
Abstract
Insulin resistance arising from Non-Alcoholic Fatty Liver Disease (NAFLD) stands as a prevalent global ailment, a manifestation within societies stemming from individuals’ suboptimal dietary habits and lifestyles. This form of insulin resistance emerges as a pivotal factor in the development of type 2 diabetes mellitus (T2DM). Emerging evidence underscores the significant role of hepatokines, as hepatic-secreted hormone-like entities, in the genesis of insulin resistance and eventual onset of type 2 diabetes. Hepatokines exert influence over extrahepatic metabolism regulation. Their principal functions encompass impacting adipocytes, pancreatic cells, muscles, and the brain, thereby playing a crucial role in shaping body metabolism through signaling to target tissues. This review explores the most important hepatokines, each with distinct influences. Our review shows that Fetuin-A promotes lipid-induced insulin resistance by acting as an endogenous ligand for Toll-like receptor 4 (TLR-4). FGF21 reduces inflammation in diabetes by blocking the nuclear translocation of nuclear factor-κB (NF-κB) in adipocytes and adipose tissue, while also improving glucose metabolism. ANGPTL6 enhances AMPK and insulin signaling in muscle, and suppresses gluconeogenesis. Follistatin can influence insulin resistance and inflammation by interacting with members of the TGF-β family. Adropin show a positive correlation with phosphoenolpyruvate carboxykinase 1 (PCK1), a key regulator of gluconeogenesis. This article delves into hepatokines’ impact on NAFLD, inflammation, and T2DM, with a specific focus on insulin resistance. The aim is to comprehend the influence of these recently identified hormones on disease development and their underlying physiological and pathological mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The extensive impact of insulin resistance caused by non-alcoholic fatty liver disease (NAFLD) is highlighted by its global prevalence. This condition is a result of people’s less-than-ideal food choices and lifestyle trends [1]. Among the many risk factors for developing type 2 diabetes mellitus (T2DM), insulin resistance due to NAFLD stands out [2]. It has been established that hepatokines are liver-secreted hormone-like substances. Despite their role in metabolism; recent findings suggest the participation of some these hepatokines, as endocrine messengers in the development of insulin resistance and ultimately, type 2 diabetes. [3]. Hepatokines constitute a category of substances released by the liver that function in the regulation of metabolism beyond hepatic tissues. Their principal roles include influencing adipocytes, pancreatic cells, muscles, and the brain, thereby exerting a substantial impact on body metabolism through signaling to target tissues [4, 5].
Within this comprehensive review, we delve into the intriguing realm of the most important hepatokines, each wielding a distinctive influence. These remarkable substances wield their effects by modulating the quantity of liver insulin receptors, exemplified by activin E [6], orchestrating a surge in glucose overload within liver or muscle cells, as demonstrated by Fibroblast growth factor 21 (FGF21) [7]. Furthermore, hepatokines such as Angiopoietin-like proteins (ANGPTLs) [8] orchestrate a metabolic shift towards fat.
As well as, hepatokines could potentially influence the molecular mechanisms that result in inflammation within the liver, adipose tissues, or muscles, for instance like Growth/Differentiation Factor-15 (GDF15), intricately navigate inflammatory pathways. This exploration illuminates the captivating interplay of hepatokines, unraveling their multifaceted roles in pathophysiological mechanisms [9]. In this regards, this article endeavors to unravel the intricacies of hepatokines, delving into their impact on NAFLD, inflammation, and ultimately, T2DM, with a specific emphasis on the intricate dynamics of insulin resistance. Through this exploration, we aim to elucidate the influence of these recently unveiled hormones on the pathogenesis of the mentioned diseases, offering valuable insights into their physiological mechanisms.
Insulin resistance, inflammation and hepatokines
As mentioned before, new research indicates that hepatokines may play a role as endocrine messengers in the progression of insulin resistance and, ultimately, the development of T2DM [3]. Insulin resistance denotes a diminution in the responsiveness of tissues to insulin, a hormone pivotal in facilitating glucose movement to muscles, adipose tissues, and the liver [10]. The impediment of insulin signal transduction, primarily attributed to mutations and polymorphisms in insulin receptors [11], stands as a predominant cause of insulin resistance. Another mechanism that might lead to insulin resistance is disruption of lipid storage in the skeletal muscles and liver. This insulin resistance has been linked to a number of metabolic disorders, including T2DM, atherosclerosis, and NAFLD [12].
