
Abstract
The modification of EGFR aptamer (Apt 1) and HER2 aptamer (Apt 2) with gold nanoparticles (AuNPs) is reported to obtain probe I (Apt 1-AuNPs) and probe II (Apt 2-AuNPs). Taking Eca109, KYSE510, and KYSE150 cells as models, the sandwich scattering system of probe I-cell-probe II was formed by the recognition of tumor markers by the aptamer modified probe, and the resonance Rayleigh scattering (RRS) spectra were investigated. The results showed that the scattering system can be used to quantitatively detect the Eca109 cell lines in the range 5.0×10 to 5.0×105 cells·mL−1 with a detection limit of 15 cells· mL−1.The system can also detect the KYSE510 cell lines in a linear range of 5.0×10 to 5.0×105 cells·mL−1 with a detection limit of 18 cells·mL−1 and the KYSE150 cell lines in a linear range of 3.0×10 to 5.0×105 cells·mL−1 with a detection limit of 12 cells·mL−1. To demonstrate the potential application of the RRS method for real sample analysis, cells were spiked into blank serum samples at concentrations from 1.0×102 to 1.0×105 cells·mL−1. The recovery was between 97.0% and 102.3%, and the RSD was between 1.1% and 4.9%, confirming the feasibility of the proposed method for ESCC cell determination.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Esophageal squamous cell carcinoma (ESCC) is an aggressive malignancy, with a high incidence and poor prognosis. In China, ESCC remains the fourth leading cause of cancer-related death, and the 5-year survival rate of ESCC patients who undergo surgery is only 30–40% [1]. Therefore, early and accurate diagnosis is essential to improve the prognosis and survival rate of ESCC patients.
In the early stages of cancer, a small number of cancer cells detach from the primary tumor site and enter the blood, pleural fluid, and other body fluids [2]. These exfoliated cancer cells are exceedingly rare, but their number is highly correlated with the severity of symptoms [3]. If the detection of cancer cells with high accuracy becomes possible, cancer diagnosis will become feasible through a simple blood test, and early diagnosis and treatment effectiveness can also be measured by comparing cancer cell numbers [4].
At present, histochemistry [5], immunohistochemistry [6], flow cytometry [7], and other techniques such as immune-magnetic beads [8], electrochemical [9], surface enhanced Raman scattering (SERS) [10] have been applied to the detection of cancer cells. Histochemical staining suffers from the preparation of additional sections beyond the original hematoxylin- and eosin-stained slides, as well as additional staining steps, which together add cost, time, and workflow complications [11]. The preparation, fixation, and antigen extraction of immunohistochemical cytopathological sections take a long time, so the analysis of body fluid samples based on this method is more complicated [12]. Compared with immunohistochemistry, flow cytometry overcomes the difficulty of evaluation and enables sensitive analysis of a variety of cell surface and intracellular markers, but requires expensive reagents and instruments [13]. Electrochemical methods need to prepare complex modified electrodes, which are difficult to regenerate and have poor reproducibility [14]. Due to the difficulty in collecting surface analysis of irregular samples on rigid substrates, SERS detection often required complex sample pretreatment steps [15]. Therefore, it is still very urgent to explore a simple, low-cost, practical method that can detect low-abundance tumor cells in specimens with high sensitivity and specificity.
In recent years, aptamers have received extensive attention. Aptamers are small RNA or single-stranded DNA with a specific three-dimensional conformation, with a length of 20–80 bases. Similar to the antigen-antibody reaction, aptamers specifically recognize targets in a shape complementarity mode, and are widely used in new drug discovery [16], drug delivery [17], cancer cell detection [18], and bioimaging [19]. The aptamer-based tumor cell detection has the advantages of flexible design and low cost. In recent years, our research team has used aptamer-modified gold nanoparticles as probes to realize the detection of esophageal cancer tumor markers and tumor cells by resonance Rayleigh scattering (RRS) spectroscopy [20, 21]. However, we found that the single aptamer-modified probe has the disadvantages of poor selectivity and low targeting efficiency, which limits its development in detection and treatment applications.
In nature, biological systems often use a multivalent cooperative interaction approach, in which multiple ligands on one biological entity simultaneously bind to receptors for high affinity and superselectivity [22]. Similarly, probes modified with multiple aptamers can effectively increase the effective concentration of ligands at the target site, resulting in stronger affinity than that of single-ligand-modified probes [23], resulting in faster and more efficient detection of target cells.
Human epidermal growth factor receptor proteins HER1 (EGFR) and HER2 belong to the same receptor tyrosine kinase family, which activates intracellular signaling pathways in response to extracellular signals [24]. EGFR and HER2 hyperexpression in the tumors of patients with ESCC was ascertained to be an important marker for the analysis of the clinical features of ESCC. There was an association between elevated levels of EGFR and HER2 in the tumors of ESCC patients with the presence of vascular tumor invasion and with the poor outcome of the disease [25]. For example, Wang et al. [26] investigated EGFR expression and gene amplification status in 193 patients with ESCC by immunohistochemistry and fluorescence in situ hybridization (FISH), and found that of the 193 patients, 95 (49.2%) patients showed EGFR overexpression (3+), and 47 (24.4%) patients harbored EGFR FISH positivity. Zhang et al.’s study found that HER2 protein was overexpressed in most esophageal squamous carcinoma tissues (60/145, 41.4%) [27]. There are also studies indicating that EGFR was overexpressed in 80.9% (76/94) of the ESCC samples, while 24.5% (23/94) of the samples overexpressed HER2. EGFR and HER2 co-overexpression was detected in 22.3% of samples (21/94) [28].
