = 0.803, cpDNA HT = 0.635) was higher than the mean intrapopulation genetic diversity (nrITS HT = 0.643, cpDNA HT = 0.602) (Table 2). When a permutation test (repeated 1000 times) was performed on the geographic distribution patterns of haplotype variations of T. chinensis, it was found that the NST values for both nrITS and cpDNA were not significantly higher than the GST values (Table 2), which indicates the absence of significant molecular phylogeographic structures in T. chinensis populations.
Analysis of molecular variance (AMOVA) performed on the nrITS sequence revealed that the genetic variation of Tamarix chinensis mainly occurred within populations (80.89%), with only 11.15% of genetic variation occurring between populations (Table 3). The results of AMOVA performed on the cpDNA sequence showed that genetic variation was almost non-existent between populations (<1%), i.e. it occurred almost entirely within populations (98.26%) (Table 3). These results are consistent with the conclusion that significant molecular phylogeographic structures are absent in T. chinensis populations.
Using the uniparentally (maternally) inherited cpDNA sequence and biparentally inherited nrITS sequence, the Mantel test was performed to assess isolation by distance (IBD) in populations to determine whether genetic distance between populations was significantly correlated with geographic distance between populations. The results for nrITS indicated that a significant positive correlation existed between the genetic differentiation coefficient and geographic distance (r = 0.399, P < 0.001), i.e. an increase in the geographic distance between populations led to a significant increase in genetic distance. However, a significant correlation was not found between the genetic differentiation coefficient and geographic distance for the cpDNA sequence (r = 0.103, P = 0.17).
Analysis of population demographic history
Neutrality tests were performed on the cpDNA sequence of Tamarix chinensis. Results revealed negative and significant Tajima's D and Fu's Fs values, including the Tajima's D and Fu's Fs values of the east region group (Table 4). By contrast, the Tajima’s D value for the nrITS sequence was negative but not significant. These results, combined with the curves obtained through mismatch distribution analysis (MDA), indicate that population expansion events may have occurred recently (Fig. 3). The star-like radiation topology in the haplotype network described above also suggest the occurrence of recent rapid expansion in T. chinensis populations.

Mismatch distribution analysis (MDA) plots for nrITS (A, B, C) and cpDNA (a, b, c). x axes = Pairwise sequence differences.
Distribution inference by ecological niche modelling
The inferred current and past (LGM) distributions of Tamarix chinensis are illustrated in Fig. 4. The AUC values based on both training and test presence data for the present and LGM were higher than expected (0.995 and 0.993, 0.996 and 0.995, respectively), demonstrating good model performance. Because the distribution ranges of T. chinensis were largely affected by Mean Temperature of Coldest Quarter and Precipitation of Warmest Quarter (Table 5), it showed that an extremely cold and dry climate was not suitable for this species. If the climatic niche of T. chinensis was conserved throughout its evolutionary history, then the shift in the distribution range of halophytes or xerophytes might reflect how the dry and saline ecosystem responded to climate change. The simulations revealed that during the cooling of the LGM, the distribution range of T. chinensis fragmented and retreated in the middle region (Shanxi Province) (Fig. 4B). During the warming of the LIG, T. chinensis showed significant habitat expansion (Fig. 4C). In a warmer scenario in the near future, we predict that the habitat of T. chinensis will expand into higher latitude and higher altitude regions, such as Liaoning province, Neimeng province and north of Shanxi (Fig. 4D). The distribution patterns during the warmer climate in both the past and potentially the future, strongly indicate that these regions may be at great risk of aridification and salinisation under global warming.

Predicted distributions of Tamarix chinensis based on ecological niche modelling using Maxent. Predicted distributions are shown for A the present time; B the LGM period (21,000 years ago); C the LIG period (120,000 years ago); D the future.
