
Abstract
The work describes a fluorescence-based study for mapping the highest affinity truncated aptamer from the full length sequence and its integration in a graphene oxide platform for the detection of Salmonella enteriditis. To identify the best truncated sequence, molecular beacons and a displacement assay design are applied. In the fluorescence displacement assay, the truncated aptamer was hybridized with fluorescein and quencher-labeled complementary sequences to form a fluorescence/quencher pair. In the presence of S. enteritidis, the aptamer dissociates from the complementary labeled oligonucleotides and thus, the fluorescein/quencher pair becomes physically separated. This leads to an increase in fluorescence intensity. One of the truncated aptamers identified has a 2-fold lower dissociation constant (3.2 nM) compared to its full length aptamer (6.3 nM). The truncated aptamer selected in this process was used to develop a fluorometric graphene oxide (GO) based assay. If fluorescein-labeled aptamer is adsorbed on GO via π stacking interaction, fluorescence is quenched. However, in the presence of target (S. enteriditis), the labeled aptamers is released from surface to form a stable complex with the bacteria and fluorescence is restored, depending on the quantity of bacteria being present. The resulting assay has an unsurpassed detection limit of 25 cfu·mL−1 in the best case. The cross reactivity to Salmonella typhimurium, Staphylococcus aureus and Escherichia coli is negligible. The assay was applied to analyze doped milk samples for and gave good recovery. Thus, we believe that the truncated aptamer/graphene oxide platform is a potential tool for the detection of S. Enteritidis.

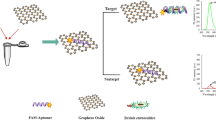
Fluorescently labelled aptamer against Salmonella enteritidis was adsorbed on the surface of graphene oxide by π-stacking interaction. This results in quenching of the fluorescence of the label. Addition of Salmonella enteritidis restores fluorescence, and this effect is used for quantification of this food-borne pathogen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Food safety is one of the major concerns for the public health in the developed and developing countries [1]. Microorganisms such as bacteria, viruses, fungi and parasites are considered as foodborne pathogens which can enter into the human bodies via contaminated foods causing the infections [2]. Foodborne diseases are caused mainly by bacteria such as Salmonella enteritis, Salmonella Typhimurium, Staphylococcus aureus, Listeria monocytogenes, Campylobacter jejuni, Bacillus cereus and Shiga-toxin producing E. coli strains (non-O157 STEC). These infections can lead to hospitalization and even to death [3]. Salmonella Typhimurium and Salmonella enteritis are the most commonly identified species from the contaminated food. Human salmonellosis is mainly caused by Salmonella enterica subspecies enterica serovar Enteritidis (S. Enteritidis). Poultry is the main reservoir for S. Enteritidis and therefore, eggs and chicken meats are the main source for S. Enteritidis related human infections. Salmonella enteritidis infection leads to fever, abdominal cramps and diarrhea after 12 to 72 h of post-infection. Several methods have been used for the detection of salmonella species including traditional culture method [4], polymerase chain reaction (PCR) [5], enzyme-linked immunosorbent assay (ELISA) [6] and loop mediated isothermal amplification (LAMP) [7]. Though the traditional culture method is the gold standard, it is time consuming, require well-experienced technicians and expensive. Immunological methods have been also used. However, they have some disadvantages such as the cross reactivity, high production cost, low stability and the need of pre-processing steps. PCR methods were also employed for the detection of salmonella using the bacteria specific primers. Real-time PCR (RT-PCR) is used for the quantitative pathogen detection using DNA fluorescent dyes as probes [2, 8]. Sophisticated instruments such as liquid/gas chromatography coupled with mass spectrophotometer have been also used for analysis of the pathogens. However, these bulky and expensive instruments cannot be routinely used for the point-of-care on-site pathogen detection.
Researchers are showing special interest towards aptamers. Aptamers are considered as chemical antibodies which can selected against various analyses for clinical, food and environmental applications [9]. Aptamers are short synthetic DNA or RNA sequences which can specifically capture their analytes with high affinity (Kds in the nanomolar to picomolar range). Specific aptamers for proteins, metal ions, bacteria, viruses and small molecules have been selected using In vitro selection protocol. Aptamers have been widely exploited in biosensors development showing many advantages over antibodies such as the high stability, low cost, ease of chemical synthesis and modification. The typical aptamer usually consists of 40 to 100 nucleotides that can at the favorable situations folds to secondary or tertiary structures forming binding packet to bind to a target molecule and form stable target-aptamer complex.