Insulin resistance may manifest at multiple junctures along the intricate insulin signaling pathway, exerting a direct influence on the modulation of glucose levels, protein synthesis, and the onset of T2DM along with its associated complications. The intricacies of insulin resistance are elucidated through diverse molecular mechanisms [2, 13, 14].
On the other hand, hepatokines are acknowledged as a category of hormones instrumental in the physiological modulation of inflammation. The ensuing instances elucidate three scenarios wherein hepatokines influence the molecular pathways underpinning inflammation and insulin resistance. Illustratively, fetuin-A (FetA) facilitates the development of lipid-induced insulin resistance through its role as an endogenous ligand for TLR-4 [15,16,17,18]. Moreover, this protein exerts a pronounced impact on expediting the assimilation of exogenous fatty acids into cellular triglycerides [19]. Functioning as an upstream modulator, FetA plays a regulatory role in the polarization of M1 macrophages and the activation of TLR4 induced by free fatty acids in adipocytes. Consequently, FetA emerges as a potential novel therapeutic target for addressing T2DM mellitus and inflammation associated with obesity [20, 21]. Recent investigations have associated FGF21 with a spectrum of anti-inflammatory effects, contributing to the expanding body of research elucidating its advantageous implications for metabolic processes [7, 21]. Notably, FGF21 mitigates inflammation in diabetes by impeding the nuclear translocation of nuclear factor-κB (NF-κB) in adipocytes and adipose tissue under insulin-resistant conditions, concurrently augmenting glucose metabolism [7, 22].
Hepatic function and secretion: insights into essential dynamics
The energy metabolism of mammals intricately hinges upon nutrient intake, wherein the liver assumes a pivotal responsibility for assessing and orchestrating the utilization of these nutrients across various tissues. Glucose, along with other monosaccharides, stands as the predominant substrate for metabolism in living organisms. The liver, in its regulatory capacity, modulates blood glucose levels through dynamic interactions with the pancreas, which releases insulin to sustain a consistent blood glucose concentration. Furthermore, during phases of satiety, the liver actively assimilates blood glucose, converting and storing it in the form of glycogen [23].
Findings from proteomic studies involving both human subjects and rodents indicate that around 40% of the proteins originate from hepatic sources [24, 25]. With the help of hormones like hepatokines, myokines, adipokines, and neurokines, the liver is able to interact with other organs. These hormones elicit physiological effects, contributing to conditions such as atherosclerosis and insulin resistance [26]. The dynamic modulation of hepatokine levels during the evolution and manifestation of metabolic disorders significantly influences metabolic processes. Understanding the dynamic interactions between hepatokines and other organs is essential not only for unraveling their diverse physiological roles and potential implications in systemic health, but also for revealing metabolic disorders significantly influences metabolic processes [5, 27]. In this regard, the following information, summarized in Table 1, furnishes a comprehensive overview of diverse hepatokines and their influence on critical physiological aspects such as insulin resistance, liver function, lipid metabolism, metabolic conditions, and lifestyle factors.
Angiopoietin-like proteins (ANGPTL)
They derive their nomenclature from the resemblance of their protein domains to the angiopoietin family. These glycoproteins, secreted by hepatocytes, operate as vascular growth factors. Notably, a coiled-coil domain in their N-terminal facilitates subunit binding and the assembly of homo-oligomers. Furthermore, they feature a fibrinogen-like domain in their C-terminal, which binds to the Tie2 receptor, endowing them with a structural similarity to angiopoietin proteins [28, 29]. As a physiological function, these hepatokines assume a pivotal role in the modulation of white adipose tissue (WAT) function and the regulation of the lipoprotein lipase gene (LPLs) expression [30]. Located on the inner surface of capillaries, lipoprotein lipase is accountable for the hydrolysis of triglycerides into fatty acids, subsequently releasing them into the tissues that lipoproteins aim to reach [31]. The Golgi system experiences regulation by ANGPTL1-8, exerting inhibitory effects on LPLs expression at the post-translational level [30]. Within the ANGPTL protein family, ANGPTL2, 4, and 8 predominantly participate in inflammatory processes, while other members can also contribute to the overall inflammatory milieu [32].