Discussion
In this study, we detected a moderate level of genetic variation for Tamarix chinensis. The total nucleotide diversity (HT) of the cpDNA sequence was 0.635, which was lower than that of 13 seed plants used as maternally inherited markers in China (Qiu et al. 2011). However, the total nucleotide diversity (HT) of the nrITS was 0.803, which was moderate compared with other seed plants in China, such as Primula obconica Hance (HT=0.994), Achyranthes bidentata Blume (HT=0.791) (Miao et al. 2017). Tamarix chinensis is a widespread species with hermaphrodite or rarely dioecious flowers. It has an out-crossing breeding system, large geographical distribution, and relatively long evolutionary history causing a moderate level of genetic diversity.
Both cpDNA and ITS data showed no significant population differentiation within Tamarix chinensis. AMOVA results revealed that almost all genetic variation exists within the populations, which is consistent with the results obtained by Zhao (2006), Li (2007), Zhang (2011) and Liang et al. (2018a). The genetic structures of populations are influenced by multiple factors such as plant breeding system, population sizes and isolation period (Hamrick et al. 1992). Two reasons may cause the weak population and group differentiation of T. chinensis. Firstly, T. chinensis flowers two to three times a year and produces a large number of seeds. The mature seeds of T. chinensis are small with long unicellular hairs on the beak only (Gaskin 2003) and are usually dispersed by wind or water. High seed-mediated gene flow frequently happens between the populations, confirmed by the non-significant isolation by geographical distance based on cpDNA. Secondly, incomplete lineage sorting occurs as a result of the low evolutionary rates of cpDNA. Given these low cpDNA evolutionary rates, most of the derived chlorotypes diverge at approximately 1.05 Ma (Miao et al. 2017). The recolonisation of T. chinensis resulted from recent population expansion and insufficient time has elapsed for new chlorotypes to become fixed in the populations. Consequently, most populations of T. chinensis share common ancestral chlorotypes.
Furthermore, we found relatively stronger population differentiation based on nrITS rather than that of cpDNA. In most angiosperms, cpDNA is uniparentally (maternally) inherited (seeds), while nrITS is biparentally inherited (pollen and seeds). Seed flow is often greater than pollen flow. As mature T. chinensis seed is usually dispersed by wind or water, it leads to more efficient seed dispersal than pollen dispersal. These trends cause weak population differentiation of the cpDNA. Zhang (2011) reported that the pollen-seed mobility (r) of T. chinensis was 7.16. r was equal to 17 that was generally considered to be a medium mobility according to the Ennos model (Petit et al. 2003), so 7.16 was less than half the medium migration rate, indicating that the historically accumulated seed flow had greater effect. The higher mutation rate of nrITS is another factor that caused the relatively deep separation of T. chinensis populations. Lastly, nrITS is sensitive to environmental stress, so genetic variation is relatively lower in similar habitats.
In this study, the haplotype trees of both nrITS and cpDNA haplotypes exhibited a H1-centred radiation differentiation pattern. According to the theory proposed by Arbogast (2002), the ancestral haplotype was located at the centre of the haplotype tree, which indicated that H1 was an ancient haplotype. In addition, we found that the intrapopulation genetic diversity of cpDNA in the northwest region group was higher than that of the east region group. Therefore, populations in the northwest region were considered ancestral populations, while the current historical pattern of the east region was from expansion events. Tamarix chinensis populations underwent long-distance dispersal and migration from west to east China, which resulted in retention of the H1 haplotype in the east region. The neutrality tests and mismatch analysis performed on cpDNA also confirmed that significant expansion occurred in populations in the northwest region. The distribution pattern of T. chinensis populations is deduced as follows: seeds dispersed from the northwest in flowing water, leading to strong gene flow between populations. This is in accordance with the result of Liang et al. (2018a), in which the expansion of T. chinensis was from the upper to lower reaches of the Yellow River during the Quaternary period. Further, the seeds of T. chinensis can germinate under moist conditions (Jiang et al. 2012; Gaskin 2003), and their salt and alkaline tolerance enabled the establishment of populations in the Yellow River or its tributaries, flooded land and coastal tidal flat.
