
Abstract
Orostachys (Crassulaceae) is a small genus of succulent plants having a predominantly East Asian distribution. Recent DNA sequence comparisons revealed polyphyletic nature of the genus and found distant relationship between its infrageneric taxa. Here we present the first molecular phylogeny of Orostachys subsection Appendiculatae based on a large number of ITS rDNA sequences representing most currently recognized members of the subsection and utilizing secondary structure information. Ribosomal spacer was a highly informative marker and provided a phylogenetic signal sufficient to resolve relationships at different scales, from affinities between species to a fine geographic structure in broadly sampled species. It was also conservative enough to allow unambiguous alignment and construction of consensus secondary structure models for ITS1 and ITS2. These models displayed a number of molecular synapomorphies defining most lineages established in our analyses. We revealed a major split in the subsection placing three species, O. spinosa, O. japonica and O. chanetii, into a strongly supported clade to the exclusion of O. thyrsiflora. Phenotypically distinct monotypic genus Meterostachys was also resolved as a part of the subsection’s clade and showed affinity to O. thyrsiflora. Our data suggested that morphology-based species concept for O. thyrsiflora requires reassessment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Orostachys Fisch. (Crassulaceae DC.) comprises ca. 20–25 species having a predominantly East Asian distribution and likely origin (Ohba 1978, 1990, 2005; Byalt 1999, 2000). Rosette vegetative morphology and a terminal dense thyrsoid-racemose inflorescence characterize the genus members. Although a rosette formed by basal cauline leaves is a frequent growth habit in distantly related Crassulacean lineages, the spadix-like inflorescence unambiguously differentiates Orostachys members therefore the genus is thought to be one of the best circumscribed in the subfamily Sedoideae (Ohba 1978). Its members grow mostly on rock crevices and dry slopes, but can also be found in meadows and even open woods. Some species contain valuable bioactive compounds and are used as traditional oriental medicinal herbs (Krasnov et al. 1979; Vegetative resources of the USSR 1990; Sung et al. 2002; Yoon et al. 2005; Jung et al. 2007; Ryu et al. 2010; Lee et al. 2011, 2014).
The genus comprises two sections, Orostachys and Schoenlandia Ohba, differing in inflorescence and flower morphology. The nominative section accommodates most species and is split into two subsections, Orostachys and Appendiculatae (Boriss.) Ohba, which are distinguished by blunt or cuspidate leaves, respectively. Molecular data has revealed only a distant relationship between all three Orostachys infrageneric taxa, placing Schoenlandia as a sister to the genus Sinocrassula Berger (Thiede and Eggli 2007), Orostachys subsect. Appendiculatae as a sister to Meterostachys Nakai and embedding subsect. Orostachys clade among Hylotelephium Ohba species (Mayuzumi and Ohba 2004; Gontcharova et al. 2006, 2008). Thus, sequence comparisons supported the view of some students treating Schoenlandia as a genus Kungia Ohba based on haplostemonous flowers and thyrsoid inflorescence (Fu and Ohba 2001; Ohba 2005; Thiede and Eggli 2007). The non-monophyly of section Orostachys and the affinities of the subsections were somewhat unexpected. Traditionally, difference in the leaf tip morphology between the subsections was seen to be a less important feature than that in growth habit between Hylotelephium and subsect. Orostachys and in inflorescence and floral features between Meterostachys and the subsect. Appendiculatae (Mayuzumi and Ohba 2004). All the lineages mentioned above were strongly supported in all analyses of the clade Hylotelephium (Mayuzumi and Ohba 2004; Gontcharova et al. 2006, 2008; Gontcharova and Gontcharov 2009), showing remarkable diversity of vegetative and particularly reproductive parts for a relatively small group (ca. 50–60 spp.).
The most recent taxonomic account recognized six species (O. cartilaginea Boriss., O. chanetii (H.Lév.) A.Berger, O. fimbriata (Turcz.) Berger, O. japonica Maxim., O. spinosa (L.) Meyer, and O. thyrsiflora Fisch.) in subsect. Appendiculatae differing in details of the leaf apex, plant size, flower color and features of the flower parts (Ohba 2005). In this treatment, a broad species concept was followed and a large number of taxa were reduced to synonyms. Based on the same characteristics and their combinations, Byalt (1999) accepted eleven species and infraspecific taxa. Whether these taxa represent distinct evolutionary entities or if the morphological diversity within the subsection is a product of a frequent local adaptation or phenotypic plasticity, remains unclear. For example, in many eastern populations, O. spinosa occurs strictly as individual monocarpic rosettes, whereas in more continental areas the same species shows only a polycarpic growth habit with numerous crowded vegetative rosettes (Bezdeleva 1995; Gontcharova 2006).
No molecular studies have been specifically focused on the phylogenetic structure of subsect. Appendiculatae to date. In large-scale analyses based on nuclear and chloroplast non-coding sequences the subsection was represented by the same three species but only internal transcribed spacer (ITS) sequence comparisons could resolve relationship between them. Orostachys spinosa and O. fimbriata formed a moderately supported clade with O. japonica as a sister (Mayuzumi and Ohba 2004; Gontcharova et al. 2006). The close relationship between Meterostachys and the subsection members was established with both nuclear and chloroplast sequences, but trnL-F failed to resolve the branching pattern between these taxa. There is evidence that a single accession per species may be not sufficient to uncover intraspecific polymorphism in Orostachys that could influence phylogeny (Kozyrenko et al. 2013). It was shown that even in subsect. Orostachys which is characterized by only minor ITS rDNA sequence divergence (Gontcharova et al. 2008), some populations harbor more than one ITS ribotype and the ribotypes grouping contradicts the morphology-based species delimitation.
