
Abstract
Leguminosae, Polygalaceae, Quillajaceae and Surianaceae together comprise the order Fabales. Phylogenetic relationships within Fabales remain an unsolved problem even though interfamilial relationships have been examined in a number of studies using different sampling approaches and both molecular and morphological data. In this study, we gather information from the nuclear 26S rDNA region as well as previously published data from the sqd1, matK and rbcL regions. Phylogenetic analyses were performed by maximum parsimony, maximum likelihood and Bayesian inference. Overall, the best-supported topology for the relationships among families within the order places the pair of Leguminosae and Polygalaceae as sister to the pair of Quillajaceae and Surianaceae. However, our approximately unbiased (AU) test of the combined data results has shown that none of the seven different topologies rejected. Furthermore, three topologies were not significantly different from each other. Therefore, similar to the previous studies, this study did not find well-supported dichotomous relationships among the four Fabales families. The Fabales topology was very sensitive to both data choice and the phylogenetic methods used, which may indicate a rapid-near-simultaneous evolution of the four Fabales families. Our results also show that while nuclear sqd1 can be helpful as a complementary region, both the nuclear sqd1 and rDNA 26S regions could be problematic when analyzed individually.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The last decades have seen an exponential increase in molecular phylogenetic studies of angiosperms and emerging consensus at higher levels. The order Fabales Bromhead was one of the most surprising angiosperm clades to result from early studies of interfamilial relationships. Since four families of Fabales are very diverse morphologically, (APG III 2009; Bello et al. 2009); until DNA sequence data became available, most of classification systems placed only the Leguminosae (Fabaceae) Juss. In the order Fabales, while the other families now placed in the order, Polygalaceae Hoffmanns. & Link, Surianaceae Arn., and Quillajaceae D. Don, appeared in different taxonomic groups (Bello et al. 2009).
Molecular studies and fossil evidence suggest an ancient origin and rapid radiation for Fabales (e.g.,., Crane et al. 1990; Zi-Chen et al. 2004; Lavin et al. 2005; Pigg et al. 2008; Bello et al. 2009) (note that the unconfirmed fossils of Polygalaceae and Surianaceae, and there is still the possibility of incomplete fossil record of Fabales). The monophyly of the order is strongly supported by several studies (e.g., Bello et al. 2009, 2012; APG IV 2016), but the overall phylogenetic relationships across the order and position of the root remain controversial; a situation common in higher-level phylogenetic studies of ancient, rapid radiations. (Bello et al. 2009). Previous studies which have recovered different interfamilial topologies for Fabales have used different DNA regions and have very different and unbalanced taxon sampling (e.g., Crayn et al. 1995; Doyle et al. 2000; Savolainen et al. 2000; Soltis et al. 2000; Kajita et al. 2001; Persson 2001; Wojciechowski et al. 2004; Lavin et al. 2005; Forest et al. 2007; Bruneau et al. 2008; Soltis et al. 2011). Phylogenetic instability has been attributed not only to the putative rapid radiation in the early history of the order, but also to sampling directed above (i.e.,. angiosperms) or below (i.e., Leguminosae, Polygalaceae) the ordinal level (Bello et al. 2009). Nevertheless, even studies focused on Fabales could not yield robust relationships for the order (Table 1).
The most comprehensive studies addressing the phylogeny of order Fabales were by Bello et al. (2009, 2012). In their first study, five different topologies were recovered using maximum parsimony (MP) and Bayesian analysis (BI) based on the rbcL and matK plastid regions (Table 1). The Shimodaira-Hasegawa test (Shimodaira and Hasegawa 1999) they conducted favored a resolved topology over a polytomy, but none of the five possible topologies outlining the relationships between the four families of Fabales received a significantly better likelihood. In all analyses, Fabales and each of its component families were monophyletic and support values were mostly very high for all these clades. However, all five topologies for interfamilial relationships within the order received low-to-moderate support, an observation common to many rosid orders and attributed to rapid, early radiation within Fabales (Bello et al. 2009; Wang et al. 2009). Furthermore, Bello et al. (2009) reported that the stem age estimate for Leguminosae, Polygalaceae and the pair Surianaceae + Quillajaceae have very similar ages, which would support the idea of a rapid radiation in the early history of the order.
In their second study (Bello et al. 2012), two hypotheses emerged from the combination of 66 morphological characters with previously published rbcL and matK plastid regions. The morphological characters described floral development and anatomy, and MP and BI analyses were used to explore three data sets which differed in the proportion of missing data and in the choice of outgroup taxa (Table 1). The two recovered topologies were (((S + Q)L)P) and (L + P)(S + Q), with the latter only recovered from BI analyses of the most densely sampled matrices (Table 1). The most frequently recovered topology, (((S + Q)L)P) was considered the most likely in the light of morphology, in spite of low-to-moderate support from both MP and BI analyses.
Despite the attention phylogenetic relationships within Fabales has received, a well-supported interfamilial topology remains elusive. This unresolved phylogeny problem of Fabales also causes unanswered evolutionary questions such as estimating diversification rates (e.g., Smith et al. 2011; Koenen et al. 2013) and understanding trait evolution and biogeography. Therefore, an unambiguous phylogenetic answer for the four Fabales families is required. Moreover, the genomic markers used to date in phylogenetic reconstructions within the order have mostly been from the plastid genome. However, the prevailing view is that nuclear and plastid DNA sequence data are needed to fully understand flowering plant evolutionary history, because nuclear regions can provide insights into hybridization, polyploidy and reticulation (Sang 2002; Álvarez and Wendel 2003). Therefore, in the present study, 26S rDNA sequence data are explored alongside previously published sqd1 data from the nuclear genome, and matK and rbcL data from the plastid genome.
sqd1 (UDP sulfoquinovose synthase gene) is a low copy nuclear gene and it is one of the five conserved orthologue set (COS) markers highlighted in a survey of universally amplifiable markers; it is 267 base pairs (bp) long in Angiosperm families, easy to align due to the lack of indels and highly parsimony informative (Li et al. 2008). Babineau et al. (2013) screened the phylogenetic utility of 19 low copy nuclear genes for caesalpinoid legumes, and they highlighted that the sqd1 region has a potential for familial to tribal-level resolution with almost 30% of parsimony informative characters.
The 26S nuclear ribosomal DNA (rDNA) has been used in several phylogenetic studies (e.g., Fan 2001; Soltis et al. 2001; Zanis et al. 2003; Weitemier et al. 2015; Xu et al. 2015). It has potentially many advantages for phylogenetic reconstruction: (1) it consists of both variable and conserved regions suitable for closely and distantly related taxa; (2) it has very high copy numbers making amplification generally easy with mostly universal primers (Baldwin et al. 1995; Bailey et al. 2003; Weitemier et al. 2015; Xu et al. 2015); and (3) like all nuclear loci, it is biparentally inherited providing insights into hybrid parentage, polyploidy events and reticulation (Álvarez and Wendel 2003). However, some drawbacks were also reported related to its high copy number, such as intra-individual and intra-genomic variation with multiple copy types found within individuals, often incomplete and bidirectional homogenization of copy types, incomplete concerted evolution, paralogy problems, secondary structures, high GC content and the presence of potentially non-functional pseudogene sequences (Hillis and Dixon 1991; Baldwin 1992; Baldwin et al. 1995; Soltis and Soltis 1998; Alvarez and Wendel 2003; Bailey et al. 2003). Among them the view on inclusion/exclusion of pseudogenes changes from one study to another (Bailey et al. 2003). While some authors exclude potential pseudogenes due to alignment or long-branch attraction concerns (LBA; Felsenstein 1978), others include them to address issues related to the potential reticulate evolution of taxa. Many approaches such as pairwise comparisons and tree-based methods were applied to detect these pseudogenes (e.g., Hughes et al. 2002).
Despite the apparent early enthusiasm for the 26S gene and its potential in phylogenetics, the 26S rDNA region’s popularity fell due to the increased interest for low-copy nuclear genes and the low phylogenetic signal subsequently reported for the region (Soltis et al. 2011). The extent of how above-mentioned issues affect phylogenetic reconstruction varies among groups of organisms. For example, phylogenetic studies rated the inclusion of the 26S conserved rDNA sequences from useful (e.g.,. Fan 2001; Neyland 2002; Soltis et al. 2011) to inconsistant (e.g., Ro et al. 1997; Muellner et al. 2003).