In this paper, we synthesized two types of aptamer-modified gold nanoparticles (probe I and probe II) that separately target two types of esophageal cancer markers: EGFR and HER2. Three different types of ESCC cells, i.e., Eca109 (EGFR+), KYSE510 (HER2+), and KYSE150 (EGFR+ and HER2+), were used to interact with the probe, and the RRS assay was performed. Based on this, a new quantitative analysis strategy for the RRS of esophageal cancer cells was proposed.
Experimental section
Materials and reagents
Chloroauric acid (HAuCl4, analytical grade, 99.9%) and sodium citrate (analytical grade, 99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China), and streptavidin (≥13 units/mg protein ) was purchased from Sigma.
The aptamers and oligonucleotide probes used in this study were obtained from Shanghai Sangon Biological Engineering Technology & Services Co., Ltd. (Shanghai, China) and have the following sequence:
EGFR aptamer: 5′-Biotin-CCGCTTTATTGTTAATTAAGTTTTATATTTCGCA CAACACACAACAATCAATATC-3′ (Apt1, 55 bases, 5′ biotin modified)
HER2 aptamer: 5′-Biotin-GCAGCGGTGTGGGGGCAGCGGTGTGGGGGCAGC GGTGTGGGGTTTTT-3′ (Apt2, 47 bases, 5′ biotin modified)
Control DNA sequence for Apt1: 5′-Biotin-AAAAAACCTGCAAATCCGTAAA
GGGTTTCGTATTTGGAACCAACCTTAAGCTAGT-3′ (DNA1, 55 bases, 5′ biotin modified)
Control DNA sequence for Apt2: 5′-Biotin-TTAACCAATTTTTCGGGGAAACT
ACTGACTCAGTAAGGTTTACTGGG-3′ (DNA2, 47 bases, 5′ biotin modified)
Roswell Park Memorial Institute (RPMI) 1640 growth medium, Dulbecco’s modified Eagle medium (DMEM), fetal bovine serum, trypsin-EDTA (25%), and penicillin–streptomycin were purchased from Gibco BRL Life Technologies (USA). All other chemicals and the reagents used were of analytical AR grade.
Determination principle
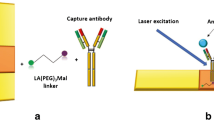
The preparation principle of the cell probe is shown in Scheme 1a. Gold nanoparticles (AuNPs) were prepared by reducing chloroauric acid with sodium citrate, and then streptavidin (SA) was modified on the AuNPs to obtain SA-AuNPs. By the highly specific binding of avidin and biotin, a biotinylated aptamer (Biotin-Apt) was connected to SA-AuNPs to obtain the cell probe.

Schematic illustration of the aptasensor sandwich assay of cancer cells by RRS
The principle of RRS measurement is shown in Scheme 1b. When the probe is mixed with the cell, the probe can target cancer cells because the modified Apt binds specifically to the marker protein on the cell. At the same time, cancer cells aggregate through probe bridging to generate scattering particles, which leads to the enhancement of the RRS signal.
Preparation and characterization of the probes
Preparation of AuNPs
AuNPs were prepared by reducing HAuCl4 solution with sodium citrate [29]. Specifically, 50.0 mL of 1.0 mM HAuCl4 solution was heated to boiling for 15 min, and then 5.0 mL of 38.8 mM sodium citrate solution was quickly added under vigorous stirring. The solution turns dark blue in 20 s and wine red after 60 s. After the color of the solution remains unchanged, the solution was heated and refluxed for 20 min. After cooling to room temperature, the prepared AuNP solution was stored in a 4 °C refrigerator for further use.
Preparation of SA-AuNPs
The pH of the AuNP solution was adjusted to 7.1 with 0.2% K2CO3, and then 20.0 μL of 1.0 mg·mL−1 SA solution was added to every 1.0 mL of AuNPs, vortexed for 1 min, and reacted at room temperature for 30 min. An appropriate amount of 5% bovine serum albumin (BSA) was added and shaken for 10 min to block the unbound site. Unreacted SA and BSA were removed by centrifugation at 12000 rpm for 30 min. The precipitate was redispersed with pH 7.4 phosphate buffer and stored in a refrigerator at 4 °C.
Preparation of the probe
Taking an appropriate amount of SA-AuNP solution, 10.0 μL of 100.0 μM biotinylated EGFR aptamer (Biotin-Apt 1) or biotinylated HER2 aptamer (Biotin-Apt 2) was added to each 1.0 mL of SA-AuNP solution, and reacted at room temperature for 30 min to obtain probe I and probe II respectively. The unbound Biotin-Apt was removed by centrifugation at 12000 rpm for 30 min. The precipitate was redispersed with pH 7.4 phosphate buffered solution and stored in a refrigerator at 4 °C.
Characterization of the probes
One milliliter of AuNPs, SA-AuNPs, probe, and determination system solution was taken and diluted to 5.0 ml with pH 7.4 phosphate-buffered solution; phosphate-buffered solution was taken as the reference, and the absorption spectrum of the sample was recorded on the UV-Vis spectrophotometer.