This contribution presents the first molecular phylogeny of the Orostachys subsect. Appendiculatae. We use the sequence data of the internal transcribed spacer (ITS) of nuclear ribosomal DNA to test whether genetic data support the recognition of current morphospecies in the subsection or not and whether the morphologically distinct Meterostachys is a sister to subsect. Appendiculatae or it is a part of this lineage. ITS rDNA is a valuable marker for plant phylogenetic reconstruction at different taxonomic levels (Bailey et al. 2003; Feliner and Rossello 2007; Calonje et al. 2009; Poczai and Hyvönen 2010) and has been successfully applied in the family Crassulaceae (Jorgensen and Frydenberg 1999; Gehrig et al. 2001; Jorgensen and Olesen 2001; Mort et al. 2002, 2005; Fairfield et al. 2004; Carrillo-Reyes et al. 2009; Yost et al. 2013) and specifically in the Hylotelephium lineage (Mayuzumi and Ohba 2004; Gontcharova et al. 2006, 2008; Gontcharova and Gontcharov 2009; Kozyrenko et al. 2013).
Materials and methods
Taxon sampling
Four species of Orostachys subsect. Appendiculatae were sampled for this study [O. spinosa (29 populations, 64 specimens), O. japonica (16 populations, 22 specimens), O. thyrsiflora Fisch. (2 populations, 2 specimens) and O. chanetii (2 populations, 2 specimens)]. Of these 66 specimens were collected in 2009–2013 from natural habitats (1–4 specimens per population; Fig. 1) and 18 specimens were received from private and public collections (each regarded here as a population). In addition to that six Orostachys and one Meterostachys ITS sequences were retrieved from GenBank and analyzed (Table 1). Sinocrassula (2 species) and Hylotelephium pseudospectabile (Praeger) S.H.Fu were used as outgroups for phylogenetic analyses. No samples of endangered or protected species were used. Specimens were collected from public land therefore, field permits were not required.

Map of sample sites for natural populations of Orostachys spinosa (19 populations; white circle) and O. japonica (5 populations; black circle). Location details and population codes (P) correspond to those in Table 1
DNA extraction, polymerase chain reaction (PCR) and sequencing
Leaf tissue or inflorescence samples were delivered to the laboratory where they were stored in a −80 °C freezer until extraction. Total genomic DNA was extracted using DNeasy Plant Mini Kit (QIAGEN, Maryland, USA), following the manufacturer’s instruction.
To amplify complete ITS1–5.8S–ITS2 region we used universal primer pairs: 1400F (Elwood et al. 1985) and ITS055R (Marin et al. 2003) for the first round of amplification and internal primers 18Sm10 (5′-AGGAGAAGTCGTAACAAGG-3′; modified from Wen and Zimmer 1996) and ITS4R (White et al. 1990) for the second round of amplification (if necessary) and cycle sequencing. The PCR products were sequenced using a BigDye terminator v. 3.1 sequencing kit (Applied Biosystems, Maryland, USA). Sequences were analyzed in both directions using an ABI 3130 genetic analyzer (Applied Biosystems, USA). The PCR products from five specimens (see below) were cloned into the pTZ57R/T vector using InsTAclone PCR Cloning Kit™ (Fermentas, Lithuania) following the manufacturer’s instructions. Vector specific M13 primers were used to sequence the cloned fragments; ten clones per specimen were sequenced. Sequences were assembled with the Staden Package v. 1.4 (Bonfield et al. 1995) and aligned manually in the SeaView program (Galtier et al. 1996).
To determine the rDNA ribotypes a DnaSP5 v.5.10.1 package (Librado and Rozas 2009) was used. Positions with gaps were considered as a fifth character.
Data analysis
The boundaries of the loci and structural domains in ITS rDNA region were determined by comparison with published Crassulacean sequences and ITS secondary structure data (Gontcharova and Gontcharov 2004; Gontcharova et al. 2006). Secondary structures of ITS1 and ITS2 regions were predicted using M. Zuker web server (Zuker 2003; http://www.bioinfo.rpi.edu/~zukerm/rna/) by screening for thermodynamically optimal and suboptimal structures using the default values. Results for the various accessions were compared to reveal the folding pattern common to them all. Consensus secondary structure models of ITS1 and ITS2 were constructed in 4SALE program (Seibel et al. 2006).
Aligned sequence matrix (available at http://purl.org/phylo/treebase/phylows/study/TB2:S16515) was analyzed using Maximum Likelihood (ML) and Maximum Parsimony (MP) methods implemented in PAUP* 4.0b10 (Swofford 2002) by a heuristic search with tree-bisection-reconnection (TBR) branch swapping and 10 random sequence addition replicates, and Bayesian inference (BI) method in MrBayes 3.1.2 (Huelsenbeck and Ronoquist 2001). The GTR+I+G model was selected as the optimal setting for ML analyses by jModelTest 2.1.1 (Darriba et al. 2012). The support for branches was calculated by bootstrap analyses with 100 (ML) or 1,000 (MP) replications of heuristic search (Felsenstein 1985). Bayesian analysis was performed using two parallel Markov Chain Monte Carlo runs, each with 2,000,000 generations under default settings. Prior to consensus calculation, the initial 500,000 generations were discarded as “burn in”. Bootstrap percentage (BP) below 50 % and Posterior Probabilities (PP) less than 0.95 were not considered.