The matK plastid region is one of the most frequently employed genes in phylogenetic analyses (e.g., Hilu et al. 2003; Luckow et al. 2003; Wojciechowski et al. 2004; Lavin et al. 2005; Kim and Kim 2011; Wanntorp et al. 2011; Kim et al. 2013; LPWG 2017). It was shown, not only for Leguminosae but also for Fabales, that this plastid gene successfully resolves many relationships with high support due to its high substitution rate (Lavin et al. 2005; Bello et al. 2009; LPWG 2017). Similarly, the rbcL region is another commonly sequenced plastid gene for Fabales. While the use of this gene for Fabales was not recommended (Bello et al. 2009), nor was it as useful as matK for Leguminosae (Lavin et al. 2005), the possibility of it contributing to a robust combined analysis should not be ruled out.
In the present study, a broader outgroup sampling compared to previous studies of Fabales was employed to reduce tree imbalance artefacts (Smith 1994), and particularly to reduce problems associated with LBA (Felsenstein 1978) by breaking long branches between the ingroup and outgroup. The 34 outgroup taxa used here were chosen to represent each family from seven Fabidae orders. Additionally, as well as combining new nuclear sequence data and previously published nuclear and plastid regions, these regions were compared to investigate possible incongruence between them. Lastly, three analytical methods MP, maximum likelihood (ML) and BI were used to investigate how these approaches perform with the new data sets.
Materials and methods
Taxon sampling
Total genomic DNA samples used in Forest (2004) were newly sequenced here for 26S rDNA. The National Center for Biotechnology Information (NCBI/GenBank) accession numbers for previously published and newly produced DNA sequences are provided in “Appendix,” including 70 26S rDNA sequences. The taxon sampling list is organized according to the most recent classification system (e.g., Gagnon et al. 2016 and LPWG 2017). We included 34 taxa from seven different orders of Fabidae as outgroup taxa.
DNA extraction, amplification and sequencing
Approximately 950 bp of the 5′-end of the 26S rDNA gene was amplified using primers N-nc26S1 and 950rev (Kuzoff et al. 1998). Amplification was performed using the following program: 2 min at 94 °C, 32 cycles of 45 s at 94 °C, annealing at 55 °C for 1 min, 1.5 min at 72 °C, and a final extension of 5 min at 72 °C. When PCR product yields were too low, one of the following additional steps was performed: (1) an increase in number of cycles (e.g., up to 35 cycles); (2) an additional PCR run using identical parameters as above repeated with 8 to 10 cycles; (3) three identical non-modified reactions pooled together on the same column for the cleaning step. All PCR products were purified with the QIAquick PCR purification kit (Qiagen inc.) and eluted in EB buffer (10 mM Tris). Complementary strands were sequenced on an ABI 377 or ABI 3100 automated sequencer following the manufacturer’s protocols. The same primers were used for amplification and for the cycle sequencing reactions. Seventy previously unpublished 26S rDNA sequences were included (Forest 2004), and 15 were downloaded from GenBank (“Appendix”). A total of 85 samples were included, 43 from Leguminosae, 17 from Polygalaceae, four from Surianaceae and 21 outgroup taxa representing diverse Fabidae orders. Unfortunately, 26S region could not be amplified for Quillaja.
Since sequencing results do not clearly indicate the presence of paralogous copies and/or pseudogenes (e.g., no significant double peaks in chromatograms), this has not been investigated further here for the 26S nuclear gene region.
Phylogenetic analyses and model selection
Sequences were assembled and aligned using the Geneious alignment option in Geneious Pro 4.8.4 (Kearse et al. 2012) with the automatic pairwise alignment tool and subsequently edited manually. Equivocal base calling at the beginning and end of assembled complementary strands were trimmed. All indels were scored as missing data. Eight different combined analyses were performed to explore the results obtained with the newly produced 26S and published sqd1 nuclear partitions separately and in combination with published matK and rbcL sequences (sqd1 alone, 26S alone, 26S + sqd1 combined, matK + rbcL combined, sqd1 + matK combined, 26S + sqd1 + matK combined, sqd1 + matK + rbcL combined, and 26S + sqd1 + matK + rbcL combined); details of each analysis are presented in Table 2. The substitution models for each of the individual genes were estimated using jModelTest2.1.10 (Guindon and Gascuel 2003; Darriba et al. 2012).
Maximum parsimony analysis was performed using PAUPRat (parsimony ratchet searches using PAUP*; (Sikes and Lewis 2001) as implemented on the CIPRES portal ((Miller et al. 2010); https://www.phylo.org/). Heuristic searches were performed with 1,000 replicates with tree-bisection-reconnection (TBR) branch swapping and a maximum of 1,000 best trees kept. All characters were equally weighted and unordered. Strict consensus trees were generated using PAUP and all the best trees found.
Maximum likelihood analysis was performed using RAxML version 8 (Stamatakis 2014) as implemented on the CIPRES portal ((Miller et al. 2010); https://www.phylo.org/). The GTRGAMMA model was applied to each partition individually, and default maximum likelihood search options were selected with 1000 bootstrap replicates. The best scoring trees with bootstrap values were saved.
Bayesian analyses were conducted using MrBayes 3.2.7a (Ronquist et al. 2012) as implemented on the CIPRES portal ((Miller et al. 2010); https://www.phylo.org/). The same GTR + G + I model of molecular evolution as for ML was applied. MrBayes was run with four (one cold and three heated) Monte Carlo Markov chains (MCMC) and for 100 million generations, sampling one tree in every 1,000 generations. This was repeated twice as independent runs, and the resulting parameter files were jointly visualized in Tracer (Rambaut and Drummond 2003) to ensure convergence. Among the 100,000 trees thus obtained, the first 25,000 trees (25%) were discarded as “burn-in”, and a maximum credibility tree and associated posterior probabilities were compiled using the remaining 75,000 trees and the “halfcompat” option of the “sumt” command. Images of the phylogenetic trees were produced using the Interactive Tree of Life (iTOL) online tool (https://itol.embl.de/) (Letunic and Bork 2016).
Alternative topology testing
The approximately unbiased (AU) (Shimodaira and Hasegawa 1999) test was used to evaluate the alternative phytogenetic relationships of the four Fabales families. For each alternative topology, P values were calculated by W-IQ-TREE (https://iqtree.cibiv.univie.ac.at/, Trifinopoulos et al. 2016) by using 10,000 bootstrap replicates and our 26S + sqd1 + matK + rbcL combined alignment.
Results
The GTR + G + I model of molecular evolution was selected as the most suitable for each of the individual genes. In the following sections, the results of the ML and BI analyses are highlighted with MP topology summaries presented in Table 3 alongside those obtained from the ML and BI analyses. Only bootstrap support values above 50% or posterior probabilities above 0.95 are discussed. Alignment details for all datasets are also summarized in Table 2 (Online resource 1–8).
Fabales is found to be monophyletic in all analyses based on sqd1 (MP, ML and BI), but interfamilial relationships other than the Leguminosae-Polygalaceae pair were not resolved (Table 3, Online resource 9). Polygalaceae is monophyletic in all analyses, and Xanthophyllum sp. is retrieved as sister to the remainder of the family. Within the monophyletic Leguminosae, all six newly recognized subfamilies are also monophyletic, except in the MP analyses in which subfamily Papilionoideae is paraphyletic. For the analyses performed with the 26S rDNA region alone (Online resource 10), both Fabales and its constituent families were resolved as monophyletic in the ML analysis (only 57%) and BI analysis (posterior probability of 1.0), but not in the MP analysis. However, the position of both Detarium (a member of subfamily Detarioideae) and Acrocarpus (a member of subfamily Caesalpinioideae) within Caesalpinioideae and Papilionoideae, respectively, was never seen in any previous analyses (e.g., LPWG 2017),
In the nuclear 26S + sqd1 ML analysis (Online resource 11), except Caesalpinioideae and Detarioideae, the remaining subfamilies were monophyletic. However, in the plastid matK + rbcL ML analysis, the phylogenetic relationships of the six subfamilies support the new classification of the LPWG (2017), all the subfamilies were monophyletic (Online resource 12). In both analyses (matK + rbcL and 26S + sqd1), Leguminosae was sister to Polygalaceae (with only 60% bootstrap support compared to 68% from the nuclear regions analysis). Quillajaceae was sister to Surianaceae with 85% bootstrap support in the plastid ML analysis, while in the nuclear tree the position of these two families was not resolved. Lastly, in contrast to highly supported monophyletic Fabales (100%) in the plastid tree, in the nuclear tree the monophyly of the order Fabales was supported by only 71% bootstrap support.