Fluorescence-based aptasensors are usually designed to undergo structure switching or conformational changes of the aptamers upon binding with the target [10]. However, it is not easy to predict the occurrence of conformation change within the full length aptamer. Moreover, slicing the non-binding region of the aptamer favors the formation of stronger complex with the target [11]. For instance, the affinity of the truncated aptamer of vascular endothelial growth factor (VEGF165) increases by 200 fold compared to the wild type aptamer [12]. We have also reported 17 fold increment in the affinity of the truncated anti-progesterone aptamer compared to the parental aptamer [13]. Therefore, mapping the binding site within the aptamer sequence represent an important step for increasing the affinity and the conformation change of the aptamer. Graphene oxide (GO) is two dimensional carbon nanomaterial which showed unique properties due to its electronic configuration, large surface area and high dispersion capability in water [14]. These unique properties of GO make it an ideal material for the development of environment-friendly and low-cost biosensor platforms [15]. Graphene oxide is a good energy acceptor. Therefore, it has been used as a sensing platform for the detection of different biomolecules using on and off fluorescence assays [16, 17]. Graphene oxide- based aptasensors were reported for the detection of thrombin and ATP. In these assays, the fluorescence of the fluorescent aptamer was quenched by the GO and then restored in the presence of the target analyte.
In this paper, we used different fluorescence assays for selecting the highest affinity truncated aptamer for S. enteritidis. Then, we report an aptamer-based fluorescence GO analytical method for the sensitive detection of S. enteritidis.
Experimental
Materials and methods
Salmonella Typhimurium, St (ATCC14028), Salmonella Enteritidis, Se (ATCC13076), Staphylococcus aureus, Sa (ATCC6538) and Escherichia coli, Ec (ATCC8739) were purchased from American Type Culture Collection (Manassas, VA). Phosphate buffered saline (PBS) tablets were purchased from Sigma-Aldrich. Prior to use, the frozen culture was activated in trypticase soy broth (TSB, Oxoid, Hampshire, UK) at 37 °C with two consecutive transfers after 18 ± 20 h incubation periods. The culture was centrifuged at 10000 X g for 10 min at 4 °C and washed twice with trypticase soy broth. Cells suspensions were prepared and adjusted by optical density (OD) at 600 nm to 0.5, that is equivalent to 108 cfumL−1. Then, the cells were serially diluted in PBS (pH 7.4). Graphene oxide dispersion was purchased from Dropsens Inc. (Asturias, Spain). HPLC purified labeled and unlabeled oligonucleotides (Table 1) were purchased from Metabion International (Planegg, Germany). The DNA oligos were dissolved in ultra pure Milli-Q water to make the stock solutions and stored at −20 °C until further use. The DNA solutions used in the experiments were diluted in binding buffer. The fluorescein-labelled oligonucleotides were protected from the light while performing the experiments.
Fluorescence measurements
All the fluorescence measurements for the fluorescein labelled aptamers, the complementary oligonucleotides and the aptamer-beacon were performed using Nanodrop ND3300 fluorospectrometer (Thermo Scientific, Canada). The samples were excited in the blue light (470 ± 10 nm) and the emission was monitored at 515 nm. All the measurements were recorded in PBS buffer (pH 7.4) at room temperature in tripcate unless otherwise mentioned. The fluorescence spectra are the average of two measurements.
Designing the aptamer sequences
The 54 nucleotide anti- S. Enteritidis aptamer sequence was chosen from previously published work (Kolovskaya et al. 2013). The truncation of this aptamer was made based on the mfold software secondary structure. The on and off fluorescence assays of duplex and aptamer beacon have been designed to find out the short sequence binding region of the aptamer. As shown in Table 1, we used three different designs. The first design, two aptamer beacons at 5′ and 3′ ends (SE54MB1 and SE54MB2) of the full length aptamer by the addition or deletion of few nucleotides either at 5’or 3′ end. The aptamer-beacons are labelled with fluorescein (Flu) and quencher (BHQ1) at 5’and 3’ends of the stem, respectively. The second design, is the 29 mer sequence from the middle of the aptamer which contains part of both apta-beacons (SE54T). This design was used for the competitive displacement of aptamer complementary sequences (SE54TC1 and SE54TC2) labeled with fluorescein and the BHQ1 at 5′ and 3′ ends, respectively. The fluorescein and quencher become in close proximity to each other upon hybridization with the truncated aptamer. The third design is the same 29 mer sequence with the fluorescein label (SE54TF) for the GO experiments.