Another topic to discuss is the refugia hypothesis for Tamarix chinensis. Previous studies of the warm-temperate zone in China produced two hypotheses: the microrefugia hypothesis and the macrorefugia hypothesis. These two adaptive strategies in response to climate change may result from different levels of climate sensitivity and migration ability in different species of plants. Platycarya strobilacea Siebold & Zucc. (Chen et al. 2012), Forsythia suspensa Vahl (Fu et al. 2014), Cotinus coggygria Scop. (Wang et al. 2014) agree with the “microrefugia hypothesis”, which suggests that species persisted in situ during the glacial period with an enlarged distribution area, contrary to the hypothesis of long distance southward migration or large-scale range contraction. On the other hand, Cercidiphyllum japonicum Siebold & Zucc. (Qiu et al. 2011), and Bupleurum longiradiatum Turcz. (Zhao et al. 2013) agree with the “macrorefugia hypothesis”, which suggests that Quaternary refugia isolation promoted allopatric speciation. No significant population differentiation and the significant sudden population expansion within T. chinensis show discrepancy from both hypotheses. Liang et al. (2018a) considered that this species experienced the greatest expansion during the LIG and a retraction during the LGM. In this work, the nrITS haplotype analysis results revealed that H2, H3, and H5 were derived from H1, they formed the dominant haplotypes of the east region and were hardly distributed in the northwest region. In particular, the Fen River Basin possessed the highest haplotype diversity among various areas of the east region. In cpDNA, some populations possessed single haplotypes, including LZ (H5), JB (H6), DB (H7, H8), LF (H6, H11), ZZ (H13), WZ (H14), SY (H15), and CY (H16). Fen River Basin possessed the most unique haplotypes. The ENM results indicated that fragmentation occurred in the habitats of T. chinensis during the warm and wet climate of the interglacials, while clustering of the distribution regions of T. chinensis populations occurred during the cold and dry climate of the glacials, with populations mainly distributed in Shaanxi, Shanxi, and Hebei. During the onset of the LGM, although the distribution areas of T. chinensis were not covered by ice sheets, populations could only survive in refugia due to the glacial-induced reduction in land surface temperatures. The populations of the glacial refugia are distinctly characterised by high levels of genetic diversity, the highest haplotype richness, and widest distribution of ancient haplotypes. Therefore, we conclude that glacial refugia exist in the midstream of the Yellow River. Interestingly, most of the refugia were located in warm-temperate zones, small cryptic refugia exist in the northwest, such as LZ. Moreover, the MDA results of cpDNA showed that all populations in the east region had undergone expansion, and the ENM results indicated that post-glacial expansion occurred in the entire population. Therefore, it can be deduced that T. chinensis retreated on a large scale into refugia during the glacial period and the sudden population expansion from refugia during the interglacial period and recolonisation to suitable habitats agreed with the “EC hypothesis”. This finding is in agreement with Miao et al. (2017), who found that Achyranthes bidentata tracked climatic oscillations by a large range of southward retreat into three main refugia during the glacial period, followed by sudden northward expansion from these refugia in the post-glacial period.
References
Aizawa, M., Kim, Z. S. & Yoshimaru, H. (2012). Phylogeography of the Korean pine (Pinus koraiensis) in northeast Asia: inferences from organelle gene sequences. J. Pl. Res. 125 (6): 713 – 723.
Alvarado-Serrano, D. F. & Knowles, L. L. (2014). Ecological niche models in phylogeographic studies: applications, advances and precautions. Molec. Ecol. Resources 14 (2): 233 – 248.
Arbogast, B. S. (2002). Estimating divergence times from molecular data on phylogenetic and population genetic timescales. Ann. Rev. Ecol. Syst. 33: 707 – 740.
Chen, S. C., Zhang, L., Zeng, J., Shi, F., Yang, H., Mao, Y. R. & Fu, C. X. (2012). Geographic variation of chloroplast DNA in Platycarya strobilacea (Juglandaceae). J. Syst. Evol. 50 (4): 374 – 385.