A certain volume of sample was analyzed for 12% PAGE gel, electrophoresis, and the gel was removed and stained with GelRed or Coomassie blue. Gels were scanned with the Amersham Imager 600 Gel Imaging System, and quantitative analysis was performed using Image Quant TL 8.1 software.
A transmission electron microscopy (TEM) sample was prepared with a copper mesh (200 mesh) coated with a carbon support film. A drop of the nanoparticle solution was dropped on the copper mesh placed on the filter paper, and the solvent was evaporated to dryness.
Cell sources and culture
The human esophageal cancer cell lines KYSE150 (simultaneously overexpressing EGFR and HER2) and KYSE510 (HER2 overexpression) were purchased from the Cell Resource Center of Shanghai Academy of Life Sciences, Chinese Academy of Sciences; the human esophageal cancer cell line Eca109 (EGFR overexpression) was purchased from the Cell Resource Center, Peking Union Medical College (which is the headquarters of National Infrastructure of Cell Line Resource, NSTI) (Beijing, China).
Mouse embryonic fibroblast NIH3T3 (EGFR and HER2 low expression) was donated by the Central Laboratory of Changzhi Medical College.
KYSE150, KYSE510, and Eca109 cell culture: RPMI 1640 completed whole medium, medium containing 10% fetal bovine serum, 100 μg·mL−1 streptomycin, and 100 U·mL−1 penicillin, was grown in a 37 °C, 5% CO2 cell incubator. NIH3T3 cell culture: DMEM medium containing 10% fetal bovine serum, 100 μg·mL−1 streptomycin, and 100 U·mL−1 penicillin was cultured in a 37 °C, 5% CO2 cell incubator.
Probe and cell binding and RRS detection
The cells in the logarithmic growth phase were removed, the medium was discarded, and the cells were rinsed with a small amount of phosphate-buffered solution. After trypsinization, the supernatant was removed by centrifugation, and the cells were redispersed with 1.0 mL phosphate-buffered solution to obtain a cell suspension, which was counted with a cell counter. One milliliter of probe I was placed into a 10.0-mL colorimetric tube, different volumes of cell suspension were added, then 1.0 mL of probe II was added, and the tube was shaken to mix. After incubation in a 37 °C water bath for 20 min, the mixture was diluted to 5.0 mL with phosphate-buffered solution and mixed carefully. The samples were simultaneously scanned at λem=λex(Δλ=0nm) on a fluorescence spectrophotometer, and the RRS spectrum of the system was recorded. The RRS intensity of the measurement system (IRRS) and the reagent blank (I0RRS) at its maximum wavelength (λmax) was recorded, ΔIRRS=IRRS − I0RRS。
Dark-field microscopy detection
Take the cells in the logarithmic growth phase, the medium was discarded, and the cells were rinsed with a small amount of phosphate-buffered solution. After trypsinization, the supernatant was removed by centrifugation, and the cells were redispersed with an appropriate amount of complete medium, seeded at an appropriate density in a 6-well plate pre-placed with cell slides (φ25 mm, TC treated), and cultured in a cell incubator at 37 °C and 5% CO2 for 24 h. The 6-well plate was removed to observe the adherence and growth of the cells. If the cells were in good condition and the density was moderate, the medium was removed, and the cells were carefully rinsed with phosphate-buffered solution three times. Cells were incubated with probe I, probe II, and probe I+probe II for 30 min in a 37 °C, 5% CO2 cell incubator. After incubation, the probe solution was aspirated and the cells were rinsed with a small amount of phosphate-buffered solution to remove the unreacted probe. Cell slides were removed, and carefully placed on glass slides, and dark-field microscopy (DFM) images were observed with an Olympus BX53 microscope equipped with a dark-field condenser.
Results and discussion
Characterization of the probes
The absorption spectrum of AuNPs (Fig. 1a) showed an absorption peak at 520 nm, indicating that well-dispersed spherical AuNPs were prepared. TEM (Figure S1a) showed that the prepared AuNPs had an average particle size of 13.0±2.25 nm and were uniformly dispersed.

(a) Absorption spectra of the measurement system. (b) Photograph showing the colorimetric change of AuNPs, SA-AuNPs, probe I, probe II, and probe I-KYSE150-probe II (from left to right). Concentration of cell: 5×104cell/mL, pH 7.4
The concentration of AuNPs can be calculated according to the method provided by the literature [30] according to the following formula combined with particle size and absorbance:
where c is the concentration of AuNPs, A450 is the absorbance of AuNPs at λ = 450 nm, and ε450 is the molar absorption coefficient. TEM showed that the diameter of the as-synthesized AuNPs was approximately 13 nm. According to the literature, ε450 (d = 13 nm) was 1.39×108 M−1cm−1. From the absorption spectrum of AuNPs (Fig. 1a), it can be seen that the A450 is 0.504. Therefore, the concentration of AuNPs is approximate:
Similarly, it can be seen from the absorption curves of probe I and probe II in Fig. 1 a that the A450 of the two probes is approximately 0.365, and it can be calculated that the concentrations of the two probes are both 2.6 nM.