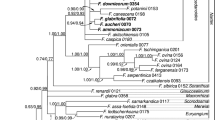
The 26S + sqd1 + matK + rbcL ML analysis yielded monophyletic Fabales (100%), Fabales families, Leguminosae subfamilies and Polygalaceae tribes (Fig. 1). While a (L + P)(Q + S) topology was observed with moderate bootstrap support (90% bootstrap support for (L + P) and 88% bootstrap support for (Q + S)). Within Leguminosae, all six subfamilies were monophyletic. Within monophyletic Polygalaceae (100%), Xanthophylleae was sister to the remainder of the family.

Maximum likelihood tree of 26S + sqd1 + matK + rbcL analysis. Outgroup taxa, Polygalaceae, Surianaceae, Quillajaceae and Leguminosae with six subfamilies (Cercidoideae, Detarioideae, Duparquetioideae, Dialioideae, Caesalpinioideae and Papilionoideae) are indicated. Bootstrap values are indicated below branches
The addition of 26S rDNA data to the other data sets did not yield higher support or better resolution (Tables 3 and 4). In contrast to 83% bootstrap support for the (L + P) clade in the sqd1 ML tree, this clade was supported with 68% bootstrap support in the sqd1 + 26S ML analysis. Similarly, the addition of 26S nuclear data to the sqd1 + matK and sqd1 + matK + rbcL did not yield better results. When matK is added, generally higher support values were obtained for all analyses, however when the rbcL is added, slightly lower values were observed (Tables 3 and 4).
Lastly, our approximately unbiased (AU) test analysis showed that ((L + P)(S + Q)) topology (1) was not significantly better than the other hypotheses (Table 5).
Discussion
Our results have shown that, while the sqd1 nuclear region may not be helpful in solving Fabales phylogeny problems on its own due to reduced support for interfamilial relationships, it can be used in combination with other regions such as matK. On the other hand, there was no difference with regard to phylogenetic relationships between analyses including 26S and those excluding it. While our sequencing results do not clearly indicate the presence of paralogous copies and/or pseudogenes (please note that this has not been investigated in depth here with additional analyses), it is possible that our 26S dataset includes paralogous copies and/or pseudogenes which are causing Caesalpinioideae and Papilionoideae to be represented as non-monophyletic. Indeed, similar results were reported by a recent study (Maia et al. 2014) using both 26S and 18S nuclear regions in an angiosperm-wide study (e.g., non-monophyletic Fabales, Leguminosae and Polygalaceae). Furthermore, lack of support across the majority of nodes in the 26S tree, especially for Leguminosae, is another concern (Online resource 11), which could be linked to the conserved nature of the region (Kuzoff et al. 1998). Therefore, the inclusion of 26S in any phylogenetic study should assess possible paralogy problems, as well as how its contribution to support and topology is compared to analyses excluding it.
Our results have shown that both the topology and the root of the order change according to choice of genes and the analytical methods (Table 3), which was also common in the previous studies that focussed on Fabales. Moreover, two possible topologies were recovered from our analyses, (L + P)(Q + S) obtained for most analyses, and (((L + P)S)Q) for MP analyses of 26S + sqd1 (Table 3). Overall, our results indicate that the ((L + P) (S + Q)) topology is the most likely; which is the same topology that was recovered from the BI analyses of matK and matK + rbcL by (Bello et al. 2009) and again from the BI analyses of matrix A and C of (Bello et al. 2012) (Table 1). However, similar to the previous studies (e.g., Forest 2004; Bello et al. 2009, 2012), it was found that both ML and BI analyses yielded low support values for the interfamilial relationships within Fabales. Furthermore, none of the seven different topologies were rejected by the AU test of our combined data, and the first three topologies were not significantly different from each other (Table 4). Indeed, this may indicate that the phylogenetic signal in the internal branches of Fabales is very weak that it is open to any small changes, which is a common feature of rapid radiations (Rota-Stabelli and Telford 2008; Roberts et al. 2009). However, Fabales is not one of the hard polytomy cases reported to date (Bello et al. 2009), in which the genes that are used may not have any phylogenetic signal for the internal branches (Braby et al. 2005; Whitfield and Kjer 2008; Kodandaramaiah et al. 2010).
Lack of resolution is a common problem across Angiosperms in general (e.g., Zeng et al. 2014; Huang et al. 2015; LPWG 2017) and there are several common reasons underlying not only unresolved rapid radiations but most phylogenetic problems, such as, gene tree incongruence due to biological events (e.g., whole genome duplication (WGD), hybridization, introgression, horizontal gene transfer, incomplete lineage sorting (ILS), extinction) (e.g., Koenen et al. 2019), outgroup problems (i.e., lack of an extant outgroup/closely related outgroup or the effect of the outgroup on ingroup topology) (e.g., Huerta-Cepas et al. 2014), or just systematic errors such as taxon sampling (Thomas et al. 2013), appropriate outgroup choice (i.e., possible systematic biases related to the outgroup sequences, such as low substitution rate and not ingroup-like G + C composition) (e.g., Rota-Stabelli and Telford 2008), LBA (e.g., Qui et al. 2001), inadequate data and inaccurate model implementation (e.g., Reddy et al. 2017; Morgan et al. 2013).
A recent study has shown that the root of Leguminosae is particularly difficult, due to several WGD events, a combination of short internal and long external branches (i.e., extinction and rapid divergence, respectively), ILS and/or reticulation (Koenen et al. 2019) (please see also Cannon et al. 2015 and Wong et al. 2017). Furthermore, it was also argued that obtaining a fully bifurcated legume tree may not be possible due to the simultaneous/near-simultaneous origin of the family (Koenen et al. 2019). Indeed, conflict is very widespread, and it is quite possible that every gene tree is incongruent with the species tree, with these incongruences being stronger for the short-internal nodes (Salichos et al. 2014), and the same evolutionary history would also be possible for the order Fabales, and even thousands of genes may not be enough to solve the Fabales phylogeny, similar to the case of Leguminosae. On the other hand, we think that LBA may not be a problem for Fabales, because in the presence of LBA the root of the group is not stable when sampling different outgroups (Qui et al. 2001), which is not the case for Fabales (e.g., Bello et al. 2009, 2012; current study). Furthermore, to overcome a possible LBA problem, we employed a broad outgroup sampling strategy (Smith 1994; Lyons-Weiler et al. 1998; Djernaes et al. 2012; Drew et al. 2014) and performed Bayesian analyses that are less vulnerable to LBA artefacts, compared to parsimony analyses (Bergsten 2005), yet both the root and topology of the tree changed according to the phylogenetic method, and genes used. However, the effect of data sampling, model implementation, outgroup choice and taxon sampling need further analyses, and future studies should focus on these possible causes for the unresolved Fabales phylogeny.
In conclusion, as with previous studies, this study did not find well-supported dichotomous relationships among the four Fabales families, which may indicate a rapid-near-simultaneous evolution of the four Fabales families. Therefore, it should not be concluded that ((L + P)(Q + S)) is the “definitive answer” for relationships within Fabales, as there is still a need for further studies to not only confirm whether ((L + P)(Q + S)) or another topology is the right answer for the order, but also to reveal the underlying reason for the unresolved phylogeny within Fabales. However, we think that this and previous studies dealing with interfamilial Fabales relationships will provide the framework for future genomic studies that address the issue. Further work is certainly needed to solve the Fabales puzzle with confidence, and to approach the underlying problem from a direction other than employing conventional phylogeny methods.