To estimate intraspecies and total pairwise distances (p-distances) between sequences MEGA v.5 (Tamura et al. 2011) was used.
Results
Eighty-four accessions were sequenced for this study. All but five O. japonica plants obtained from horticulture (P6–8, 13, 14; Table 1) produced readable ITS sequences with no polymorphism. Five PCR products yielding a partially unreadable electropherogram were cloned and 50 DNA clones carrying ITS region were sequenced. All the clones produced clear signal at the electropherogram and their sequencing revealed the presence of two allelic variants of ITS1 locus in equal proportions. These intragenomic ITS1 variants had either 8 or 7 nucleotides at the poly-C sector close to 5′ end of the spacer (Fig. 2).

Consensus secondary structure models of ITS1 and ITS2 of Orostachys subsect. Appendiculatae based on Mfold predictions for 84 sequences obtained for this study. Arrows indicate synapomorphic changes for respective species. In the box, variability in homopolymeric region of ITS1 revealed by direct sequencing and cloning from some O. japonica accessions and respective ribotypes are shown
The total length of the ITS region ranged from 607 (P49) to 614 bp (P47, see below). ITS1 and ITS2 were almost identical in length and GC content, 225.77 ± 1.48 bp, 62.60 ± 1.60 % and 225.03 ± 1.29 bp, 63.0 ± 1.60 %, respectively. The 5.8S rDNA sequence region had a conserved length of 161 bp and somewhat lower GC content than in spacers, 54.42 ± 0.75 %. In GenBank sequence AB480590 (P47, Table 1) the 5.8S gene was 162 bp long due to an extra G at position 20. Otherwise the entire ITS region in this accession was identical to that in P20.
Sequence conservation was relatively high across the data set. ITS1 had 152 (67.2 %), ITS2 had 168 (74.6 %) and 5.8S–154 (95.6 %) universally conservative positions. That allowed the unambiguous alignment of the entire ITS region with only a few gaps (624 positions; 147 parsimony informative ITS1-88, 5.8S-6, ITS2-53) and the generation of consensus secondary structure models of ITS1 and ITS2 in subsect. Appendiculatae (Fig. 2). Four hundred and one homologous characters present in all spacer sequences analyzed here were numbered separately for each spacer and these numbers were used for referring to specific positions in the text. These positions were classified into five categories: 100 % conserved nucleotides, highly conserved positions with only one unique change within subsect. Appendiculatae, moderately conserved positions with two changes, variable positions with ≥3 changes, and insertions characteristic for some taxa (Fig. 2).
Both spacers formed four-helix structures interconnected by unpaired nucleotides (spacers) (Fig. 2). In ITS2, the spacers between helices were relatively short, with only 18 % of nucleotides being not a part of the helical domains. In ITS1, stretches of unpaired nucleotides were longer and accounted for ca. 37 % of the spacer length. The core structure of the ITS1 comprised a relatively long (nt 1–42) single-stranded region at the 5′ end, 15 bp (nt 43–85) long stem 1, 1 nt spacer between stems 1 and 2, 12 bp (nt 87–122) stem 2, 15 nt (positions 123–137) and 16 nt (positions 152–168) spacers flanking stem 3 (5 bp, nt 138–151) and the longest stem 4 (18 bp, nt 169–216) followed by the 10 nt long terminal spacer (Fig. 2).
The ITS2 secondary structure model displayed several structural landmarks typical for angiosperms: relatively large hairpin loop (positions 20–29) of the stem 1 (positions 7–43), 11 bp long stem 2 (positions 46–77) harboring U–U and C-A bulge (positions 50–73 and 51–72, respectively), long (≥32 bp; 84–185 nt) stem 3 containing the conserved sequence GGUGGU at the 5′ end (positions 118–123; Fig. 2). Numerous Compensatory Base Changes (CBCs) and hemi-Compensatory Base Changes (hCBCs) found in the structured ITS alignment supported our folding patterns. Even the most conserved elements of ITS2, stems 2 and 3, contained 15 hCBCs (2 and 13, respectively) but there were only 2 CBCs in stem 3 (92G-C178 → A-U and 123U-A145 → G-U, O. thyrsiflora sequences; Fig. 2).
Even at the initial alignment steps it was evident that autapomorphic character states or indels differentiated some GenBank sequences from the respective conspecific accessions. Moreover, O. thyrsiflora sequences AB480590 and AB088577 differed significantly with new sequences obtained for the same species and showed more similarity to O. spinosa accessions. Further analyses revealed that GenBank sequences AB088578 and AB480587 contained positions and/or indels altering generally conserved secondary structure folding produced for a large number of accessions. To avoid the introduction of a false phylogenetic signal caused by possible sequencing errors, we omitted these sequences from further analyses and retained in the data set only those identical or generally agreeing with our ITS secondary structures models (e.g. AB480590 having extra G in 5.8S).