Figure 1 a shows that when the AuNPs are modified with SA, the position of the absorption peak is red-shifted by 4 to 524 nm. After SA-AuNPs combined with Biotin-Apt to obtain the probe, its absorption peaks were located at 536 nm (probe I) and 532 nm (probe II), respectively. The red shift may be due to the change of the surface dielectric constant of AuNPs and the change of the thickness of the adsorption layer on the surface of AuNPs during the modification process [31], and the decrease of the peak intensity is due to the loss of gold nanoparticles during centrifugation. No significant broadening of the absorption peak was observed after surface modification, indicating that no significant aggregation of the particles occurred. The TEM results (Figure S1b, c, d) also confirmed that no aggregation of nanoparticles occurred during probe preparation.
The hydrodynamic diameters and zeta potentials of AuNPs, SA-AuNPs, probe I, and probe II were measured at pH 7 to further verify the presence of aptamer on the nanoparticles. Table S1 shows that the hydrodynamic diameter of AuNPs is smaller than SA-AuNPs and probes. This phenomenon might be explained by two reasons: firstly, modification of SA and Biotin-Apt onto the surface of AuNPs increases the particle size; secondly, because the contribution of the Biotin-Apt to the hydrodynamic size of the functionalized nanoparticles [32].
The zeta potentials of AuNPs dispersed in water are negatively charged because the nanoparticles are coated by citrate ion which possess three carboxyl groups [33]. The more negative ζ-potential of both SA-AuNP and probes compared to citrate-stabilized AuNPs ζ-potential (Table S1) contributes to improving the stability of functionalized nanoparticles dispersions. Nanoparticles modified with biotinylated oligonucleotides are spontaneously redispersed because of steric and electrostatic repulsion provided by the introduction of negatively charged oligonucleotide moieties.
When the probe was mixed with cells, the absorption spectrum of the probe underwent a large-scale red shift, accompanied by the disappearance of characteristic peaks (Fig. 1a), indicating that the nanoparticles were aggregated [34]. Fig. 1 b shows that the colors of the AuNPs, SA-AuNPs, probe I, and probe II solutions are basically the same, showing a wine red, indicating that the gold nanoparticles are in a uniform dispersion state, while the color of the probe I-KYSE150-probe II solution changes to blue-purple, showing that the probe binds to cells and the gold nanoparticles aggregate.
The probes were characterized by SDS-PAGE. Figure S2a is stained with Coomassie brilliant blue; lane 1 is the protein molecular weight standard; lane 4 is AuNPs with no protein band; lane 2 is SA, with a protein band at 66kDa (the molecular weight of SA is approximately 66 kDa); lane 3 is SA-AuNPs, and a protein band appeared at the same position as SA, indicating that SA was attached to the surface of AuNPs.
Figure S2b and c stained with GelRed. Figure S2b is the electrophoresis picture of probe I, in which lane 1 is the DNA ladder, which is used to mark the number of bases of Apt, lane 2 is SA-AuNPs, lane 5 is AuNPs, and neither shows bands; lane 3 is Apt 1, which has a band at 55 bases (Apt 1 contains 55 bases); lane 4 is probe I, and a band appears at the same position as Apt 1, indicating that Apt 1 is modified on SA-AuNPs. Figure S2c is the electrophoresis picture of probe II, in which lane 1 is DNA ladder, lane 2 is SA-AuNPs, lane 5 is AuNPs, no bands are displayed, lane 3 is Apt 2 in which a band appeared at 47 bases (Apt 2 contains 47 bases), lane 4 is probe II, and a band appears at the same position as Apt 2, indicating that Apt 2 is modified on SA-AuNPs.
The hydrodynamic diameter of SA is approximately 5 nm [35, 36]. According to the method provided in the literature [37], the SA was modeled as cylinders (5 nm in diameter) and assumed to be radially assembled on the AuNPs to form a monolayer with the maximum packing density to estimate the number of SA that can theoretically be attached to the surface of AuNPs. According to the above assumptions, the projected area of SA can be calculated:
The surface area of gold nanoparticles is:
The number of SA that can be connected to the surface of AuNPs is:
Therefore, if each AuNP is completely covered by SA, there should be 27 SA molecules on the surface. SA is a tetrameric protein with four identical subunits arranged symmetrically in D2, and each subunit has a binding site for biotin, while SA binds to the surface of AuNPs, causing two of these sites to be hidden, so it is expected that each SA molecule can bind up to two Biotin-Apt molecules [32], so it can be estimated that each AuNP particle can theoretically bind to approximately 54 Apt molecules at most.
Using Image Quant TL software, the number of Apt molecules attached to the surface of AuNPs was calculated by gel image analysis. The results show that there are about 48 Apt 1 molecules on each AuNP in probe I; there are about 51 Apt2 molecules on each AuNP in probe II, which are consistent with the theoretical simulation results. The higher number of connected Apts in probe II may be since the number of bases in Apt 2 is smaller than that of Apt 1, so the size of a single Apt 2 molecule is smaller.
In order to evaluate the stability of the probe, probe I and probe II were stored at 4 °C for 15 days, and the response for cells was investigated using RRS every day (Figure S3). The nanoparticles remained well dispersed and did not aggregate within 15 days at 4 °C, and the RSD of its intensities was<1.7 %, which revealed the good stability of the probe.