Collins, W. D., Bitz, C. M., Blackmon, M. L., Bonan, G. B., Bretherton, C. S., Carton, J. A., Chang, P., Doney, S. C., Hack, J. J., Henderson, T. B., Kiehl, J. T., Large, W. G., Mckenna, D. S., Santer, B. D. & Smith, R. D. (2006). The community climate system model version 3 (CCSM3). J. Climate 19 (11): 2122 – 2143.
Comes, H. P. & Kadereit, J. W. (1998). The effect of quaternary climatic changes on plant distribution and evolution. Trends Pl. Sci. 3 (11): 432 – 438.
Clement, M. D., Posada, D. & Crandall, K. A. (2000). TCS: a computer program to estimate gene genealogies. Molec. Ecol. 9 (10): 1657 – 1659.
Evanno, G. S., Regnaut, S. J. & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molec. Ecol. 14 (8): 2611 – 2620.
Excoffier, L. & Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molec. Ecol. Resources 10 (3): 564 – 567.
Fu, Y. X. (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147 (2): 915 – 925.
Fu, Z. Z., Li, Y. H., Zhang, K. M. & Li, Y. (2014). Molecular data and ecological niche modeling reveal population dynamics of widespread shrub Forsythia suspensa (Oleaceae) in China's warm-temperate zone in response to climate change during the Pleistocene. BMC Evol. Biol. 14: 114.
Gao, X. M., Ma, K. P. & Chen, L. Z. (2001). Species diversity of some deciduous broad-leaved forests in the warm-temperate zone and its relations to community stability. Acta Phytoecol. Geobot. Sin. 25: 553 – 559.
García-Vázquez, A., Pinto Llona, A. C. & Grandal-d'Anglade, A. (2017). Post-glacial colonization of Western Europe brown bears from a cryptic Atlantic refugium out of the Iberian Peninsula. Hist Biol. 31 (5): 618 – 630.
Gaskin, J. F. (2003). Tamaricaceae. In: K. Kubitzki & C. Bayer (eds), The Families and Genera of Vascular Plants, V: 363 – 368. Springer-Verlag, Berlin.
Gaskin, J. F. & Schaal, B. A. (2002). Hybrid Tamarix widespread in U.S. invasion and undetected in native Asian range. Proc. Natl. Acad. Sci. U.S.A. 99 (17): 11256 – 11259.
Hamrick, J. L., Godt, M. J. W. & Sherman-Broyles, S. L. (1992). Factors influencing levels of genetic diversity in woody plant species. In: W. T. Adams, S. H. Strauss, D. L. Copes & A. R. Griffin (eds), Population Genetics of Forest Trees. Forestry Sciences 42: 95 – 124. Springer, Dordrecht. https://doi.org/10.1007/978-94-011-2815-5_7
Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature 405 (6789): 907 – 913.
Hewitt, G. (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos. Trans., Ser. B. 359 (1442): 183 – 195.
Horsak, M., Limondin-Lozouet, N., Jurickova, L., Granai, S., Horackova, J., Legentil, C. & Lozek, V. (2019). Holocene succession patterns of land snails across temperate Europe: East to west variation related to glacial refugia, climate and human impact. Palaeogeogr. Palaeoclimatol. Palaeoecol. 524: 13 – 24.
Hu, L. J., Uchiyama, K., Shen, H. L., Saito, Y., Tsuda, Y. & Ide, Y. (2008). Nuclear DNA microsatellites reveal genetic variation but a lack of phylogeographical structure in an endangered species, Fraxinus mandshurica, across North-east China. Ann. Bot. (Oxford) 102 (2): 195 – 205.
Jiang, Z., Chen, Y. & Bao, Y. (2012). Population genetic structure of Tamarix chinensis in the yellow river delta, China. Pl. Syst Evol. 29 8 (1): 147 – 153.
Li, L., Abbott, R., Liu, B., Sun, Y., Li, L., Zou, J.,Wang, X., Miehe, G. & Liu, J. (2013). Pliocene intraspecific divergence and Plio-Pleistocene range expansions within Picea likiangensis (Lijiang spruce), a dominant forest tree of the Qinghai-Tibet Plateau. Molec. Ecol. 22 (20): 5237 – 5255.