Determination of the binding affinity of the truncated aptamer
After selecting the truncated aptamer that contains the binding region to the target, the dissociation constant (Kd) of that sequence (SE54T) was determined using fluorescence assay. 106 cfu.mL-1of S. Enteritidis cells were first incubated in the media overnight. Then washed with PBS. Different concentrations of fluorescein-labeled SE54T aptamer solutions in 100 μl PBS were heated to 95 °C for 5 min, cooled to 4 °C for 10 min, kept at room temperature for 10 min and then incubated with the adsorbed cell on the wells for 1 h.
Then, the wells were washed with 500 μl PBS for two times, then suspended in 100 μl PBS and the fluorescence measurements were recorded. The saturation curve was plotted as the SE54T aptamer concentration versus fluorescence intensities and the Kd was calculated using non-linear regression fitting of the curve.
Aptabeacon and competitive fluorescence displacement assays
For the aptamer-beacons assay, the aptamers were first heated to 95 °C for 5 min, cool to 4 °C for 10 min and kept at room temperature for 10 min. Then, increased number of S. Enteritidis cells were incubated with 100 nM solutions of aptamer beacon and the fluorescence intensity of each sample was recorded. In the competitive fluorescence displacement assay, a mixture of 500 nM solutions of each truncated DNA aptamer and equal concentrations of their 5’fluorescein-labelled and 3’ BHQ1 labelled complementary sequences in PBS were kept at 90 °C water bath and then cooled slowly to room temperature for 3 h. The slow cooling processes make the perfect duplex and bring the fluorescein-BHQ1 pair at the closest distance possible. 100 nM of hybridized duplex were incubated with different concentrations of S. Enteritidis ranging from 102 to 107 cfu-mL-1 for 30 min. The fluorescence intensity of each sample was recorded. The specificity of the sequence SE54T sequence was tested by incubating the duplex with 104 cfu.mL-1 of similar bacteria associated with S. Enteritidis such as S. Typhimurium, Staph. Aureus and E Coli.
Graphene oxide/aptamer-based analytical platform
The concentration of the aptamer was optimized in order to get a significant fluorescence intensity to 25 nM (data not shown that was fixed in the subsequent experiments. For the GO concentration optimization, 25 nM of aptamer was titrated against different GO solutions with concentrations ranging from 0 to 50 μg.mL−1. The aptamer solutions were incubated at room temperature with the GO to ensure maximum adsorption on the GO surface. For the detection experiments, different concentrations (from 102 to 107) of S. enteritidis in phosphate buffered saline were incubated with the optimized GO/aptamer mixture (20 μg.mL−1 of GO and 25 nM of aptamer) for 30 min at room temperature and the fluorescence spectrum was recorded for each sample. The selectivity of the GO/aptamer platform was tested by incubating it with 104 cfu.mL-1 of different bacterial cells associated with S. Enteritidis such S. Typhimurium, Staph. Aureus and E Coli.