RRS spectra
The RRS spectrum of the assay system is shown in Fig. 2. The unmodified AuNPs, SA-AuNPs, probes, and cells all have weak RRS signals. When the probe interacts with different cells, different results appear:

RRS spectra of the signal probe measurement system (a). RRS spectra of sandwich-type measurement system (b). The concentration of cell: 100 cell·mL−1, pH 7.4
-
(1)
When the probe interacts with the corresponding cells, for example, the surface of probe I is modified with EGFR-specific Apt, which can specifically bind to Eca109 (EGFR+), resulting in a significant increase in the RRS signal. The same situation exists between HER2 Apt-modified probe II and HER2-overexpressing KYSE510 cells, indicating that Apt-modified AuNP probes can specifically bind to target proteins, thereby realizing quantitative detection of cancer cells.
-
(2)
Because the probes can interact with different protein sites on the cell surface, probe I and probe II can bind to esophageal cancer cell KYSE150, which overexpresses both EGFR and HER2 proteins, causing a significant increase in the RRS signal. It was also found that the RRS intensity was as follows: probe I-KYSE150 < probe I-Eca109, probe II-KYSE150 < probe II-KYSE510. This indicates that the probe has a strong specificity and a stronger effect on the cell type that overexpresses the corresponding marker, which may be because a single marker overexpressing cells can provide more recognition positions.
-
(3)
When the probe interacts with cells with low expression of corresponding markers, such as probe I with KYSE510 cells, and probe II with Eca109 cells, the RRS signal level is very low, indicating that the probe is highly selective for cells.
Figure 2 b shows that the simple mixing of the two probes (probe I + probe II) does not cause changes in the RRS signal, and the mixed analysis of the two probes has different effects on different cells:
-
(1)
The effect of mixed probes on the same type of cells is stronger than that of single probes. The RRS intensity was as follows: probe I-Eca109 < probe I-Eca109-probe II, probe II-KYSE510<probe I-KYSE510-probe II, probe I-KYSE150<probe I-KYSE150-probe II, probe II-KYSE150<probe I-KYSE150-probe II, indicating that mixed probe assay has higher sensitivity than the single probe assay. The reason may be that although cells only significantly overexpress a certain marker, there may also be a small amount of expression of other markers, and the simultaneous action of the two probes can bind more cancer cells, thereby amplifying the signal.
-
(2)
The sandwich-type structure has a better effect on the determination of cell types expressing two markers at the same time. The RRS intensity was as follows: probe I-Eca109-probe II<probe I-KYSE150-probe II, probe I-KYSE510-probe II<probe I-KYSE150-probe II. This should be because KYSE150 cells overexpress EGFR and HER2 at the same time, which can provide suitable binding sites for different probes to form a sandwich-type structure, thereby forming large-volume scattering particles. However, Eca109 and KYSE510 cells can only provide limited targets for probe binding, which is not conducive to the generation of sandwich structures.
-
(3)
Mixed probe analysis can detect more cell types, which can be either cell overexpressing a certain marker, or cells expressing two markers at the same time.
The results of the RRS assay showed that the mixed analysis strategy using the two probes could effectively improve the sensitivity of the analysis and expand the range of detection methods.
Dark-field microscopy detection (see the Supplementary Information)
Optimization of the reaction conditions (see the Supplementary Information)
Sensitivity of the method
Under the optimized reaction conditions, different concentrations of cells were reacted with probe I+probe II, and their RRS intensity ΔI was measured. Fig. 3 shows the RRS spectra of Eca109 (a), KYSE510 (b), and KYSE150 (c) cell interactions with the probe. It can be seen from the figure that the maximum scattering wavelength of the assay system occurs at 325 nm, and the logarithm of the cell concentration of the cells is proportional to the RRS intensity in a certain range.

RRS intensities of different concentrations of cells (cells·mL−1) reacted with probes (a) Eca109, (b) KYSE510, and (c) KYSE150
Take ΔI as the ordinate and the logarithm of the cell concentration (log[c]) as the abscissa to draw a standard curve. The relevant parameters are shown in Table 1 and Table S2.
It can be seen from Table 1 that all three kinds of cells have a good linear relationship, among which the KYSE150 cell has a wider linear range and a lower detection limit.
Selectivity of the method
To investigate the specificity of the probe, two random sequences were used as the control sequences of the aptamer, DNA1 instead of Apt1, and DNA2 instead of Apt2, and AuNPs were modified with random sequences to obtain control probes control 1 and control 2.
To investigate the cell selectivity of the probe, the mouse embryonic fibroblast NIH3T3, which expresses both low EGFR and HER2, was used as a control cell, and the cells were incubated with the probe. The result is shown in Figure S8:
-
(1)
The interaction of control 1 and control 2 with cells cannot cause an increase in RRS signals, while the binding of Apt-modified probes to corresponding cells will lead to a significant increase in RRS signals, indicating that Apt-modified probes have strong specificity.
-
(2)
The combination of NIH3T3 cells with all the probes did not lead to the increase of RRS, indicating that the probes are selective for cells, and only the cells that overexpress the corresponding marker proteins can bind to the corresponding probes.
The above results were verified by the DFM assay. The probes were reacted with different cells under the same conditions and observed under a microscope. The DFM results (Fig. 4) showed that when Eca109, KYSE510, and KYSE150 cells were combined with the probe, a large number of probes accumulated on the cell surface, accompanied by bright golden scattered light [38, 39]. However, when the NIH3T3 cells interacted with the probe, no obvious scattered light appeared on the cell surface, and only dark yellow scattered light from the cytoplasm and organelles in the cell membrane was detected by DFM.