References
Álvarez I, Wendel JF (2003) Ribosomal ITS sequences and plant phylogenetic inference. Molec Phylogenet Evol 29:417–434. https://doi.org/10.1016/S1055-7903(03)00208-2
Angiosperm Phylogeny Group (2009) An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot J Linn Soc 161:105–121. https://doi.org/10.1111/j.1095-8339.2009.00996.x
Angiosperm Phylogeny Group (2016) An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J Linn Soc 181:1–20. https://doi.org/10.1111/boj.12385
Babineau M, Gagnon E, Bruneau A (2013) Phylogenetic utility of 19 low copy nuclear genes in closely related genera and species of caesalpinioid legumes. S African J Bot 89:94–105. https://doi.org/10.1016/j.sajb.2013.06.018
Bailey CC, Carr TG, Harris SA, Hughes CE (2003) Characterization of angiosperm nrDNA polymorphism, paralogy, and pseudogenes. Molec Phylogen Evol 29:435–455. https://doi.org/10.1016/j.ympev.2003.08.021
Baldwin BG (1992) Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants: an example from the Compositae. Molec Phylogen Evol 1:3–16. https://doi.org/10.1016/1055-7903(92)90030-K
Baldwin BG, Sanderson MJ, Porter JM, Wojciechowski MF, Campbell CS, Donoghue MJ (1995) The ITS region of nuclear ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann Missouri Bot Gard 82:247–277. https://doi.org/10.2307/2399880
Bello MA, Bruneau A, Forest F, Hawkins JA (2009) Elusive relationships within order Fabales: phylogenetic analyses using matK and rbcL sequence data. Syst Bot 34:102–114. https://doi.org/10.1600/036364409787602348
Bello MA, Rudall PJ, Hawkins JA (2012) Combined phylogenetic analyses reveal interfamilial relationships and patterns of floral evolution in the eudicot order Fabales. Cladistics 28:393–421. https://doi.org/10.1111/j.1096-0031.2012.00392.x
Bergsten J (2005) A review of long-branch attraction. Cladistics 21:163–193. https://doi.org/10.1111/j.1096-0031.2005.00059.x
Braby MF, Trueman JW, Eastwood R (2005) When and where did troidine butterflies (Lepidoptera: Papilionidae) evolve? Phylogenetic and biogeographic evidence suggests an origin in remnant Gondwana in the Late Cretaceous. Invert Syst 19:113–143. https://doi.org/10.1071/IS04020
Bruneau A, Mercure M, Lewis GP, Herendeen PS (2008) Phylogenetic patterns and diversification in the caesalpinioid legumes. This paper is one of a selection of papers published in the Special Issue on Systematics Research. Botany 86:697–718. 10.1139/B08-058
Cannon SB, Mckain MR, Harkess A, Nelson MN, Dash S, Deyholos MK, Peng Y, Joyce B, Stewart CN Jr, Rolf M, Kutchan T (2015) Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Molec Biol Evol 32:193–210. https://doi.org/10.1093/molbev/msu296
Crane P, Manchester S, Dilcher D (1990) Fossil leaves and well-preserved reproductive structures from the Fort Union Formation (Paleocene) near Almont. North Dakota, USA. Fieldiana Geol 20:1–63
Crayn DM, Fernando ES, Gadek PA, Quinn CJ (1995) A reassessment of the familial affinity of the Mexican genus Recchia Mocino & Sessé ex DC. Brittonia 47:397–402. https://doi.org/10.2307/2807568
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nature Meth 9:772–772. https://doi.org/10.1038/nmeth.2109
Djernaes M, Klass KD, Picker MD, Damgaard J (2012) Phylogeny of cockroaches (Insecta, Dictyoptera, Blattodea), with placement of aberrant taxa and exploration of out-group sampling. Syst Entomol 37:65–83. https://doi.org/10.1111/j.1365-3113.2011.00598.x
Doyle JJ, Chappill JA, Bailey CD, Kajita T (2000) Towards a comprehensive phylogeny of legumes: evidence from rbcL sequences and non-molecular data. In: Herendeen PS, Bruneae A, Pollarda PS (eds) Advances in legume systematics, Part 9. Royal Botanic Gardens, Kew, London, pp 1–20
Drew BT, Ruhfel BR, Smith SA, Moore MJ, Briggs BG, Gitzendanner MA, Soltis PS, Soltis DE (2014) Another look at the root of the angiosperms reveals a familiar tale. Syst Biol 63:368–382. https://doi.org/10.1093/sysbio/syt108
Fan CX, Qiu-Yun (2001) Phylogenetic relationships within Cornus (Cornaceae) based on 26S rDNA sequences. Amer J Bot 88:1131–1138. https://doi.org/10.2307/2657096
Felsenstein J (1978) Cases in which parsimony or compatibility methods will be positively misleading. Syst Biol 27:401–410. https://doi.org/10.1093/sysbio/27.4.401
Forest F (2004) Systematics of Fabales and Polygalaceae, with emphasis on Muraltia and the origin of the Cape flora. University of Reading, Reading
Forest F, Chase MW, Persson C, Crane PR, Hawkins JA (2007) The role of biotic and abiotic factors in evolution of ant dispersal in the milkwort family (Polygalaceae). Evolution 61:1675–1694. https://doi.org/10.1111/j.1558-5646.2007.00138.x
Gagnon E, Bruneau A, Hughes CE, de Queiroz LP, Lewis GP (2016) A new generic system for the pantropical Caesalpinia group (Leguminosae). PhytoKeys 71:1–160. https://doi.org/10.3897/phytokeys.71.9203
Guindon S, Gascuel O (2003) A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst Biol 52:696–704. https://doi.org/10.1080/10635150390235520
Hillis DM, Dixon MT (1991) Ribosomal DNA: molecular evolution and phylogenetic inference. Quart Rev Biol 66:411–453. https://doi.org/10.1086/417338
Hilu KW, Borsch T, Müller K, Soltis DE, Soltis PS, Savolainen V, Chase MW, Powell MP, Alice LA, Evans R (2003) Angiosperm phylogeny based on matK sequence information. Amer J Bot 90:1758–1776. https://doi.org/10.3732/ajb.90.12.1758
Huang C-H, Sun R, Hu Y, Zeng L, Zhang N, Cai L, Zhang Q, Koch MA, Al-Shehbaz I, Edger PP, Pires JC, Tan D-Y, Zhong Y, Ma H (2015) Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Molec Biol Evol 33:394–412. https://doi.org/10.1093/molbev/msv226
Huerta-Cepas J, Marcet-Houben M, Gabaldón T (2014) A nested phylogenetic reconstruction approach provides scalable resolution in the eukaryotic Tree of Life. PeerJ PrePrints 2:e223v1. https://doi.org/10.7287/peerj.preprints.223v1
Hughes CE, Bailey CD, Harris SA (2002) Divergent and reticulate species relationships in Leucaena (Fabaceae) inferred from multiple data sources: insights into polyploid origins and nrDNA polymorphism. Amer J Bot 89:1057–1073. https://doi.org/10.3732/ajb.89.7.1057
Kajita T, Ohashi H, Tateishi Y, Bailey CD, Doyle JJ (2001) rbcL and legume phylogeny, with particular reference to Phaseoleae, Millettieae, and allies. Syst Bot 26:515–536
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kim DK, Kim JH (2011) Molecular phylogeny of tribe Forsythieae (Oleaceae) based on nuclear ribosomal DNA internal transcribed spacers and plastid DNA trnL-F and matK gene sequences. J Pl Res 124:339–347. https://doi.org/10.1007/s10265-010-0383-9
Kim JS, Hong JK, Chase MW, Fay MF, Kim JH (2013) Familial relationships of the monocot order Liliales based on a molecular phylogenetic analysis using four plastid loci: matK, rbcL, atpB and atpF-H. Bot J Linn Soc 172:5–21. https://doi.org/10.1111/boj.12039