Results and discussion
Generally, the DNA library sequences used for the SELEX screen should have certain length (between 40 and 100 nucleotides) in order to increase its diversity and maximize the probability of selecting high affinity aptamers for the target. However, not all the nucleotides in the aptamer are usually involved in the critical binding with the target molecules. In most cases, the parts of the aptamer sequence which forms stem-loop structure, G-quartet, bulges and/or pseudoknots are involved in the direct binding with the target molecules [18,19,20]. Some extra nucleotides can be essential for supporting the contact between the target molecule and the aptamer. However, the rest of the aptamer sequence which is not involved in the binding can destabilizing the aptamer-target complexes [21]. Moreover, long aptamers are not usually compatible with biosensors which works based on conformation change. Therefore, post SELEX modification of aptamer is needed for the development of sensitive biosensors [22]. Kolovskaya el al. [23] have selected high affinity and specificity aptamers for S. Enteritidis using in vitro selection from a pool of 2 × 1015 DNA sequences. The Kd of the full length aptamers were consisting of 80 nucleotides including the primer-binding sites at 3’and 5′ ends was 80 nM. However, after elimination of 25 nucleotides from 5’end including the primer binding site, the remaining sequence which consists of 54 nucleotides has shown improved affinity with a kd of 6.3 nM. The anti-bacterial activity of the shorter aptamer was increased to 42% compared to the full length aptamer (9%) [23]. In this study, we aim to do further truncation in order to improve the affinity and exploit the shorter sequence in a GO fluorescence-based assay for the detection of S. Enteritidis.
Selection of the highest affinity truncated aptamer
The secondary structure obtained from the mfold software of the full length aptamer is shown in Fig. 1a. In order to find out the short aptamer sequence that contains the binding domain to the target, we split the SE54 aptamer into three different parts. Two stem-loop structures (SE54MB1 and SE54MB2) from both ends and a truncated part from the middle region were investigated in this study. For the stem-loop design, we added or modified few nucleotides in the stem region to make the perfect stem. Then, the two ends of the stem were labelled with fluorescein and BHQ1 to form aptamer beacon. The aptamer-beacon, SE54MB1 consist of 16 nts (3–18) from the 5’region of the full aptamer SE54and SE54MB2 is 22mer (27–48) from 3′region of SE54. Out of 22 nucleotides of the aptamer SE54MB2, 15 nucleotides represent the constant primer binding site. No significant change in the fluorescence intensity of both SE54MB1 and SE54MB2 after incubation with S. Enteritidis bacteria was observed (data not shown). These results indicate that both SE54MB1 and SE54MB2 parts did not undergo a conformation change that can interrupt the fluorescein-quencher pair in the presence of S. Enteritidis. In other words, we concluded from this experiment that the nucleotides for SE54MB1 and SE54MB2 parts were not enough for target binding and some other nucleotides from the middle part are essential for the binding. In fact, it is expected that the constant region of the aptamer (primer-binding region) does not contribute to the binding properties of the aptamer or has minimal involvement compared to the overall structure of the aptamer.

The secondary structure prediction of the full length aptamer sequence (SE54) and the truncated aptamer SE54T using mfold software. The blue and green scissors shows the truncation of stem and loop molecular beacon at 5′ (position 3 to 18) and 3′ (position 27 to 48) ends of the parental aptamer. The black scissors shows the truncation of the middle part of the aptamer (position 12 to 40)
After we figured out that the two hairpins at both ends of the aptamers did not bind to the target, we then investigated the sequence taken from the middle (SE54T). As shown in Fig. S1, Two short complementary sequences of the SE54T aptamer forming fluorescein and quencher pair were used as reporters for the target binding to the aptamer. The reporting complementary sequences were hybridized to SE54T to form DNA duplex. The design was done in such a way for the fluorescein and the quencher to be in close proximity to each other when the duplex is formed. Upon addition of the target molecule, the aptamer binds to the target and changes its conformation leading to complete or partial displacement of either one or both complementary sequences. These changes lead to physical separation of fluorescein and BHQ1 which can be detected via the increase in the fluorescence intensity. Figure 2 shows the fluorescence spectra of fluorescein labelled complementary sequence (Fig. 2a, black), after duplex formation (blue) and the fluorescence recovery after S. Enteritidis binding (red). When equal concentration of SE54T and the reporter sequences SE54C1 and SE54C2 are duplexed, 75% of the fluorescence intensity was quenched compared to the free fluorescein labelled SE54C1, indicating the perfect duplex formation. The decrease in the fluorescence intensity reveals that the fluorescein and the BHQ1 are in close contact to each other as required in the fluorescence displacement assay. As shown in Fig. 2b, the fluorescence intensity increased drastically by the addition of S. Enteritidis cells, implying that the fluorescein and the BHQ1 were separated due to either dissociation of one or both complementary sequences. We calculated the limit of detection (LOD) from this assay to be 30cfumL-1. These results indicate that the SE54T is involved in the S. Enteritidis binding. It was also found that this truncated part, SE54T has a stem-loop secondary structure as shown in Fig. 1b which might form the binding pocket for the S. Enteritidis surface protein. We also investigated the specificity of this truncated aptamer against Salmonella Typhimurium, Staphylococcus aureus and Escherichia coli and the results have shown high specificity (Fig. 2c). Therefore, we selected this truncated part for our GO platform to develop a fluorescence-based analytical assay.