Dark-field microscopic images of the selective binding of the probe to cells. Scale bar: 20 μm for all images
Application of the method
Taking the human serum sample without cancer cells as a blank, using the standard addition method, different concentrations of cancer cells were added to the blank serum. The prepared serum sample was combined with the probe, and its RRS intensity was measured. Each sample was measured 3 times, and then the recovery and RSD% were calculated. Tables 2, 3, and 4 are the results of the spiked recovery experiment of Eca109 and KYSE510 KYSE150 cells in serum samples. The recovery rate was between 97.0% and 102.3%, and the RSD was between 1.1% and 4.9%, indicating that the matrix effect of serum has little effect on the determination of cancer cells by the RRS method, and the established method can be used for the quantitative detection of cancer cells in serum samples.
Conclusion
In this paper, gold nanoparticles were modified with EGFR aptamer (Apt1) and HER2 aptamer (Apt2) to obtain probe I and probe II. ESCC cancer cells aggregated through the connection of probes to generate large-volume scattering particles, which resulted in the increase of RRS signals, thus establishing a RRS detection method for cancer cells based on the probe I-cell-probe II sandwich structure.
The cellular entry mechanism of aptamer-modified gold nanoparticles probe can depend on the type of cells being studied and the specific aptamer sequence used for modification. Because cells will readily recognize certain proteins, it is hypothesized that the interaction of antisense oligonucleotide-modified gold nanoparticle agents with proteins is a possible mechanism of recognition and their subsequent internalization [40]. When aptamers are attached to the surface of gold nanoparticles, they can facilitate the binding and uptake of the nanoparticles by target cells through receptor-mediated endocytosis mechanisms.
Because different types of cancer cells may coexist in patients with esophageal cancer, if a single-target probe is used for detection, some cancer cells will be missed, and even false negative results will occur, resulting in missed diagnosis. Multitarget mixed probe can effectively improve the detection rate of cancer cells and reduce the risk of missed diagnosis.
However, considering the complexity of actual clinical samples, our existing research is far from meeting the needs of clinical testing. We still need a much greater understanding of how to control surface architecture in order to stabilize and maximize the assay response. Continued optimization of different parameters is necessary to monitor cancer cell lines in point of care complex environments. Therefore, we have now started the practical application research of this method in whole blood samples of patients with ESCC. The experiment was meant to mimic a real clinical sample, which normally would contain thousands of different species. On the other hand, we also begin to study the potential of the probe as fluorescent probes for biomedical imaging, and to confirm the capability of the designed probe accumulation in tumor tissues through the targeted delivery of aptamer by ex vivo imaging.
Further optimization of this method may prove necessary for applications in clinical diagnostics; nevertheless, this proof of concept has demonstrated the potential applicability of this method for rapid cellular and molecular detection. The enhanced selectivity of our assay has shown promise for extraction and detection of target cells from whole blood, and additional investigation may prove this method valuable for the detection of numerous molecular analytes from blood or other complex biological mixtures. We believe that the assay has enormous potential for application of cancer cell detection from clinical samples.
References
Wang C, Wang J, Chen Z, Gao Y, He J (2017) Immunohistochemical prognostic markers of esophageal squamous cell carcinoma: a systematic review. Chin J Cancer 36(1):1–17. https://doi.org/10.1186/s40880-017-0232-5
Yang L, Yan X, Chen J, Zhan Q, Hua Y, Xu S, Li Z, Wang Z, Dong Y, Zuo D (2021) Hexokinase 2 discerns a novel circulating tumor cell population associated with poor prognosis in lung cancer patients. Proc Natl Acad Sci U S A 118(11):e2012228118. https://doi.org/10.1073/pnas.2012228118
Chen J, Xu J, Wan T, Deng H, Li D (2021) High-sensitive detection of small-cell lung cancer cells based on terminal deoxynucleotidyl transferase-mediated extension polymerization aptamer probe. ACS Biomater Sci Eng 7(3):1169–1180. https://doi.org/10.1021/acsbiomaterials.0c01633
Fukuyama S, Kumamoto S, Nagano S, Hitotsuya S, Yasuda K, Kitamura Y, Iwatsuki M, Baba H, Ihara T, Nakanishi Y (2021) Detection of cancer cells in whole blood using a dynamic deformable microfilter and a nucleic acid aptamer. Talanta 228:122239. https://doi.org/10.1016/j.talanta.2021.122239