Li, R. (2007). Study on develop of EST-SSR primer and genetic structure of Tamarix chinensis Lour. Nanjing Forestry University.
Liang, H. Y., Feng, Z. P., Pei, B., Li, Y. & Yang, X. T. (2018a). Demographic expansion of two Tamarix species along the Yellow River caused by geological events and climate change in the Pleistocene. Sci. Rep. 8 (1): 60.
Liang, H. Y., Liu, C., Li, Y., Wang, Y., Kong, Y., Quan, J. & Yang, X. (2018b). Low population genetic differentiation in two Tamarix species (Tamarix austromongolica and Tamarix chinensis) along the Yellow River. Genetica 147 (1): 13 – 22.
Librado, P. & Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 (11): 1451 – 1452.
Miao, C. Y., Yang, J., Mao, R. L. & Li, Y. (2017). Phylogeography of Achyranthes bidentata (Amaranthaceae) in China's warm-temperate zone inferred from chloroplast and nuclear DNA: insights into population dynamics in response to climate change during the Pleistocene. Pl. Molec. Biol. Rep. 35 (1): 166 – 176.
Muellner-Riehl, A. N. (2019). Mountains as evolutionary arenas: patterns, emerging approaches, paradigm shifts, and their implications for plant phylogeographic research in the Tibeto-Himalayan region. Frontiers Pl. Sci. 10: 195.
Ni, J., Harrison, S. P., Prentice, I. C., Kutzbach, J. E. & Sitch, S. (2006). Impact of climate variability on present and Holocene vegetation: A model-based study. Ecol. Modelling 191 (3 – 4): 469 – 486.
Park, B. & Donoghue, M. J. (2019). Phylogeography of a widespread eastern North American shrub, Viburnum lantanoides. Amer. J. Bot. 106 (3): 389 – 401.
Petit, R. J., Aguinagalde, I., Beaulieu, J. L., Bittkau, C., Brewer, S., Cheddadi, R., Ennos, R., Fineschi, S., Grivet, D., Lascoux, M., Mohanty, A., Müller-Starck, G., Demesure-Musch, B., Palmé, A., Martín, J. P., Rendell, S. & Vendramin, G. G. (2003). Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300 (5625): 1563 – 1565.
Phillips, S. J., Anderson, R. P. & Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecol Modelling 190 (3 – 4): 231 – 259.
Pons, O. & Petit, R. J. (1996). Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144 (3): 1237 – 1245.
Qiu, Y. X., Fu, C. X. & Comes, H. P. (2011). Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world's most diverse temperate flora. Molec. Phylogenet. Evol. 59 (1): 225 – 244.
Rogers, A. R. & Harpending, H. (1992). Population-growth makes waves in the distribution of pairwise genetic differences. Molec. Biol. Evol. 9 (3): 552 – 569.
Schoenswetter, P. & Schneeweiss, G. M. (2019). Is the incidence of survival in interior Pleistocene refugia (nunataks) underestimated? Phylogeography of the high mountain plant Androsace alpina (Primulaceae) in the European Alps revisited. Ecol. Evol. 9 (7): 4078 – 4086.
Shangguan, T. L., Li, J. P. & Guo, D. G. (2009). Advance in mountain vegetation ecology in the warm-temperate zone of China. J. Mountain Sci. 27: 129 – 139.
Song, W., Cao, L. J., Li, B. Y., Gong, Y. J., Hoffmann, A. A. & Wei, S. J. (2018). Multiple refugia from penultimate glaciations in East Asia demonstrated by phylogeography and ecological modelling of an insect pest. BMC Evol. Biol. 18: 152.
Sun, Y., Li, L., Li, L., Zou, J. & Liu, J. (2015). Distributional dynamics and interspecific gene flow in Picea likiangensis and P. wilsonii triggered by climate change on the Qinghai-Tibet Plateau. J. Biogeogr. 42 (3): 475 – 484.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 (3): 585 – 595.