Altogether, 91 Orostachys sequences were analyzed further. In this data set the DnaSP5 v.5.10.1 program identified 26 unique ribotypes (Table 1). As expected, the most sampled species (64 accessions) O. spinosa was characterized by the highest number of ribotypes, 14. Of these, eight ribotypes were unique for a specific population (1–3 sequences) and six cases of ribotype sharing between populations were observed. The most frequent R13 was found in 17 individuals from seven populations distributed in Altai Mts. (P23–25), Southern Urals (P21, 22) and two samples from the horticulture (P44, 45). In O. japonica 23 sequences yielded nine ribotypes (Table 1). In this species, six out of 16 populations harbored a unique ribotype (1–3 sequences) each. Three ribotypes, R3, R4, and R5 were shared by 3 (P4, 11, 16), 6 (P5–8, 13–15), and 5 (P6–8, 13, 14), respectively, Japanese and Korean natural and horticultural populations (Table 1). Orostachys thyrsiflora and O.chanetii, each produced a unique ribotype (Table 1). No ribotype sharing between species was observed and no population harbored more than one ribotype except for populations P6–8, 13, and 14 characterized by intrangenomic polymorphism (R4 and R5).
Overall divergence (p-distances) between 91 Orostachys sequences was relatively low (0.0439 ± 0.0045) and unevenly distributed across the data set. The lowest divergence was found within O. japonica (0.0136 ± 0.0029) while O. spinosa sequences were twice as divergent (0.0269 ± 0.0045). In O. thyrsiflora and O. chanetii this difference was even more profound, 0.0174 ± 0.0051 and 0.0732 ± 0.0114, respectively.
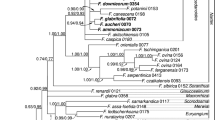
The maximum likelihood phylogenetic tree based on 26 ribotype sequences and three outgroups resolved four strongly supported species clades in subsect. Appendiculatae, O. spinosa (98 %BP, 1.00 PP), O. japonica (97–98 % BP, 1.0 PP), O.chanetii (91 % BP in ML, 1.0 PP), and O. thyrsiflora (100 % BP, 1.00 PP; Fig. 3). Meterostachys sikokiana showed affinity to O. thyrsiflora and their moderately supported clade (79 % BP in ML) was resolved as sister to a strongly supported (98 % BP, 1.0 PP) lineage comprising O. spinosa, O. japonica and O. chanetii. The branching pattern in this three-species clade remained unresolved. The ribotype-rich O. spinosa and O. japonica clades were further significantly structured. In O. spinosa two subclades were evident, a robust lineage comprising ribotypes found in the Amur River basin and the Sea of Japan coast (R16–20, 24; Eastern subclade) and a moderately supported (75 % BP in ML) assemblage of six ribotypes from the western part of the species distribution range (Western subclade). The latter also included two ribotypes from northeast Asia, Magadan (R22) and Shantar Islands (R21). It should be noted that two GenBank (AB480590 and AB088577) and most horticulture (P46–50) accessions, assigned to O. thyrsiflora, were found in this subclade of O. spinosa and showed no affinity to two O. thyrsiflora sequences obtained in this study (Fig. 3).

ML phylogenetic tree (GTR+I+Г model) based on 26 ITS rDNA ribotypes (624 aligned nucleotide positions, 147 are parsimony informative) for subsect. Appendiculatae species, Meterostachys and three outgroup taxa. Bootstrap values greater than 50 % (ML/MP) and posterior probabilities >0.95 are given above/below the branches. Branches with 100 % support and 1.00 PP are shown bold-face. Ribotype (R) and population (P) codes are the same as in Table 1. Synapomorphic substitutions (in boxes) are linked to the branches of their evolutionary origin
In the subclades, ribotypes were also arranged according to their origin. Terminal clades comprised plants from Primorsky Territory of Russia (R19, 20, 24; 86–88 % BP, 0.95 PP), Middle Amur River Basin (R16–18; 78–87 % BP, 0.99 PP), northeast Asia (R21, 22, 81–85 % BP, 1.0 PP), Altai and Ural Mts. (R13, 26; 100 % BP, 1.0 PP) and the Lake Baikal area (R12, 14, 15, 27; 99 % BP, 1.0 PP). Similarly, O. japonica ribotypes found in the Korean Peninsula and Japan (R3-5, 8, 9; 66–72 % BP) were distinct from those originating from Russia and northeast China (R 6, 7, 10, 11; 98 % BP, 1.0 PP).
ITS sequences provided a number of characteristics that could be regarded as molecular synapomorphies (Telford 2002; Marin et al. 2003) of all major clades established in our analyses (Figs. 2, 3). All members of the subsection including Meterostachys shared four substitutions that differentiated them from other representatives of the Hylotelephium lineage (Sinocrassula, Hylotelephium, and Orostachys subsect. Orostachys; 40 sequences). These are non-pairing U125 position in the spacer between stems 2 and 3, G144 in the terminal loop of the stem 3 of ITS1, G at the position 95 allowing 95G-C175 pair in the stem 3 and G208 in the terminal loop of the stem 4 of ITS2 (Figs. 2, 3). The major clades in the subsection, O. thyrsiflora/Meterostachys and O. spinosa/O.japonica/O. chanetii, were characterized by a single synapomorphic change each, A → G at position 221 of ITS1 and A → U at position 104 of ITS2, respectively. Two synapomorphies in ITS2 (hCBC C-G → 84U-G185 and A → G222 change) characterized O. spinosa and three synapomorphic substitutions differentiated representatives of its Eastern subclade (substitution G → A205 disrupting pair formation, hCBC G-U → 210A-U176 in ITS1 and hCBC C-G → 49U-G74 in ITS2; Figs. 2, 3). Orostachys japonica accessions shared two synapomorphic insertions in the distal part of the stem 4 of ITS 1, A between positions 195–196 potentially allowing formation an additional 190U-A195-1 pair and C between the positions 197 and198 forming a side bulge. In addition, two synapomorphic substitutions were found for this species, A → U at the position 212 of ITS1 also allowing additional 212U-G174 pair and U → C change in the fifth nucleotide of ITS2 (Fig. 2).