Kodandaramaiah U, Pena C, Braby MF, Grund R, Muller CJ, Nylin S, Wahlberg N (2010) Phylogenetics of Coenonymphina (Nymphalidae: Satyrinae) and the problem of rooting rapid radiations. Molec Phylogen Evol 54:386–394. https://doi.org/10.1016/j.ympev.2009.08.012
Koenen EJM, De Vos JM, Atchison GW, Simon MF, Schrire BD, De Souza ER, De Queiroz LP, Hughes CE (2013) Exploring the tempo of species diversification in legumes. S African J Bot 89:19–30. https://doi.org/10.1016/j.sajb.2013.07.005
Koenen EJ, Ojeda DI, Steeves R, Migliore J, Bakker FT, Wieringa JJ, Kidner C, Hardy OJ, Pennington RT, Bruneau A, Hughes CE (2019) Large-scale genomic sequence data resolve the deepest divergences in the legume phylogeny and support a near-simultaneous evolutionary origin of all six subfamilies. New Phytol 225:1355–1369. https://doi.org/10.1111/nph.16290
Kuzoff RK, Sweere JA, Soltis DE, Soltis PS, Zimmer EA (1998) The phylogenetic potential of entire 26S rDNA sequences in plants. Molec Biol Evol 15:251–263. https://doi.org/10.1093/oxfordjournals.molbev.a025922
Lavin M, Herendeen PS, Wojciechowski MF (2005) Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst Biol 54:575–594. https://doi.org/10.1080/10635150590947131
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucl Acids Res 44:W242–W245. https://doi.org/10.1093/nar/gkw290
Li M, Wunder J, Bissoli G, Scarponi E, Gazzani S, Barbaro E, Saedler H, Varotto C (2008) Development of COS genes as universally amplifiable markers for phylogenetic reconstructions of closely related plant species. Cladistics 24:727–745. https://doi.org/10.1111/j.1096-0031.2008.00207.x
LPWG (2017) A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny. Taxon 66: 44–77. https://doi.org/10.12705/661.3
Luckow M, Miller JT, Murphy DJ, Livshultz T (2003) A phylogenetic analysis of the Mimosoideae (Leguminosae) based on chloroplast DNA sequence data. In: Klitgaard BB, Bruneau A (eds) Advances in legume systematics, Part 10. Higher Level Systematics. Royal Botanic Gardens, Kew, pp 197–220
Lyons-Weiler J, Hoelzer GA, Tausch RJ (1998) Optimal outgroup analysis. Biol J Linn Soc 64:493–511. https://doi.org/10.1111/j.1095-8312.1998.tb00346.x
Maia VH, Gitzendanner MA, Soltis PS, Wong GK-S, Soltis DE (2014) Angiosperm phylogeny based on 18S/26S rDNA sequence data: constructing a large data set using Next-Generation sequence data. Int J Pl Sci 175:613–650. https://doi.org/10.1086/676675
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the gateway computing environments workshop (GCE), 14 Nov 2010, New Orleans, LA, pp 1–8. https://doi.org/10.1109/GCE.2010.5676129
Morgan CC, Foster PG, Webb AE, Pisani D, McInerney JO, O’Connell MJ (2013) Heterogeneous models place the root of the placental mammal phylogeny. Molec Biol Evol 30:2145–2156. https://doi.org/10.1093/molbev/mst117
Muellner AN, Samuel R, Johnson SA, Cheek M, Pennington TD, Chase MW (2003) Molecular phylogenetics of Meliaceae (Sapindales) based on nuclear and plastid DNA sequences. Amer J Bot 90:471–480. https://doi.org/10.3732/ajb.90.3.471
Neyland R (2002) A phylogeny inferred from large-subunit (26S) ribosomal DNA sequences suggests that Burmanniales are polyphyletic. Austral Syst Bot 15:19–28. https://doi.org/10.1071/SB01001
Persson C (2001) Phylogenetic relationships in Polygalaceae based on plastid DNA sequences from the trnL-F region. Taxon 50:763–779. https://doi.org/10.2307/1223706
Pigg KB, Devore ML, Wojciechowski MF (2008) Paleosecuridaca curtisii gen. et sp. nov., Securidaca-Like Samaras (Polygalaceae) from the Late Paleocene of North Dakota and their significance to the divergence of families within the Fabales. Int J Pl Sci 169:1304–1313. https://doi.org/10.1086/591981
Qiu YL, Lee J, Whitlock BA, Bernasconi-Quadroni F, Dombrovska O (2001) Was the ANITA rooting of the angiosperm phylogeny affected by long-branch attraction? Molec Biol Evol 18:1745–1753. https://doi.org/10.1093/oxfordjournals.molbev.a003962
Rambaut A, Drummond A (2003) Tracer: a program for analysing results from Bayesian MCMC programs such as BEAST & MrBayes. University of Edinburgh, UK. Available at: https://beast.bio.ed.ac.uk/Tracer
Reddy S, Kimball RT, Pandey A, Hosner PA, Braun MJ, Hackett SJ, Han KL, Harshman J, Huddleston CJ, Kingston S, Marks BD (2017) Why do phylogenomic data sets yield conflicting trees? Data type influences the avian tree of life more than taxon sampling. Syst Biol 66:857–879. https://doi.org/10.1093/sysbio/syx041
Ro K-E, Keener CS, McPheron BA (1997) Molecular phylogenetic study of the Ranunculaceae: utility of the nuclear 26S ribosomal DNA in inferring intrafamilial relationships. Molec Phylogen Evol 8:117–127. https://doi.org/10.1006/mpev.1997.0413
Roberts TE, Sargis EJ, Olson LE (2009) Networks, trees, and treeshrews: assessing support and identifying conflict with multiple loci and a problematic root. Syst Biol 58:257–270. https://doi.org/10.1093/sysbio/syp025
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MRBAYES 3.2: Efficient Bayesian phylogenetic inference and model selection across a large model space. Syst Biol 61:539–542. https://doi.org/10.1093/sysbio/sys029
Rota-Stabelli O, Telford MJ (2008) A multi criterion approach for the selection of optimal outgroups in phylogeny: recovering some support for Mandibulata over Myriochelata using mitogenomics. Molec Phylogen Evol 48:103–111. https://doi.org/10.1016/j.ympev.2008.03.033
Salichos L, Stamatakis A, Rokas A (2014) Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Molec Biol Evol 31:1261–1271. https://doi.org/10.1093/molbev/msu061
Sang T (2002) Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit Rev Biochem Molec Biol 37:121–147. https://doi.org/10.1080/10409230290771474
Savolainen V, Chase MW, Hoot SB, Morton CM, Soltis DE, Bayer C, Fay MF, De Bruijn AY, Sullivan S, Qiu Y-L (2000) Phylogenetics of flowering plants based on combined analysis of plastid atpB and rbcL gene sequences. Syst Biol 49:306–362. https://doi.org/10.1093/sysbio/49.2.306
Shimodaira H, Hasegawa M (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Molec Biol Evol 16:1114–1116. https://doi.org/10.1093/oxfordjournals.molbev.a026201
Sikes D, Lewis P (2001) Beta software, version 1. PAUPRat: PAUP* implementation of the parsimony ratchet. Distributed by the authors. Department of Ecology and Evolutionary Biology, University of Connecticut, Storrs
Smith AB (1994) Rooting molecular trees: problems and strategies. Biol J Linn Soc 51:279–292. https://doi.org/10.1111/j.1095-8312.1994.tb00962.x
Smith SA, Beaulieu JM, Stamatakis A, Donoghue MJ (2011) Understanding angiosperm diversification using small and large phylogenetic trees. Amer J Bot 98:404–414. https://doi.org/10.3732/ajb.1000481
Soltis PS, Soltis DE (1998) Molecular evolution of 18S rDNA in angiosperms: implications for character weighting in phylogenetic analysis. In: Soltis PS, Soltis DE (eds) Molecular systematics of plants II. Springer, New York, pp 188–210. https://doi.org/10.1007/978-1-4615-5419-6_7