Thompson, J. D., Gibson, T. J., Plewniak, F. & Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25 (24): 4876 – 4882.
Tian, B., Liu, R., Wang, L., Qiu, Q., Chen, K. & Liu, J. (2009). Phylogeographic analyses suggest that a deciduous species (Ostryopsis davidiana Decne., Betulaceae) survived in northern China during the Last Glacial Maximum. J. Biogeogr. 36 (11): 2148 – 2155.
Tiffney, B. H. & Manchester, S. R. (2001). The Use of geological and paleontological evidence in evaluating plant phylogeographic hypotheses in the Northern Hemisphere Tertiary. Int. J. Pl. Sci. 162 (S6): S3 – S17.
Wan, D. S., Feng, J. J., Jiang, D. C., Mao, K. S., Duan, Y. W., Miehe, G. & Opgenoorth, L. (2016). The Quaternary evolutionary history, potential distribution dynamics, and conservation implications for a Qinghai-Tibet Plateau endemic herbaceous perennial, Anisodus tanguticus (Solanaceae). Ecol. Evol. 6 (7): 1977 – 1995.
Wang, W., Tian, C. Y., Li, Y. H. & Li, Y. (2014). Molecular data and ecological niche modelling reveal the phylogeographic pattern of Cotinus coggygria (Anacardiaceae) in China's warm-temperate zone. Pl. Biol. 16 (6): 1114 – 1120.
Xia, M., Tian, Z., Zhang, F., Khan, G., Gao, Q., Xing, R., Zhang, Y., Yu, J. & Chen, S. (2018). Deep intraspecific divergence in the endemic herb Lancea tibetica (Mazaceae) distributed over the Qinghai-Tibetan plateau. Frontiers Genet. 9: 492.
Yu, G., Chen, X., Ni, J., Cheddadi, R., Guiot, J., Han, H., Harrison, S. P., Huang, C., Ke, M., Kong, Z., Li, W., Liew, P., Liu, J., Liu, K. B., Prentice, I. C., Qui, W., Ren, G., Song, C., Sugita, S., Sun, X., Tang, L., Campo, E. V., Xia, Y., Xu, Q., Yan, S., Yang, X., Zhao, J. & Zheng, Z. (2000). Palaeovegetation of China: a pollen data-based synthesis for the mid-Holocene and last glacial maximum. J. Biogeogr. 27 (3): 635 – 664.
Zeng, Y. F., Liao, W. J., Remy, J. P. & Zhang, D. Y. (2011). Geographic variation in the structure of oak hybrid zones provides insights into the dynamics of speciation. Molec. Ecol. 20 (23): 4995 – 5011.
Zhang, R. H. (2011). Study on the genetic variation of Tamarix chinensis Lour. Nanjing Forestry University.
Zhao, C., Wang, C. B., Ma, X. G., Liang, Q. L. & He, X. J. (2013). Phylogeographic analysis of a temperate-deciduous forest restricted plant (Bupleurum longiradiatum Turcz.) reveals two refuge areas in China with subsequent refugial isolation promoting speciation. Molec. Phylogenet. Evol. 68 (3): 628 – 643.
Zhao, J. K. (2006). Study on genetic structure of Tamarix chinensis Lour. in natural populations in Yellow River Delta. Nanjing Forestry University.
Funding
This study was financially supported by the Science and Technology Innovation Funds of Gansu Agricultural University-Scientific research start-up funds for openly-recuited doctors (2017RCZX-19).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 37 kb)
Rights and permissions
About this article

Cite this article
Sun, L., Liu, G., Lu, Y. et al. Molecular data and ecological niche modelling reveal the phylogeographic pattern of the widespread shrub Tamarix chinensis Lour. (Tamaricaceae) in China. Kew Bull 75, 41 (2020). https://doi.org/10.1007/s12225-020-09899-z
Accepted:
Published:
DOI: https://doi.org/10.1007/s12225-020-09899-z