Discussion
Analyses of a large number of ITS rDNA sequences representing most currently recognized members of Orostachys subsect. Appendiculatae and utilizing secondary structure information, resolved the first comprehensive phylogeny of the subsection. We revealed a major split in the subsection placing three species, O. spinosa, O. japonica and O. chanetii, into a clade, strongly supported by a high significance threshold and synapomorphic substitutions in the secondary structure of the spacers, to the exclusion of O. thyrsiflora. The latter species showed affinity to the monotypic genus Meterostachys, which was also a part of the subsection’s clade (Fig. 3). Earlier ITS rDNA sequence comparisons revealed a sister relationship between Meterostachys and subsect. Appendiculatae, (Mayuzumi and Ohba 2004), but in our phylogeny Meterostachys was embedded among Orostachys species with its ITS sequence being less divergent from O. spinosa, O. japonica and O. chanetii than those of O. thyrsiflora (results not shown). Screening of the structured sequence alignment revealed that Meterostachys shares at least four molecular synapomorphies with members of the subsection (see “Results”). Furthermore a synapomorphic substitution A → G221 (ITS1; Fig. 2) unites Meterostachys and O. thyrsiflora.
Thus, in contrast to previous studies, which corroborated the generic status of Meterostachys, the results presented here indicated that it could be a part of subsect. Appendiculatae. Due to the rosette habit and similar color of the petals, Meterostachys was regarded as a member of the genus Orostachys by Ohwi (1953). Ohba (1978) argued that differences between the two genera in inflorescence morphology (bracteates cymose in Meterostachys and thyrsoid-paniculate to paniculate in Orostachys) and basally connate carpels in Orostachys warranted their independent status and his view was accepted in most recent taxonomic treatments of the family (Eggli et al. 1995; Ohba 2001, 2005). Indeed, there is no phenotypic feature, except for a rosette life form and cuspidate mostly cartilaginous leaf tip, uniting subsect. Appendiculatae and Meterostachys. Neither of these characteristics could be regarded as synapomorphic for the Meterostachys/subsect. Appendiculatae clade because they are typical for many unrelated Crassulacean lineages. It should be noted that Meterostachys and its closest relative in the subsection, O. thyrsiflora, not only have contrasting morphology but different geographical patterns also. The current distribution of Meterostachys is limited to Korean Peninsula and Japan (with a single record for Sichuan, China; Byalt 1997), whereas O. thyrsiflora occurs more westward, from Mongolia to the Southern Urals.
The polyphyly of the traditional genus Orostachys is beyond doubt (Mayuzumi and Ohba 2004; Gontcharova et al. 2006, 2008; Gontcharova and Gontcharov 2009) and requires relevant taxonomic adjustments. Subsection Appendiculatae could be raised to the genus level under a new name, or merged with Meterostachys having priority, or retained in the enlarged genus Orostachys embracing Meterostachys and Hylotelephium. Molecular data presented here favors unity of subsect. Appendiculatae and Meterostachys but this affinity requires further conformation with additional Meterostachys accessions, O. fimbriata samples, more molecular and preferably phenotypic markers.
ITS rDNA sequences
ITS rDNA was a highly informative marker for Orostachys subsect. Appendiculatae and provided a phylogenetic signal sufficient to resolve relationships at different scales, from affinities between species to a fine geographic structure in two broadly sampled species, O. spinosa and O. japonica (Fig. 3). It was also conservative enough to allow unambiguous alignment and construction of consensus secondary structure models for ITS1 and ITS2 provided with universal numbering system (Fig. 2), a useful tool for in-depth analyses of ITS rDNA evolution (Caisová et al. 2011). Moreover, these models displayed a number of molecular synapomorphies defining most lineages established in our analyses (Figs. 2, 3). These features could be important for the taxonomy of highly polymorphic groups of plants having no apparent phenotypic synapomorphies.
Our data revealed unequal ITS rDNA evolutionary rates in subsect. Appendiculatae. O. chanetii and O. spinosa were characterized by increased sequence divergence when compared with O. japonica and O. thyrsiflora. In O. thyrsiflora this difference could be attributed to a limited number of sequences analyzed, but not in O. japonica, represented by 23 accessions covering its entire distribution range. High ITS sequence diversity in subsect. Appendiculatae contrasts with results obtained recently for the type subsection of the genus, Orostachys, where only seven highly similar ribotypes were identified in a comparable data set (4 species, 86 accessions; Kozyrenko et al. 2013). There is a chance that our direct sequencing approach revealed only a fraction of the ITS diversity present in the group studied. Increasing evidence suggests that intra-genomic rDNA variants are frequent and abundant in most plant individuals (Denduangboripant and Cronk 2000; Bailey et al. 2003; Xiao et al. 2010; Peng et al. 2010; Song et al. 2012; Simon et al. 2012; Matyasek et al. 2012). Such polymorphism was found in a homopolymeric region of ITS1 in three O. japonica horticultural accessions (ribotype 4 and 5; Figs. 2, 3) but it had no implication for the resulting tree topology. Recent sequence-tagged pyrosequencing-based studies suggested that intra-genomic variability was markedly smaller than intra-specific or inter-specific divergence and likely has no or little effect on phylogeny (Chen et al. 2012; Song et al. 2012).