Soltis DE, Soltis PS, Chase MW, Mort ME, Albach DC, Zanis M, Savolainen V, Hahn WH, Hoot SB, Fay MF, Axtell M, Swensen SM, Prince LM, Kress WJ, Nixon KC, Farris JS (2000) Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot J Linn Soc 133:381–461. https://doi.org/10.1111/j.1095-8339.2000.tb01588.x
Soltis DE, Kuzoff RK, Mort ME, Zanis M, Fishbein M, Hufford L, Koontz J, Arroyo MK (2001) Elucidating deep-level phylogenetic relationships in Saxifragaceae using sequences for six chloroplastic and nuclear DNA regions. Ann Missouri Bot Gard 88:669–693. https://doi.org/10.2307/3298639
Soltis DE, Smith SA, Cellinese N, Wurdack KJ, Tank DC, Brockington SF, Refulio-Rodriguez NF, Walker JB, Moore MJ, Carlsward BS (2011) Angiosperm phylogeny: 17 genes, 640 taxa. Amer J Bot 98:704–730. https://doi.org/10.3732/ajb.1000404
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Thomas JA, Trueman JW, Rambaut A, Welch JJ (2013) Relaxed phylogenetics and the Palaeoptera problem: resolving deep ancestral splits in the insect phylogeny. Syst Biol 62:285–297. https://doi.org/10.1093/sysbio/sys093
Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ (2016) W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucl Acids Res 44:W232–W235. https://doi.org/10.1093/nar/gkw256
Wang H, Moore MJ, Soltis PS, Bell CD, Brockington SF, Alexandre R, Davis CC, Latvis M, Manchester SR, Soltis DE (2009) Rosid radiation and the rapid rise of angiosperm-dominated forests. Proc Natl Acad Sci USA 106:3853–3858. https://doi.org/10.1073/pnas.0813376106
Wanntorp L, Gotthardt K, Muellner AN (2011) Revisiting the wax plants (Hoya, Marsdenieae, Apocynaceae): Phylogenetic tree using the matK gene and psbA-trnH intergenic spacer. Taxon 60:4–14. https://doi.org/10.1002/tax.601002
Weitemier K, Straub SC, Fishbein M, Liston A (2015) Intragenomic polymorphisms among high-copy loci: a genus-wide study of nuclear ribosomal DNA in Asclepias (Apocynaceae). PeerJ 3:e718. https://doi.org/10.7717/peerj.718
Whitfield JB, Kjer KM (2008) Ancient rapid radiations of insects: challenges for phylogenetic analysis. Annual Rev Entomol 53:449–472. https://doi.org/10.1146/annurev.ento.53.103106.093304
Wojciechowski MF, Lavin M, Sanderson MJ (2004) A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Amer J Bot 91:1846–1862. https://doi.org/10.3732/ajb.91.11.1846
Wong MM, Vaillancourt RE, Freeman JS, Hudson CJ, Bakker FT, Cannon CH, Ratnam W (2017) Novel insights into karyotype evolution and whole genome duplications in legumes. BioRxiv 099044. https://doi.org/10.1101/099044
Xu J, Xu Y, Yonezawa T, Li L, Hasegawa M, Lu F, Chen J, Zhang W (2015) Polymorphism and evolution of ribosomal DNA in tea (Camelliasinensis, Theaceae). Molec Phylogen Evol 89:63–72. https://doi.org/10.1016/j.ympev.2015.03.020
Zanis MJ, Soltis PS, Qiu YL, Zimmer E, Soltis DE (2003) Phylogenetic analyses and perianth evolution in basal angiosperms. Ann Missouri Bot Gard 90:129–150. https://doi.org/10.2307/3298579
Zeng L, Zhang Q, Sun R, Kong H, Zhang N, Ma H (2014) Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nature Commun 5:4956. https://doi.org/10.1038/ncomms5956
Zhi-Chen S, Wei-Ming W, Fei H (2004) Fossil pollen records of extant angiosperms in China. Bot Rev 70:425–458. https://doi.org/10.1663/0006-8101(2004)070[0425:FPROEA]2.0.CO;2
Acknowledgements
We would like to thank Professor Anne Bruneau (Universite de Montreal, Canada) and Dr. Colin Hughes (Universitat Zurich, Switzerland) for their valuable suggestions, and anonymous reviewers for helpful comments. The first author also thanks Professor Darren Crayn (James Cook University, Australia) for sending some samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Jim Leebens-Mack.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendices
Appendix
Taxon sampling for the phylogenetic analyses of order Fabales based on the nuclear sqd1 and 26S rDNA, and the plastid rbcL and matK. A dash indicates the region was not sampled. Information is presented in the following order: taxon name, voucher specimen of the samples worked in this study (SOURCE); GenBank accessions: sqd1, 26S, matK, rbcL.
Leguminosae. Subfamily Duparquetioideae: Duparquetia orchidacea Baill., Bruneau 1098 (K); MG431081, MG431186, EU361937.1, —. Subfamily Cercidoideae: Adenolobus garipensis (E.Mey.) Torre & Hillc., Leistuer et al. 246 (K); —, MG431188, EU361844.1, AM234268.1. A. pechuelli (Kuntze) Torre & Hillc., Oliver et al. 6527; MG431096, MG431178, JN881353.1, —. Bauhinia syringifolia (F.Muell.) Wunderlin, Weston 2449 (NSW); —, MG431187, —, AM234267.1. B. galpinii N.E.Br., Forest 347 (NBG); MG431094, MG431172, JN881366.1, AM234262.1. Brenierea insignis Humbert, Dupuy M430 (K); —, MG431166, EU361889.1, AM234269.1. Cercis canadensis L., JBM 1397–91; MG431097, MG431189, EU361912.1, U74188.1. Griffonia physocarpa Baill., Cheek 8013 (K); MG431095, MG431190, EU361961.1, AM234265.1. Subfamily Dialioideae: Dialium guianensis (Aubl.) Sandw., Klitgaard 686 (K); MG431086, —, EU361930.1, AM234245.1. Poeppigia procera Presl., Howard 5162 (MT); MG431087, —, EU362026.1, AM234246.1. Storckiella australiensis J.H.Ross & B.Hyland, Hill et al. 2096 (K); —, —, GU321970.1, AM234249.1. Zenia insignis Chun, Manos 1418 (DUKE); —, —, EU362065.1, AF308722.1. Subfamily Detarioideae: Afzelia bella Harms., Breteler 13120; MG431085, —, EU361846.1, KC628648.1. Amherstia nobilis Wall., Baker 490 (KEP); MG431084, MG431182, EU361849.1, AM234234.1. Anthonotha macrophylla P.Beauv., Wieringa 2996 (WAG); MG431083, MG431205, EU361853.1, KC628430.1. A. pynaertii (De Wild.) Exell & Hillc., Breteler 12781 (WAG); MG431063, —, EU361854.1, —. Aphanocalyx cynometroides Oliver, Wieringa 2355 (WAG); —, MG431179, —, AM234241.1. A. djumaensis (De Wild.) J.Leonard; —, —, EU361856.1, —. A. margininervatus J.Leonard, Breteler 12,346 (WAG); MG431082, —, —, —. Brownea sp., A. Pérez and P. Alvia 38,917 QCA (K); MG431069, —, AY386932.1, U74186.1. Browneopsis ucayalina Huber, Klitgaard 684 (K); MG431089, MG431185, EU361894.1, AM234233.1. Crudia gabonensis Harms, Wieringa 2585 (WAG); —, MG431167, EU361922.1, AM234230.1. Cynometra crassifolia Benth.; —, —, KF294055.1, —. C. mannii Oliv., Bruneau 1364; MG431062, MG431177, —, AM234231.1. Detarium macrocarpum Harms., Breteler 12,528 (WAG); —, MG431195, GU321969.1, AM234239.1. Goniorrhachis marginata Taub., Cara 3585, Lewis and Klitgaard 5338; MG431136, MG431183, —, —. Hymenostegia klainei Pellegr., Wieringa 2575 (WAG); MG431061, —, —, KC628501.1. H. robusta Wieringa & Mackinder; —, —, EU361976.1, —. Intsia bijuga (Colebr.) Kuntze; —, —, EU361981.1, KF496786.1. Intsia sp., Colector. 