a The fluorescence spectra of SE54C1 (black) and after forming the duplex (blue), and then after incubation with the S. Enteritidis (red). b Calibration curve, a plot of the change in the fluorescence intensity versus logarthim of the S. Enteritidis concentration. c The displacement assay response against 107 cfu.mL-1 of S. Enteritidis, Salmonella Typhimurium, Staphylococcus aureus and Escherichia coli. The samples were excited at 470 ± 10 nm and the emission intensity 515 nm was used for the plots. The error bars represent the standard deviation of 3 measurements
Determination of the dissociation constant of SE54T aptamer
As shown from the displacement assay, the 29 mer truncated aptamer, SE54T (12–40) is involved in the S. Enteritidis binding and likely undergoes conformational change upon target binding. We further proceeded to calculate the binding affinity of SE54T to the S. Enteritidis. The percentage increase in the fluorescence intensity with increasing concentration of S. Enteritidis cells was plotted (Fig. S2). The saturation curve obtained was used to calculate the Kd of the SE54T-SE complex by non-linear regression fitting. The Kd value was found to be 3 nM, which is two times higher affinity towards S. Enteritidis compared to the original SE54 aptamer (Kd of 6.3 nM), confirming that the truncated aptamer makes more stable complex with S. Enteritidis.
Graphene oxide-based aptamer assay
Since the truncated aptamer SE54T has shown conformational change after binding to S. Enteritidis as well as high affinity compared with the full apatmer sequence, we have chosen it for the GO assay. As shown in Scheme 1. the GO assay was based on simple fluorescence on/off strategy. The fluorescein labeled aptamers were adsorbed on the surface of GO by π stacking interactions. This adsorption leads to complete quenching of the fluorescence due to fluorescence resonance energy transfer (FRET) from the fluorescein to GO in the absence of target. However, in the presence of target molecule, the specific aptamer binds to its target and dissociate from the GO surface due to the conformational change of the aptamer. This leads to a rapid increase in the fluorescence intensity.

Schematic diagram represents the working principle of the graphene oxide aptamer assay
Optimization of the graphene oxide/ aptamer ratio
With the aim to optimize the concentration of GO which gives the maximum quenching efficiency of the fluorescence, different GO concentrations were initially investigated. The optimization experiments were performed for the full aptamer sequence SE54 and the selected truncated sequence SE54T. As shown in Fig. 3a and b, in the absence of GO, a strong fluorescence peak of the fluorescein labelled aptamers were observed (red curves). Addition of increasing concentrations of GO solutions ranging from 0 to 50 μg.mL−1 to the fluorescence aptamers lead to significant quenching of the fluorescence (more than 90% when the GO concentration reached 20 μg.mL−1). No further decrease in the fluorescence by adding higher GO concentration is observed as shown in Fig. 3c. Plot of fluorescence intensity ratio against concentration of GO shows liner relationship from 0 to 20 μg.mL−1 (Figs. S3, S4). This indicates an ideal off state (low background signal). Therefore, the ratio of GO to SE54 or SE54T was selected to be 20 μg.mL−1 GO for 25 nM aptamer as optimum conditions for further experiments.