Our analyses using both GenBank and newly obtained sequences once more highlighted the problem of data quality in the GenBank. From nine initially retrieved ingroup sequences, two were excluded from the analyses because they contained likely sequencing errors revealed by secondary structure predictions and preliminary phylogenetic analyses (results not shown) and for two more sequences alternative taxonomic assignment was suggested (see Table 1).
Species concept
Results of our phylogenetic analyses have several implications for understanding relationships in subsect. Appendiculata. We confirmed the distinctness of O. spinosa, O. japonica, O. chanetii and O. thyrsiflora by resolving their species clades and established distant relationship between the former species and the rest of the subsection (Fig. 3). At the same time, our data suggested that morphology-based species concepts for some species are deficient. Specifically, this relates to differentiation of O. thyrsiflora from O. spinosa and O. cartilaginea from O. japonica.
It was found that two GenBank sequences and four horticulture accessions of O. thyrsiflora were resolved in the O. spinosa clade and only two plants, also from horticulture (P49, 50), showed a distant relationship to O. spinosa, forming a robust species clade (Table 1; Fig. 3). Even if we disregard horticultural material having often questionable identification, we cannot deny possible confusion in distinguishing the two species by the professionals. The distinction between O. spinosa and O. thyrsiflora is based on petal color (yellowish vs. white to pinkish, respectively) and anther color (yellow vs. purple-red) and keeled leaves (O. thyrsiflora). These features are mostly well-preserved in the herbaria specimens and hardly likely to be overlooked in horticulture. Flower color was unknown for the specimens sequenced earlier and our misidentified horticultural plants, whereas in both “true” O. thyrsiflora accessions pink flowers were confirmed. At the same time all the specimens under discussion had keeled leaves as it is typical for O. thyrsiflora but not O. spinosa. Based on these facts, we conclude that the leaf morphology alone cannot reliably distinguish O. thyrsiflora from O. spinosa. Interestingly, the range of O. thyrsiflora largely overlaps with that of O. spinosa (from Eastern Mongolia and Eastern Sayan Mts. to the South Urals and Northern Kazakhstan) and ecological niches of the species are also similar (Byalt 1999); however, they rarely occur sympatrically. Our generally arbitrary sampling in the area where both species occur resulted in five populations of O. spinosa; specimens from three more populations were introduced into horticulture from this area earlier and were also used in this study. In the same area our collectors came across only two populations of O. thyrsiflora, which suggests the relatively rare occurrence of this species at least at the eastern and northern parts of its distribution area.
Data obtained in this study corroborated morphological observations of Gontcharova (2006) suggesting that O. cartilaginea is indistinguishable from O. japonica. She did not observe cartilaginous appendages at the leaf tip, the major characteristic on which O. cartilaginea was based, in live plants from the locus classicus of this species [Russia, Primorsky Territory, Fadeevka sentiment, Razdolnaja (Suyfun) River] and several other localities in Russia (Borissova 1939; Bezdeleva 1995; Byalt 1999). It was suggested that this appendage could be an artifact of fleshy O. japonica specimen drying. Borissova (1939) and other authors accepting O. cartilaginea mentioned that this species is often confused with O. japonica and O. fimbriata, but argue that cartilaginous/non-cartilaginous/cartilaginous with fimbriate margin, respectively, leaf tip clearly differentiate these taxa. Flower color is not a decisive characteristic to discriminate the three species (white to pinkish/white/white to reddish, respectively; Borissova 1939; Bezdeleva 1995; Byalt 1999; Fu and Ohba 2001; Ohba 2001). The locus classicus of O. cartilaginea was sampled for this study (P10; Fig. 1) and sequence comparisons revealed that ribotype R6 residing here was a part of a lineage comprising O. japonica populations from Russia (P9, 10, 19) and NE China (P20; Fig. 3).