4202; MG431060, —, —, —. Isoberlinia scheffleri (Harms) Greenway, Herendeen 16-XII-97–2 (US); —, MG431169, EU361983.1, AM234240.1. Macrolobium acaciifolium (Benth.) Benth.; —, —, —, U74191.1. M. archeri Cowan, Klitgaard 683; MG431059, —, —, —. M. bifolium (Aubl.) Pers.; —, —, EU361996.1, —. Saraca declinata (Jack) Miq., Manos 1417 (DUKE); MG431080, —, EU362033.1, JX856761.1. Tamarindus indica L., JBM 2138–76 (MT); MG431088, MG431184, EU362056.1, AB378732.1. Subfamily Caesalpinioideae: Acrocarpus fraxinifolius Arn., Manos 1416 (DUKE); —, MG431154, GU321971.1, AY904371.1. Archidendron hirsutum I.Nielsen, Douglas 625 (MEL); MG431110, MG431157, EU361860.1, AM234253.1. Caesalpinia decapetala (Roth) Alson, Herendeen and Mbago 19-XII-97–1 (US); KF379299.1, —, KF379248.1, —. C. pulcherrima (L.) Sw; KF379321.1, —, EU361906.1, U74190.1. Calliandra juzepczukii Standl.; —, —, EU812019.1, —. C. trinervia Benth., Klitgaard 622 (K); MG431072, MG431160, —, —. Calpocalyx dinklagei Harms., Breteler 15,461 (WAG); MG431107, MG431155, EU361907.1, AM234257.1. Cassia grandis L. f., Smith 2061 (MT); MG431065, —, —, —. Cedrelinga cateniformis (Ducke) Ducke, T.D. Pennington, A. Daza and A. Muellner 17,761 MOL (K) (sqd1)/Klitgaard 698 (K) (26S); MG431074, MG431159, AF521818.1, AM234256.1. Ceratoniasiliqua L., Wieringa 3341 (WAG); —, MG431194, AY386852.1, U74203.1. Chamaecrista fasciculata (Michx.) Greene; —, —, AY386955.1, U74187.1. C. nictitans (L.) Moench var. jaliscensis (Greenman) Irwin & Barnaby, Klitgaard 654; MG431098, MG431181, —, —. Colvillea racemosa Bojer; KF379329.1, —, EU361916.1, AY904425.1. Conzattia multiflora Standl.; KF379326.1, —, AY386918.2, AY904416.1. Delonix boiviniana (Baill.) Capuron, Bruneau 1365 (MT); KF379330.1, —, KF379239.1, —. D. floribunda (Baill.) Capuron, Bruneau 1393 (MT); KF379331.1, —, KF379240.1, AY904421.1. D. pumila Du Puy, Phillipson & R.Rabev., Bruneau 1411 (MT); KF379328.1, —, KF379237.1, AY904424.1. D. regia (Boj. ex Hook.) Raf; KF379327.1, —, KF379238.1, AY904419.1. D. velutina Capuron, Bruneau 1354 (MT); KF379324.1, —, KF379236.1, AY904423.1. Denisophytum madagascariense R.Vig.; KF379301.1, —, KF379246.1, —. Erythrostemon calycinus (Benth.) L.P.Queiroz, Lewis 1885 (K); —, MG431176, —, —. E. ivorense A.Chev., Breteler 15,446 (WAG); MG431092, —, EU361948.1, U74205.1. Gleditsia sinensis Lam., Haston V200305; —, —, —, AY904374.1. G. triacanthos L., JBM 2327–82 (sqd1)/ JBM 2674–95 (MT) (26S); MG431093, MG431173, EU361958.1, —. Guilandina bonduc L.; KF379298.1, —, KF379242.1, —. Gymnocladus dioica (L.) Koch, JBM 1830–72 (sqd1)/ JBM 2099–88 (MT) (26S); MG431066, MG431174, EU361966.1, U74193.1. Inga edulis Mart.; —, —, EU361980.1, —. I. nouragensis Poncy; —, —, —, JQ626021.1. Inga sp., Klitgaard 677 (K); MG431075, MG431193, —, —. Mezoneuron scortechinii F.Muell., Wieringa 4195 (WAG); MG431134, —, —, —. Mimosa colombiana Britton & Killip, A.M. Torres 21,343 (K); MG431073, —, DQ790603.1, —. M. pudica L.; —, —, —, KJ008941.1. Moullava digyna (Rottl.) E.Gagnon & G.P.Lewis, comb. nov., Lewis 2067 (K); MG431135, —, EU361902.1, —. Parkia multijuga Benth., Klitgaard 697 (K); MG431109, MG431161, EU362018.1, AM234251.1. Parkinsonia aculeata L., Spellenberg and Brouillet 12,704 (MT); KF379325.1, MG431168, —, —. P. raimondoi Brenan; —, —, —, AY904413.1. P. florida (Benth. ex A.Gray) S.Watson; —, —, AY386856.2, —. Pentaclethra macroloba (Willd.) Kuntze, B. Boyle et al. 6720 (K) (sqd1)/ DeWilde 11,496 (WAG) (26S); MG431108, MG431156, AY386904.1, —. P. macrophylla Benth.; —, —, —, AM234250.1. Poincianella palmeri (S.Watson) E.Gagnon & G.P.Lewis, comb. nov., Lewis et al. 2065 (K); MG431133, —, —, —. Pterogyne nitens Tul., Herendeen 13- XII-97–1 (US); MG431090, MG431171, EU362031.1, AY904377.1. Senna alata (L.) Roxb., Bruneau 1076 (K); MG431064, MG431180, EU362042.1, U74250.1. Tara spinosa (Molina) Britton & Rose; KF379323.1, —, —, —. Umtiza listerina T.Sim, Schrire 2602 (K); MG431091, MG431175, EU362062.1, AM234237.1. Vachellia caven (Molina) Seigler & Ebinger, JBM 386–89 (MT); —, MG431191, AF274131.1, Z70145.1. Zapoteca tetragona (Willd.) H.M.Hern., Klitgaard 649 (K); —, MG431158, AF523097.1, JQ592095.1. Subfamily Papilionoideae: Arachis hypogaea L.; FJ824608.1, —, EU307349, U74247.1. Astragalus laxmannii var. robustior (Hook.) Barneby & S.L.Welsh; —, —, —, JX848460.1. A. lusitanicus Lam., J.R. Edmondson and M.A.S. McClintock 2803 (K); MG431068, —, —, —. A. mongholicus Bunge; —, —, EF685993.1, —. Baphia nitida Afzel. ex Lodd., Bruneau s.n. (LBG); MG431103, MG431162, EU361867.1, AM234261.1. Bobgunnia fistuloides (Harms) J.H.Kirkbr. & Wiersema, Breteler 14,870 (WAG); MG431071, MG431165, EU361885.1, AM234258.1. Cadia purpurea (G.Piccioli) Aiton; —, —, JX295932.1, U74192.1. C. pubescens Bojer ex Baker, L.J. Dorr, L.C. Barnett, and R. Brooks 3279 (K); MG431104, —, —, —. Cladrastis kentukea (Dum.Cours.) Rudd; —, —, AF142694.1, —. C. sinensis Hemsl., E. Punethalengam s.n. (K); MG431105, —, —, Z95551.1. Dalbergia congestiflora Pittier; —, —, AF142696.1, —. D. hupeana Hance; —, —, —, U74236.1. D. yunnanensis Franch., Sino-British Exp. to Cangshan 1981 (K); MG431099, —, —, —. Exostyles venusta Spreng., Klitgaard 24 (K); MG431067, —, JX152591.1, —. Lecointea peruviana J.F.Macbr., B.B. Klitgaard 679 (K); MG431106, MG431163, JX295927.1, AM234260.1. Lotus corniculatus L.Cowan., R.S. MFF128 (K); MG431100, —, HM049505.1, U74213.1. Sclerolobium sp., Klitgaard 687 (K); —, MG431170, AM234242.1, —. Lupinus luteus L. (ABH 31,123); MG431101, —, HM851129.1, HM850145.1. Sophora chrysophylla (Salisb.) Seem. —, GU256432.1, —, —. S. microphylla Aiton, N.A. Smith (AK); MG431070, —, —, —. Swartzia cadiosperma Spruce ex. Benth., Klitgaard 664 (K); MG431102, MG431164, EU362053.1, AM234259.1. Wisteria sinensis (Sims) DC.; FN675910.1, —, AF142732.1, Z95544.1. Polygalaceae: Tribe Xanthophylleae: Xanthophyllum octandrum Domin, Forster 9554 (NY); —, MG431137, —, AM234229.1. Xanthophyllum sp., Coode 7760 (K); MG431076, —, EU604044.1, —. Tribe Carpolobieae: Atroxima afzeliana (Oliv. ex Chodat) Stapf, Jongkind 4281 (WAG); —, MG431150, EU604049.1, AM234175.1. Carpolobia alba G.Don., Cable 747 (K); MG431114, MG431145, EU604053.1, AM234176.1. Tribe Moutabea: Eriandra fragrans P.Royen & Steenis. R. Pullen 7234 (K); MG431115, MG431146, EU604051.1, AM234170.1. Moutabea aculeata (Ruiz & Pav.) Poepp. & Endl., Smith 1522 (US); —, MG431149, —, AM234169.1. M. guianensis Aubl.; —, —, JQ626362.1, —. Tribe Polygaleae: Bredemeyera colletioides (Phil.) Chodat, Guaglianone et al. 1587 (NY); —, MG431148, —, AM234171.1. B. floribunda Willd., Bello 742 (COL) (sqd1)/ Irwin et al. 27,995 (NY) (26S); MG431113, MG431147, EU596520.1, EU644699.1. Comesperma esulifolium (Gand.) Telford 12,350 (CANB); —, MG431192, EU596516.1, AM234179.1. Monnina xalapensis Kunth, Chase 963 (K); —, MG431151, EU604047.1, AM234184.1. Muraltia alba Levyns, Goldblatt 9515 (MO); —, MG431144, —, —. M. heisteria (L.) DC. —, —, —, AJ829698.1. M. spinosa (L.) Dumort, Chase 281 (K); —, MG431152, —, —. M. thunbergii Eckl. & Zeyh., Forest 250 (K, NBG); MG431111, —, AM889730.1, —. Polygala acuminata Willd., Wurdack 1818 (NY); —, MG431141, —, AM234195.1. P. alpicola Rupr., Chase 11,747 (K); —, MG431139, EU604041.1, AM234191.1. P. californica Nutt.; —, —, AY386842.1, —. P. chamaebuxus (L.) var. grandiflora Chase 11,323 (K); —, MG431142, —, —. P. cowellii (Britton) S.F. Blake; —, —, —, AM234199.1. P. ligustrioides A. St. Hil. Harley et al. 20,751 (K); —, MG431143, —, AM234202.1. P. senega L., Brouillet 99–11 (MT); —, MG431138, —, —. Polygala sp., Bello 48; MG431112, —, —, —. P. vulgaris L., Fay 316 (K); —, MG431140, EU604046.1, AM234193.1. Securidaca diversifolia (L.) S.F.Blake, Chase 2998 (MICH); —, —, JQ588837.1, AM234225.1. Surianaceae: Cadellia pentastylis F.Muell., Thompson and Robin s.n. (K); MG431116, MG431196, EU604056.1, L29491.1. Guilfoylia monostylis (Benth.) F. Muell., Fernando and Wannan s.n. (UNSW 21,246); —, MG431203, EU604031.1, L29494.1. Recchia mexicana Moc. & Sessé ex DC., no voucher (see Forest, 2004); —, MG431153, EU604045.1, AM234270.1. Suriana maritima L.; —, —, AY386950.1, U07680.1. Stylobasium spathulatum Desf., Latz. 13,213 (K); MG431117, MG431204, EU604032.1, U06828.1. Quillajaceae: Quillaja saponaria Molina, M.W. Chase 10,931 (K) (sqd1)/ Morgan 2146 (WS) (26S); MG431077, —, AY386843.1, U06822.1. Outgroups. Zygophyllales: Krameria ixine Lofling., Fernandez 22,529 (COK); MG431078, —, EU604050.1, EU644679.1. K. lanceolata Torr., Chase 103 (MICH); —, MG431198, —, —. Zygophyllum rosowii Bunge D1507; —, —, JF956824.1, JF944812.1. Z. xanthoxylum (Bunge) Maxim. Chase 1700 (K); —, MG431197, —, —. Celastrales: Celastrus orbiculatus Thunb., M.W. Chase 2274 (K); MG431079, AF222357.1, EF135517.1, AY788194.1. Oxalidales: Eucryphia lucida (Labill.) Baill.; —, AF036494.1, —, —. Malpighiales: Licania alba (Bernoulli) Cuatrec.; —, KJ414473.1, —, —. Viola suavis M.Bieb.; AM503808.1, —, —, —. V. chaerophylloides (Regel) W.Becker; —, —, JQ950581.1, JQ950611.1. Rosales: Colubrina arborescens (Mill.) Sarg., M.J.M. Christenhusz 5714; MG431131, —, —, —. C. asiatica (L.) Brongn.; —, DQ146521.1, —, —. Elaeagnus commutata Bernh. ex Rydb.; —, —, —, JX848456.1. Elaeagnus sp., M.W. Chase 2414 (K); MG431130, AF479235.1, —, —. E. umbellata Thunb.; —, —, AY257529.1, —. Ficus sp., Moore 315; —, —, —, EU002278.1. F. benjamina L.; FN675916.1, —, JQ773509.1, —. F. tikoua Bureau; —, JF317386.1, —, —. Fragaria × ananassa (Weston) Duchesne; —, X58118, —, U06805.1. Fragaria vesca L.; XM_004290997.1, —, AF288102.1, —. Hippophae rhamnoides L., M.J. Crawley MJC150; MG431129, JF317389.1, JF317428.1, JF317488.1. Humulus lupulus L.Fay, M.F. MFF341 (K); MG431128, AY686777.1, AY257528.1, AF206777.1. Gironniera sp., Puradyatmika 10,455 (BO, MAN, FRE, K, L, CANB, A, SING, BRI, BISH); MG431132, —, —, —. G. subaequalis Planch.; —, —, AF345319.1, AF500340.1. Malus domestica Borkh.; XM_008395413.1, —, AM042561.1, —. M. spectabilis (Aiton) Borkh.; —, —, —, JQ391363.1. Prunus armeniaca L.; FN675931.1, —, HQ235101.1, KF154869.1. P. avium (L.) L.; FN675932.1, —, AM503828.1, HQ235394.1. P. cerasus L.; FN675933.1, —, FJ899111.1, HQ235416.1. P. domestica L.; FN675934.1, —, FJ899110.1, L01947.2. P. persica (L.) Stokes; FN675912.1, AY935820.1, AF288117.1, AF411493.1. Shepherdia argentea (Pursh) Nutt., Chase 3176 (K); —, MG431201, —, AJ225787.1. S. canadensis (L.) Nutt.; —, —, KC475874.1, —. Fagales: Alnus glutinosa (L.) Gaertn; —, AF479106, KF419025.1, EU644678.1. Betula pendula Roth AM503778.1, —, AY372014.1, KF418943.1. Casuarina equisetifolia L., P.J. Edwards 4011 (K); MG431119, —, AY033837.1, AY033859.1. Juglans nigra L.; —, AF479105.1, —, U00437.1. J. regia L.Fay, M.F. et al. MFF416 (K); MG431118, —, AF118038.1, —. Morella cerifera (L.) Small; —, AF479247.1, —, —. M. nana (A.Chev.) J.Herb.; —, —, KF419020.1, —. M. quercifolia (L.) Killick, M.F. Fay s.n. (K); MG431120, —, —, —. M. rubra Lour; —, —, —, KF418924.1. Myrica gale L., M.F. Fay MFF 238 (K); MG431123, —, AY191715.1, AJ626757.1. Nothofagus alpina (Poepp. & Endl.) Oerst.; —, —, —, L13342.2. N. antarctica (G.Forst.) Oerst.; —, —, AY263924.1, —. N. obliqua (Mirb.) Oerst., M.W. Chase 33,143 (K) (1000 Plant Genomes Project); MG431121, —, —, —. Platycarya strobilacea Siebold & Zucc., Herbarium Kewense Cultivated Plants s.n. (K); MG431122, —, AY147100.1, AY263933.1. Ticodendron incognitum Gómez-Laur. & L.D.Gómez, R.K. Brummitt and R. Aizprua 21,139 (K); MG431124, —, U92855.1, AF061197.1. Trigonobalanus verticillata Forman, Chase 595 (K); —, MG431202, AB084771.1, AB084768.1. Cucurbitales: Abobra tenuifolia (Gillies ex Hook. & Arn.) Cogn., Chase 915 (K); —, MG431200, DQ536629.1, AF008961.1. Begonia glabra Aubl., Chase 945 (K); —, MG431199, —, —. B. ulmifolia Willd.; —, —, GU397115.1, —. B. metallica W.G.Sm. × Begonia sanguinea Raddi; —, —, —, L12670.1. Bolbostemma paniculatum (Maxim.) Franquet, TCMK 854 (K); MG431125, —, DQ469139.1, DQ501255.1. Corynocarpus laevigatus J.R.Forst. & G.Forst., M.W. Chase 236 (NCU); MG431126, AF479110.1, AY968448.1, AF148994.1. Cucumis sativus L.; XM_004167788.1, —, DQ536662.1, L21937.1. Datisca cannabina L., M.W. Chase 2745; MG431127, AY968410.1, AB016467.1, L21939.1.
Information on Electronic Supplementary Material
Online resource 1. sqd1 sequence alignment in nexus format.
Online resource 2. 26S sequence alignment in nexus format.
Online resource 3. sqd1 + 26S sequence alignment in nexus format.
Online resource 4. matK + rbcL sequence alignment in nexus format.
Online resource 5. sqd1 + matK sequence alignment in nexus format.
Online resource 6. sqd1 + matK + rbcL sequence alignment in nexus format.
Online resource 7. sqd1 + 26S + matK sequence alignment in nexus format.
Online resource 8. sqd1 + 26S + matK + rbcL sequence alignment in nexus format.
Online resource 9. Maximum likelihood tree of the nuclear sqd1 data set. Outgroup taxa, Polygalaceae, Surianaceae, Quillajaceae and Leguminosae are indicated.
Online resource 10. Maximum likelihood tree of the nuclear 26S data set. Outgroup taxa, Polygalaceae, Surianaceae, Quillajaceae and Leguminosae are indicated.
Online resource 11. Maximum likelihood tree of the combined nuclear sqd1 + 26S data sets. Outgroup taxa, Polygalaceae, Surianaceae, Quillajaceae and Leguminosae are indicated.
Online resource 12. Maximum likelihood tree of the combined plastid matK + rbcL data sets. Outgroup taxa, Polygalaceae, Surianaceae, Quillajaceae and Leguminosae are indicated.
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Aygoren Uluer, D., Hawkins, J.A. & Forest, F. Interfamilial relationships in order Fabales: new insights from the nuclear regions sqd1 and 26S rDNA. Plant Syst Evol 306, 66 (2020). https://doi.org/10.1007/s00606-020-01691-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00606-020-01691-7