The fluorescence spectra of 25 nM SE54 (a) and SE54T (b) after incubation with different concentrations of GO. c Plot of the fluorescence intensity change versus the GO concentration. The fluorescence spectra were recorded by exciting at 470 ± 10 nm and the emission intensity 515 nm was used for the plots. The error bars represent the standard deviation of 3 measurements
Dose response of the graphene oxide aptamer assays
The optimized GO/aptamer ratio has been then used for the sensing experiment. We compared the behavior of the full aptamer sequence with the truncated aptamer. As shown in Fig. 4a and d, the off state of fluorecein-aptamer/GO complex was turned on by the addition of the S. Enteritidis. The fluorescence intensity gradually increased with increasing the number of cells from 102 to 107 cfu.mL-1. In order to determine LOD of the two aptamer-based assays, we plotted the calibration curves as the logarithm of the number S. Enteritidis cells vs % change in the fluorescence intensity (Fig. 4b and e). Linear relationship was observed for the two assays with LOD of 38 and 25 cfu.mL-1 for the full length SE54 and SE54T, respectively. The LOD was calculated from 3STD/m, where STD is the standard deviation of the aptasensor probe when no analyte was added and m is the slope of the straight line.These results indicate that the truncated sequence gave better sensitivity compared with the full aptamer sequence. Moreover, the LOD of the truncated aptamer is almost two-fold less than the previously reported sensor fabricated with the full length aptamer. This is attributed to the improved affinity of the aptamer by removing the non-essential nucleotides from the parent aptamer which can cause steric hindrance and decrease the affinity to the target. Various detection methods used for the detection of salmonella has been compared in Table 2. It is clear that our method showed higher sensitivity than most of the reported methods [24,25,26,27,28,29,30,31,32]. Wu et al. [33] have reported a LOD of 11 cfu.mL-1 using a colourimetric aptasensor which was little below our LOD (25 cfu.mL-1). However, this method requires several steps for the synthesis of ZnFe2O4/rGO nanostructures as well as several reagents such as TMB and hydrogen peroxide to generate the color which in turn increases the time and the cost of the assay. Thus, our method is much lower in cost, less reagents and simpler.

The fluorescence spectra of SE54F/GO platform (a) and SE54TF/GO platform (d) after incubation with different concentration of S. Enteritidis (a: blank, b:102, c: 103, d: 104, e: 105, f:106, g: 107 cfu.mL-1. b and e are the calibration plots of SE54F/GO and SE54TF/GO assays, respectively. c and f are the selectivity studies for both GO aptamer assays against 107 cfu.mL-1 SA, E. coli and ST. The fluorescence spectra were recorded by exciting at 470 ± 10 nm and the emission intensity 515 nm was used for the plots. The error bars represent the standard deviation of 3 measurements
Selectivity of the graphene oxide aptamer- based assay
The selective binding of SE54TF and SE54F aptamer assays to the S. Enteritidis cells was investigated by testing the platforms against other related bacteria such as ST, E.coli and SA. No considerable increase in the fluorescence intensity was obtained by these pathogens except ST compared to the response caused by the S. Enteritidis as can be seen in Fig. 4c and f. The cross reactivity with S. Enteritidis (25% increase in the fluorescence intensity) can be due to the similar structure of both species from the same family. Therefore, we believe that the GO-based fluorescence aptamer assay represents sensitive and selective platform for the detection of S. Enteritidis. Moreover, this method has several advantages over the other reported methods for the detection of S. Enteritidis such as SPR [34], electrochemical immunosensors [35] and gold nanoparticle-based DNA sensor [36] in terms of sensitivity, simplicity, capability of high throughput screening and multiplexing by introducing different fluorescent tags.
Testing of the SE aptamer-based platform in spiked milk samples
The response of the SE54T platform was tested for the detection of S. Enteritidis from the real samples. In order to do that, we spiked 1000, 5000 and 10,000 cfu.mL-1 of S. Enteritidis in 50% milk. The recovery percentages for each sample were tabulated in Table S1. These results show the good recovery percentages of S. Enteritidis and indicate minimal interference from the milk matrix. This confirms the possible applicability of the new assay for the detection of S. Enteritidis in real samples.
Conclusions
In this work, we have performed a mapping study to identify a shorter aptamer sequence which binds with high affinity to S. Enteritidis. Reducing the length of the aptamer by eliminating the non-binding region have doubled the binding affinity. The truncated aptamer was then exploited in a GO-based competitive displacement fluorescence assay for the detection of S. Enteritidis. The new analytical platform based on the truncated aptamer showed a LOD of 25 cfu.mL-1 which was two times lower than the platform fabricated using the full sequence. Moreover, the GO/aptamer- based method showed high selectivity against other related bacteria such as Salmonella Typhimurium, Staphylococcus aureus and Escherichia coli. Our results suggest that the elimination of non-binding region of the original aptamer by truncation leads to improvement of the binding affinity and consequently, less LOD for the sensor. Good recovery percentages of S. Enteritidis were obtained from spiked milk samples using this assay. The light scattering at very high concentration of bacteria is challenging. However, this can be managed by diluting the sample. This assay is easy to use, has the capability of high throughput screening and can be multiplexed to detect other pathogens.