References
Bailey CD, Carr TG, Harris SA, Hughes CE (2003) Characterization of angiosperm nrDNA polymorphism, paralogy, and pseudogenes. Molec Phylogenet Evol 29(3):435–455
Bezdeleva TA (1995) Crassulaceae. In: Kharkevicz SS (ed) Plantae vasculares Orientis Extremi Sovietici, vol 7. Nauka, Sankt Petersburg, pp 214–235
Bonfield JK, Smith KF, Staden R (1995) A new DNA sequence assembly program. Nucl Acids Res 23:4992–4999
Borissova AG (1939) Crassulaceae. In: Komarov VL (ed) Flora of USSR. Academiae Scientiarum URSS, Leningrad 9:8–134, 471–486
Byalt VV (1997) Meterostachys sikokiana (Crassulaceae), a new species and genus in the flora of China. Bot Zhurn (Moscow & Leningrad) 82(7):128–130
Byalt VV (1999) Monograph of the genus Orostachys Fisch. (Crassulaceae). Dissertation, Botanical Institute, Russian Academy of Sciences
Byalt VV (2000) Conspectus generis Orostachys Fisch. (Crassulaceae). Novosti Sist Vyssh Rast 32:40–50
Caisová L, Marin B, Sausen N, Pröschold T, Melkonian M (2011) Polyphyly of Chaetophora and Stigeolonium within the Chaetophorales (Chlorophyceae), revealed by sequence comparisons of nuclear-endoced SSU rRNA genes. J Phycol 47:164–177
Calonje M, Martin-Bravo S, Dobes C et al (2009) Non-coding nuclear DNA markers in phylogenetic reconstruction. Pl Syst Evol 282(3–4):257–280
Carrillo-Reyes P, Sosa V, Mort ME (2009) Molecular phylogeny of the Acre clade (Crassulaceae): dealing with the lack of definitions for Echeveria and Sedum. Molec Phylogenet Evol 53(1):267–276
Chen X, Liao B, Song J, Pang X, Han J, Chen S (2012) A fast SNP identification and analysis of intraspecific variation in the medicinal Panax species based on DNA barcoding. Gene 530(1):39–43
Darriba D, Taboada G, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Meth 9:772
Denduangboripant J, Cronk QC (2000) High intraindividual variation in internal transcribed spacer sequences in Aeschynanthus (Gesneriaceae): implications for phylogenetics. Proc R Soc Lond B 267(1451):1407–1415
Eggli U, ‘t Hart H, Nyffeler R (1995) Towards a consensus classification of the Crassulaceae. In: Eggli U (ed) Evolution and systematics of the Crassulaceae. Backhuys, Leiden, pp 173–192
Elwood HJ, Olsen GJ, Sogin ML (1985) The small-subunit ribosomal RNA gene sequences from the hypotrichous ciliates Oxytricha nova and Stylonychia pustulata. Molec Biol Evol 2:399–410
Fairfield KN, Mort ME, Santos-Guerra A (2004) Phylogenetics and evolution of the Macaronesian members of the genus Aichryson (Crassulaceae) inferred from nuclear and chloroplast sequence data. Pl Syst Evol 248:71–83
Feliner GN, Rossello JA (2007) Better the devil you know? Guidelines for insightful utilization of nrDNA ITS in species-level evolutionary studies in plants. Molec Phylogenet Evol 44(2):911–919
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fu KJ, Ohba H (2001) Crassulaceae. In: Wu ZY, Raven PH (eds) Flora of China. Science Press, Beijing, Missouri Botanical Garden Press, St. Louis, 8:202–268
Galtier N, Gouy M, Gautier C (1996) Seaview and phylo-win: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci 12:543–548
Gehrig H, Gaubmann O, Marx H, Schwarzrott D, Kluge M (2001) Molecular phylogeny of the genus Kalanchoe (Crassulaceae) inferred from nucleotide sequences of the ITS–1 and ITS–2 regions. Pl Sci 160:827–835
Gontcharova SB (2006) Sedoideae, Crassulaceae of the Russian Far East flora. Dalnauka, Vladivostok
Gontcharova SB, Gontcharov AA (2004) Sequence and secondary structure evolution of ITS rDNA in the family Crassulaceae. Chromosome Sci 8:142–144
Gontcharova SB, Gontcharov AA (2009) Molecular phylogeny and systematics of flowering plants of the family Crassulaceae DC. Molec Biol 43:794–803
Gontcharova SB, Artyukova EV, Gontcharov AA (2006) Phylogenetic relationships among members of the subfamily Sedoideae (Crassulaceae) inferred from the ITS region sequences of nuclear rDNA. Russ J Genet 42:654–661
Gontcharova SB, Gontcharov AA, Stephenson R (2008) Analysis of phylogenetic relationships in the family Crassulaceae based on nucleotide sequences of ITS region of nuclear rDNA. Bot Zhurn (Moscow & Leningrad) 93:96–113
Huelsenbeck JP, Ronoquist F (2001) MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Jorgensen TH, Frydenberg J (1999) Diversification in insular plants: inferring the phylogenetic relationship in Aeonium (Crassulaceae) using ITS sequences of nuclear ribosomal DNA. Nordic J Bot 19(5):613–621
Jorgensen TH, Olesen JM (2001) Adaptive radiation of island plants: evidence from Aeonium (Crassulaceae) of the Canary Islands. Bot J Linn Soc 132:223–240
Jung HJ, Choi J, Nam JH, Park HJ (2007) Anti-ulcerogenic effects of the flavonoid-rich fraction from the extract of Orostachys japonicus in mice. J Med Food 10(4):702–706
Kozyrenko MM, Gontcharova SB, Gontcharov AA (2013) Phylogenetic relationships among Orostachys subsection Orostachys species (Crassulaceae) based on nuclear and chloroplast DNA data. J Syst Evol 51(5):578–589
Krasnov EA, Saratikov AC, Surov YP (1979) Plants of the family Crassulaceae. Tomsk University, Tomsk
Lee JH, Lee SJ, Park S et al (2011) Characterisation of flavonoids in Orostachys japonicus A. Berger using HPLC-MS/MS: contribution to the overall antioxidant effect. Food Chem 124:1627–1633