Hoshi K, Messina MS, Ohata J, Chung CY-S, Chang CJ (2022) A puromycin-dependent activity-based sensing probe for histochemical staining of hydrogen peroxide in cells and animal tissues. Nat Protoc 17(7):1691–1710. https://doi.org/10.1038/s41596-022-00694-7
Geng H, Feng C, Sun Z, Fan X, Xie Y, Gu J, Fan L, Liu G, Li C, Thorne RF (2023) Chloride intracellular channel 1 promotes esophageal squamous cell carcinoma proliferation via mTOR signalling. Transl Oncol 27:101560. https://doi.org/10.1016/j.tranon.2022.101560
Chen X, Wang J, Song L, Yu Y, Shi M, Jiang W, Liu X, He X (2022) Downregulation of fibulin-4 inhibits autophagy and promotes the sensitivity of esophageal squamous cell carcinoma cells to apatinib by activating the Akt-mTOR signaling pathway. Thorac Cancer 13(18):2592–2605. https://doi.org/10.1111/1759-7714.14595
Kuai J-H, Wang Q, Zhang A-J, Zhang J-Y, Chen Z-F, Wu K-K, Hu X-Z (2018) Epidermal growth factor receptor-targeted immune magnetic liposomes capture circulating colorectal tumor cells efficiently. World J Gastroenterol 24(3):351–359. https://doi.org/10.3748/wjg.v24.i3.351
Zhang X, Jiang X, Wang W, Luo S, Guan S, Li W, Situ B, Li B, Zhang Y, Zheng L (2023) A simple and sensitive electrochemical biosensor for circulating tumor cell determination based on dual-toehold accelerated catalytic hairpin assembly. Mikrochim Acta 190(2):65. https://doi.org/10.1007/s00604-023-05649-7
Xu Y, Zhang D, Lin J, Wu X, Xu X, Akakuru OU, Zhang H, Zhang Z, Xie Y, Wu A (2022) Ultrahigh SERS activity of the TiO2@Ag nanostructure leveraged for accurately detecting CTCs in peripheral blood. Biomater Sci 10(7):1812–1820. https://doi.org/10.1039/d1bm01821c
Fereidouni F, Todd A, Li Y, Chang C-W, Luong K, Rosenberg A, Lee Y-J, Chan JW, Borowsky A, Matsukuma K (2019) Dual-mode emission and transmission microscopy for virtual histochemistry using hematoxylin- and eosin-stained tissue sections. Biomed Opt Express 10(12):6516–6530. https://doi.org/10.1364/boe.10.006516
Tan WCC, Nerurkar SN, Cai HY, Ng HHM, Wu D, Wee YTF, Lim JCT, Yeong J, Lim TKH (2020) Overview of multiplex immunohistochemistry/ immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun 40(4):135–153. https://doi.org/10.1002/cac2.12023
Shu X, Sansare S, Jin D, Zeng X, Tong K-Y, Pandey R, Zhou R (2021) Artificial-intelligence-enabled reagent-free imaging hematology analyzer. Adv Intell Syst 3(8):2000277. https://doi.org/10.1002/aisy.202000277
Yang Y, Huang Y, Li C (2019) A reusable electrochemical sensor for one-step biosensing in complex media using triplex-forming oligonucleotide coupled DNA nanostructure. Anal Chim Acta 1055:90–97. https://doi.org/10.1016/j.aca.2018.12.031
Zhang Q, Xu G, Guo N, Wang T, Song P, Xia L (2021) In-situ synthesis of methyl cellulose film decorated with silver nanoparticles as a flexible surface-enhanced Raman substrate for the rapid detection of pesticide residues in fruits and vegetables. Materials 14(19):5750. https://doi.org/10.3390/ma14195750
Wang W, He S, Dong G, Sheng C (2022) Nucleic-acid-based targeted degradation in drug discovery. J Med Chem 65(15):10217–10232. https://doi.org/10.1021/acs.jmedchem.2c00875
Fan R, Tao X, Zhai X, Zhu Y, Li Y, Chen Y, Dong D, Yang S, Lv L (2023) Application of aptamer-drug delivery system in the therapy of breast cancer. Biomed Pharmacother 161:114444. https://doi.org/10.1016/j.biopha.2023.114444
Li J, Ren X, Zhao J, Lou X (2021) PD-L1 aptamer isolation via Modular-SELEX and its applications in cancer cell detection and tumor tissue section imaging. Analyst 146(9):2910–2918. https://doi.org/10.1039/d1an00182e
Zhang W, Zhou R, Yang Y, Peng S, Xiao D, Kong T, Cai X, Zhu B (2021) Aptamer-mediated synthesis of multifunctional nano-hydroxyapatite for active tumour bioimaging and treatment. Cell Prolif 54(9):e13105. https://doi.org/10.1111/cpr.13105
Li J, Wang J, Zhang X, Chang H, Wei W (2018) Highly selective detection of epidermal growth factor receptor by multifunctional gold-nanoparticle-based resonance Rayleigh scattering method. Sens. Actuators B Chem 273:1300–1306. https://doi.org/10.1016/j.snb.2018.07.046
Li J, Yang Y, Wang J, Zhang B, Chang H, Wei W (2018) Resonance Rayleigh scattering detection of the epidermal growth factor receptor based on an aptamer-functionalized gold-nanoparticle probe. Anal Methods 10(24):2910–2916. https://doi.org/10.1039/c8ay00860d
Fasting C, Schalley CA, Weber M, Seitz O, Hecht S, Koksch B, Dernedde J, Graf C, Knapp E-W, Haag R (2012) Multivalency as a chemical organization and action principle. Angew Chem Int Ed Engl 51(42):10472–10498. https://doi.org/10.1002/anie.201201114
Wang Y, Liu X, Wu L, Ding L, Effah CY, Wu Y, Xiong Y, He L (2022) Construction and bioapplications of aptamer-based dual recognition strategy. Biosens Bioelectron 195:113661. https://doi.org/10.1016/j.bios.2021.113661