References
Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M-A, Roy SL, Jones JL, Griffin PM (2011) Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis 17:7–15
Zhao X, Lin CW, Wang J, Oh DH (2014) Advances in rapid detection methods for foodborne pathogens. J Microbiol Biotechnol 24:297–312
Velusamy V, Arshak K, Korostynska O, Oliwa K, Adley C (2010) An overview of foodborne pathogen detection: in the perspective of biosensors. Biotechnol Adv 28:232–254
Day JB, Basavanna U, Sharma SK (2009) Development of a cell culture method to isolate and enrich Salmonella enterica serotype enteritidis from shell eggs for subsequent detection by real-time PCR. Appl Environ Microbiol 75:5321–5327
Chen J, Tang J, Liu J, Cai Z, Bai X (2012) Development and evaluation of a multiplex PCR for simultaneous detection of five foodborne pathogens. J Appl Microbiol 112:823–830
Primiceri E, Chiriaco MS, de Feo F, Santovito E, Fusco V, Maruccio G (2016) A multipurpose biochip for food pathogen detection. Anal Methods 8:3055–3060
Chen Z, Zhang K, Yin H, Li Q, Wang L, Liu Z (2015) Detection of Salmonella and several common Salmonella serotypes in food by loop-mediated isothermal amplification method. Food Sci Human Wellness 4:75–79
Law JW-F, Ab Mutalib N-S, Chan K-G, Lee L-H (2014) Rapid methods for the detection of foodborne bacterial pathogens: principles, applications, advantages and limitations. Front Microbiol 5:770
Song K-M, Lee S, Ban C (2012) Aptamers and their biological applications. Sensors (Basel, Switzerland) 12:612–631
Lau PS, Li Y (2013) Exploration of structure-switching in the design of aptamer biosensors, pp 69–92. In: Biosensors based on aptamers and enzymes. Springer
MacDonald J, Houghton P, Xiang D, Duan W, Shigdar S (2016) Truncation and mutation of a transferrin receptor aptamer enhances binding affinity. Nucleic Acid Ther 26:348–354
Kaur H, Yung LY (2012) Probing high affinity sequences of DNA aptamer against VEGF165. PLoS One 7:e31196
Alhadrami H, Chinnappan R, Eissa S, Rahamn AA, Zourob M (2017) High affinity truncated DNA aptamers for the development of fluorescence based progesterone biosensors. Anal Biochem 525:78–84
Chung C, Kim Y-K, Shin D, Ryoo S-R, Hong BH, Min D-H (2013) Biomedical applications of graphene and graphene oxide. Acc Chem Res 46:2211–2224
Jung JH, Cheon DS, Liu F, Lee KB, Seo TS (2010) A graphene oxide based immuno-biosensor for pathogen detection. Angew Chem Int Ed 49:5708–5711
Chang H, Tang L, Wang Y, Jiang J, Li J (2010) Graphene fluorescence resonance energy transfer aptasensor for the thrombin detection. Anal Chem 82:2341–2346
He Y, Wang Z-G, Tang H-W, Pang D-W (2011) Low background signal platform for the detection of ATP: When a molecular aptamer beacon meets graphene oxide. Biosens Bioelectron 29:76–81
Jayasena SD (1999) Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin Chem 45:1628–1650
Gao S, Zheng X, Jiao B, Wang L (2016) Post-SELEX optimization of aptamers. Anal Bioanal Chem 408:4567–4573
Cowperthwaite MC, Ellington AD (2008) Bioinformatic analysis of the contribution of primer sequences to aptamer structures. J Mol Evol 67:95–102
Zhou J, Soontornworajit B, Snipes MP, Wang Y (2011) Structural prediction and binding analysis of hybridized aptamers. J Mol Recognit 24:119–126
Maehashi K, Katsura T, Kerman K, Takamura Y, Matsumoto K, Tamiya E (2007) Label-free protein biosensor based on aptamer-modified carbon nanotube field-effect transistors. Anal Chem 79:782–787