Lee GS, Lee HS, Kim SH et al (2014) Anti-cancer activity of the ethylacetate fraction from Orostachys japonicus for modulation of the signaling pathway in HepG2 human hepatoma cells. Food Sci Biotechnol 23(1):269–275
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Marin B, Palm A, Klingberg M, Melkonian M (2003) Phylogeny and taxonomic revision of plastid-containing euglenophytes based on ssu rDNA sequence comparisons and synapomorphic signatures in the ssu rRNA secondary structure. Protist 154:99–145
Matyasek R, Renny-Byfield S, Fulnecek J et al (2012) Next generation sequencing analysis reveals a relationship between rDNA unit diversity and locus number in Nicotiana diploids. BMC Genom 13:722
Mayuzumi S, Ohba H (2004) The phylogenetic position of East Asian Sedoideae (Crassulaceae) inferred from chloroplast and nuclear DNA sequences. Syst Bot 29:587–598
Mort ME, Soltis DE, Soltis PS, Francisco-Ortega J, Santos-Guerra A (2002) Phylogenetics and evolution of the Macaronesian clade of Crassulaceae inferred from nuclear and chloroplast sequence data. Syst Bot 27:271–288
Mort ME, LevsenN Randle CP, Jaarsveld EV, Palmer A (2005) Phylogenetics and diversification of Cotyledon (Crassulaceae) inferred from nuclear and chloroplast DNA sequence data. Am J Bot 92(7):1170–1176
Ohba H (1978) Generic and infrageneric classification of the Old World Sedoideae (Crassulaceae). J Fac Sci U Tokyo III 12:139–198
Ohba H (1990) Notes towards a monograph of the genus Orostachys (Crassulaceae) (1). J Jap Bot 65:193–203
Ohba H (2001) Crassulaceae. In: Iwatsuki K, Boufford DE, Ohba H (eds) Flora of Japan, vol 2b. Kodasha, Tokyo, pp 10–31
Ohba H (2005) Orostachys. In: Eggli U (ed) Illustrated handbook of succulent plants: Crassulaceae. Springer, Berlin, pp 135–142
Ohwi J (1953) Crassulaceae. Flora of Japan. Shibundo, Tokyo, pp 585–592
Peng YY, Baum BR, Ren CZ et al (2010) The evolution pattern of rDNA ITS in Avena and phylogenetic relationship of the Avena species (Poaceae: Aveneae). Hereditas 147(5):183–204
Poczai P, Hyvönen J (2010) Nuclear ribosomal spacer regions in plant phylogenetics: problems and prospects. Molec Biol Rep 37(4):1897–1912
Ryu DS, Baek GO, Kim EY, Kim KH, Lee DS (2010) Effects of polysaccharides derived from Orostachys japonicus on induction of cell cycle arrest and apoptotic cell death in human colon cancer cells. BMB Rep 43(11):750–755
Seibel PN, Müller T, Dandekar T, Schultz J, Wolf M (2006) 4SALE—a tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinform 7:498
Simon UK, Trajanoski S, Kroneis T, Sedlmayr P, Guelly C, Guttenberger H (2012) Accession-specific haplotypes of the internal transcribed spacer region in Arabidopsis thaliana-a means for barcoding populations. Molec Biol Evol 29(9):2231–2239
Song J, Shi L, Li D, Sun Y, Niu Y et al (2012) Extensive pyrosequencing reveals frequent intra-genomic variations of internal transcribed spacer regions of nuclear ribosomal DNA. PLoS One 7(8):e43971. doi:10.1371/journal.pone.0043971
Sung SH, Jung WJ, Kim YC (2002) A novel flavonol lyxoside of Orostachys japonicus herb. Nat Prod Lett 16(1):29–32
Swofford DL (2002) PAUP* Phylogenetic analysis using parsimony (and other methods). Beta version 10. Sinauer Associates, Sunderland
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molec Biol Evol 28:2731–2739
Telford MJ (2002) Cladistic analyses of molecular characters: the good, the bad and the ugly. Contr Zool 71(1–3):93–100
Thiede J, Eggli U (2007) Crassulaceae DC. In: Kubitzki K (ed) The families and genera of vascular plants. Springer, Berlin, pp 83–118
Vegetative resources of the USSR (1990) Flowering plants, their chemical composition, use. Families Caprifoliaceae–Plantaginaceae. Nauka, Leningrad
Wen J, Zimmer EA (1996) Phylogeny of Panax L. (the Ginseng Genus, Araliaceae): inference from ITS sequences of nuclear ribosomal DNA. Molec Phylogenet Evol 5:167–177
White TJ, Bruns T, Lee S, Taylor J (1990) Amplifcation and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR Protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Xiao LQ, Möller M, Zhu H (2010) High nrDNA ITS polymorphism in the ancient extant seed plant Cycas: incomplete concerted evolution and the origin of pseudogenes. Molec Phylogenet Evol 55(1):168–177
Yoon NY, Min BS, Lee HK et al (2005) A potent anti-complementary acylated sterol glucoside from Orostachys japonicus. Arch Pharm Res 28(8):892–896
Yost JM, Bontrager M, McCabe SW et al (2013) Phylogenetic relationships and evolution in Dudleya (Crassulaceae). Syst Botany 38(4):1096–1104
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucl Acids Res 31:3406–3415
Acknowledgments
We thank Shamil Abdullin, Viktor Bogatov, Roman Dudkin, Konstantin Kiselev, Yuri Ovchinnikov and Valentin Yakubov for sampling natural populations of O. spinosa; Marko Doboš for sharing his persona Orostachys collection and Sun Yan for assistance in sampling in Northern China. This study was supported by a RFBR grant (12-04-01379-a).
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Mark Mort.
Rights and permissions
About this article

Cite this article
Nikulin, A.Y., Nikulin, V.Y., Gonctharova, S.B. et al. ITS rDNA sequence comparisons resolve phylogenetic relationships in Orostachys subsection Appendiculatae (Crassulaceae). Plant Syst Evol 301, 1441–1453 (2015). https://doi.org/10.1007/s00606-014-1165-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-014-1165-y