Hedner C, Borg D, Nodin B, Karnevi E, Jirstrom K, Eberhard J (2018) Expression and prognostic significance of human epidermal growth factor receptors 1, 2 and 3 in oesophageal and gastric adenocarcinomas preneoadjuvant and postneoadjuvant treatment. J Clin Pathol 71(5):451–462. https://doi.org/10.1136/jclinpath-2017-204774
Delektorskaia VV, Chemeris GI, Kononets PV, Grigorchuk AI (2010) Immunohistochemical study of epidermal growth factor receptor expression in esophageal squamous cell carcinoma. Arkh Patol 72(5):3–6
Wang X, Niu H, Fan Q, Lu P, Ma C, Liu W, Liu Y, Li W, Hu S, Ling Y (2016) Predictive value of EGFR overexpression and gene amplification on icotinib efficacy in patients with advanced esophageal squamous cell carcinoma. Oncotarget 7(17):24744–24751. https://doi.org/10.18632/oncotarget.8271
Zhan N, Dong W-G, Tang Y-F, Wang Z-S, C-l X (2012) Analysis of HER2 gene amplification and protein expression in esophageal squamous cell carcinoma. Med Oncol 29(2):933–940. https://doi.org/10.1007/s12032-011-9850-y
Hou W, Qin X, Zhu X, Fei M, Liu P, Liu L, Moon H, Zhang P, Greshock J, Bachman KE (2013) Lapatinib inhibits the growth of esophageal squamous cell carcinoma and synergistically interacts with 5-fluorouracil in patient-derived xenograft models. Oncol Rep 30(2):707–714. https://doi.org/10.3892/or.2013.2500
Turkevich J, Stevenson PC, Hillier J (1951) A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss Faraday Soc 11:55–75. https://doi.org/10.1039/df9511100055
Haiss W, Thanh NTK, Aveyard J, Fernig DG (2007) Determination of size and concentration of gold nanoparticles from UV-Vis spectra. Anal Chem 79(11):4215–4221. https://doi.org/10.1021/ac0702084
Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP (2008) Biosensing with plasmonic nanosensors. Nat Mater 7(6):442–453. https://doi.org/10.1038/nmat2162
D'Agata R, Palladino P, Spoto G (2017) Streptavidin-coated gold nanoparticles: critical role of oligonucleotides on stability and fractal aggregation. Beilstein J Nanotechnol 8:1–11. https://doi.org/10.3762/bjnano.8.1
Gulicovski JJ, Cerovic LS, Milonjic SK, Popovic IG (2008) Adsorption of itaconic acid from aqueous solutions onto alumina. J Serb Chem Soc 73(8-9):825–834. https://doi.org/10.2298/jsc0809825g
Jiang Y, Zhao H, Zhu N, Lin Y, Yu P, Mao L (2008) A simple assay for direct colorimetric visualization of trinitrotoluene at picomolar levels using gold nanoparticles. Angew Chem Int Ed Engl 47(45):8601–8604. https://doi.org/10.1002/anie.200804066
Häussling L, Michel B, Ringsdorf H, Rohrer H (1991) Direct observation of streptavidin specifically adsorbed on biotin-functionalized self-assembled monolayers with the scanning tunneling microscope. Angew Chem Int Ed Engl 30:569–572. https://doi.org/10.1002/anie.199105691
Weber PC, Ohlendorf DH, Wendoloski JJ, Salemme FR (1989) Structural origins of high-affinity biotin binding to streptavidin. Science 243(4887):85–88. https://doi.org/10.1126/science.2911722
Yeo ELL, Chua AJS, Parthasarathy K, Yeo HY, Ng ML, Kah JCY (2015) Understanding aggregation-based assays: nature of protein corona and number of epitopes on antigen matters. RSC Adv 5:14982–14993. https://doi.org/10.1039/c4ra12089b
El-Sayed IH, Huang XH, El-Sayed MA (2005) Surface plasmon resonance scattering and absorption of anti-EGFR antibody conjugated gold nanoparticles in cancer diagnostics: applications in oral cancer. Nano Lett 5(5):829–834. https://doi.org/10.1021/nl050074e
Huang Y-F, Lin Y-W, Lin Z-H, Chang H-T (2009) Aptamer-modified gold nanoparticles for targeting breast cancer cells through light scattering. J Nanopart Res 11(4):775–783. https://doi.org/10.1007/s11051-008-9424-x
Giljohann DA, Seferos DS, Patel PC, Millstone JE, Rosi NL, Mirkin CA (2007) Oligonucleotide loading determines cellular uptake of DNA-modified gold nanoparticles. Nano Lett 7(12):3818–3821. https://doi.org/10.1021/nl072471q
Funding
Great thanks for the support from the Fundamental Research Program of Shanxi Province (No. 20210302124325), Research Project of Health Commission of Shanxi Province (No. 2022112), Scientific and Technologial Innovation Programs of Higher Education Institutions in Shanxi (No. 2019L0660), and the Doctoral Scientific Research Foundation of Changzhi Medical College (No.BS201916).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The authors declare that they have no competing of interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, J., Wang, J., Ma, D. et al. Highly sensitive and specific resonance Rayleigh scattering detection of esophageal cancer cells via dual-aptamer target binding strategy. Microchim Acta 190, 248 (2023). https://doi.org/10.1007/s00604-023-05828-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-023-05828-6