Kolovskaya OS, Savitskaya AG, Zamay TN, Reshetneva IT, Zamay GS, Erkaev EN, Wang X, Wehbe M, Salmina AB, Perianova OV (2013) Development of bacteriostatic DNA aptamers for salmonella. J Med Chem 56:1564–1572
Lee S-C, Kim M-S, Yoo K-C, Ha N-R, Moon J-Y, Lee S-J, Yoon M-Y (2017) Sensitive fluorescent imaging of Salmonella enteritidis and Salmonella typhimurium using a polyvalent directed peptide polymer. Microchim Acta 184:2611–2620
Duan YF, Ning Y, Song Y, Deng L (2014) Fluorescent aptasensor for the determination of Salmonella typhimurium based on a graphene oxide platform. Microchim Acta 181:647–653
Zhu H, Zhao G, Wang SQ, Dou W (2017) Photometric sandwich immunoassay for Salmonella pullorum and Salmonella gallinarum using horseradish peroxidase and magnetic silica nanoparticles. Microchim Acta 184(6):1873–1880
Chen YY, Fang YC, Lin SY, Lin YJ, Yen SY, Huang CH et al (2017) Corona-induced micro-centrifugal flows for concentration of Neisseria and Salmonella bacteria prior to their quantitation using antibody-functionalized SERS-reporter nanobeads. Microchim Acta 184(4):1021–1028
Liu K, Yan X, Mao B, Wang S, Deng L (2016) Aptamer-based detection of Salmonella enteritidis using double signal amplification by Klenow fragment and dual fluorescence. Microchim Acta 183(2):643–649
Lei P, Tang H, Ding S, Ding X, Zhu D, Shen B et al (2015) Determination of the invA gene of Salmonella using surface plasmon resonance along with streptavidin aptamer amplification. Microchim Acta 182(1–2):289–296
Jia F, Duan N, Wu S, Dai R, Wang Z, Li X (2016) Impedimetric salmonella aptasensor using a glassy carbon electrode modified with an electrodeposited composite consisting of reduced graphene oxide and carbon nanotubes. Microchim Acta 183(1):337–344
Fei J, Dou W, Zhao G (2016) Amperometric immunoassay for the detection of Salmonella pullorum using a screen-printed carbon electrode modified with gold nanoparticle-coated reduced graphene oxide and immunomagnetic beads. Microchim Acta 183(2):757–764
Fei J, Dou W, Zhao G (2015) A sandwich electrochemical immunosensor for Salmonella pullorum and Salmonella Gallinarum based on a screen-printed carbon electrode modified with an ionic liquid and electrodeposited gold nanoparticles. Microchim Acta 182(13–14):2267–2275
Wu S, Duan N, Qiu Y, Li J, Wang Z (2017) Colorimetric aptasensor for the detection of Salmonella enterica serovar typhimurium using ZnFe2O4-reduced graphene oxide nanostructures as an effective peroxidase mimetics. Int J Food Microbiol 261:42–48
Waswa JW, Debroy C, Irudayaraj J (2006) Rapid detection of Salmonella enteritidis and Escherichia coli using surface plasmon resonance biosensor. J Food Process Eng 29:373–385
Melo AMA, Alexandre DL, Furtado RF, Borges MF, Figueiredo EAT, Biswas A, Cheng HN, Alves CR (2016) Electrochemical immunosensors for Salmonella detection in food. Appl Microbiol Biotechnol 100:5301–5312
Alocilja EC, Zhang D, Shi C (2013) AuNP-DNA biosensor for rapid detection of Serovar Enteritidis, pp 43–53. Advances in applied nanotechnology for agriculture. American Chemical Society
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The author(s) declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOCX 126 kb)
Rights and permissions
About this article

Cite this article
Chinnappan, R., AlAmer, S., Eissa, S. et al. Fluorometric graphene oxide-based detection of Salmonella enteritis using a truncated DNA aptamer. Microchim Acta 185, 61 (2018). https://doi.org/10.1007/s00604-017-2601-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00604-017-2601-9