As mentioned before, the ANGPTL protein family is involved in inflammatory processes in various bodily conditions. Although the first member of this family, ANGPTL1, has an unclear role in inflammation, it does interfere with inflammatory pathways in cancer. ANGPTL1 inhibits the integrin a1ß1/focal adhesion kinase (FAK)-Src/JAK/STAT3 signaling pathway by binding to integrin a1ß1 in liver cancer cells, thus suppressing metastasis and angiogenesis. Furthermore, ANGPTL1’s binding to integrin a1ß1 inhibits the expression of zinc finger protein SLUG, which has an inhibitory effect on lung cancer [33,34,35,36].
Moreover, ANGPTL2 plays a role in inflammatory responses and various metabolic disorders. This hepatokine, which is also released from adipose tissue, exerts a favorable impact on inflammation, obesity, and systemic insulin resistance in both humans and mice. Figure 2 shows, in a research study, recombinant ANGPTL2 was utilized to treat human umbilical vein endothelial cells, leading to the activation of Rac1 and NF-κB via integrin α5ß1, thereby enhancing the inflammatory pathway [33, 37]. Additionally, this treatment was applied to endothelial cells in atherosclerosis, where the interaction of this hepatokine with integrin α5ß1 resulted in increased degradation of IκBα and elevated secretion of pro-inflammatory cytokines and adhesive molecules such as TNF-α, interleukin 6 (IL-6), and intercellular adhesion molecule 1 (ICAM-1), as presented [33, 38, 39].
Within its structural framework, ANGPTL3 incorporates a distinctive epitope associated with lipoprotein lipase, thereby influencing the activity of lipoprotein lipase and modulating plasma triglyceride levels. The influence of ANGPTL3 on obesity, insulin resistance, and hyperlipidemia has been substantiated through rodent studies [40]. Subsequent investigations involving rodents elucidated that elevated ANGPTL3 expression correlates with increased plasma triglyceride levels and decreased levels of free fatty acids [41]. The ANGPTL3 protein was identified as a suppressor of LPLs in an in-vitro setting, resulting in consequential alterations to free fatty acid and triglyceride levels in the bloodstream [42]. Human studies have revealed that the inhibition of ANGPTL3 leads to a reduction in triglyceride levels [43]. Moreover, diabetic patients exhibit elevated serum levels of ANGPTL3 compared to non-diabetic individuals [8]. Investigations on mice have demonstrated the involvement of ANGPTL3 in the accumulation of triglycerides in white adipose tissue cells. By supplying glucose to accumulated adipocytes, ANGPTL3 KO mice have compensated for their tissue mass weight, which stayed similar to control mice [44]. Evidence suggests that the coiled-coil region of ANGPTL3 plays a pivotal role in suppressing the breakdown of very-low-density lipoproteins (VLDLs) [45]. The regulatory influence of liver X receptor (LXR) activity on ANGPLT3 concentrations has been documented as well [46]. Leptin and insulin, at the mRNA expression level, inhibit the production of ANGPTL3. Hormones like these cause hyperlipidemias and hyperfattyacidemia in those who are overweight, diabetic, or insulin-resistant [47]. Inhibiting ANGPTL3 leads to a downregulation of genes associated with the gluconeogenesis pathway and a reduction in the efficiency of insulin-mediated VLDL secretion [48]. Apart from the aforementioned functions, when ANGPTL3 binds to αVß3 integrin, it enhances the movement and attachment of endothelial cells, promoting angiogenesis and indicating the potential pro-inflammatory role of ANGPTL3. Additionally, a separate animal study demonstrated that upregulation of ANGPTL3 in mice with diabetic retinopathy led to increased levels of Bax, p53, VEGF, and inflammatory cytokines (IL-6 and IL-1B) [33, 49].
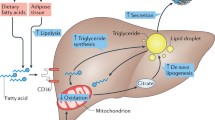
Regarding ANGPTL4, this protein displays a reduced molecular size in comparison to ANGPTL3, while yet maintaining its structural arrangement for LPL binding [50]. Its ability to increase liver cell lipid buildup contributes to T2DM [5]. This particular protein is alternatively recognized as Fasting-induced adipose factor (Fiaf) [4]. Predominantly secreted by adipose tissue and hepatocytes, ANGPTL4 levels exhibit an increase during periods of fasting. Catechamine-induced short-term fasting appears to trigger a signaling cascade that releases fatty acids and shifts metabolism toward fat use. In response to glucocorticoids, ANGPTL4 expression is stimulated during extended fasting, leading to fatty acid release. Consequently, the impact of this protein is more pronounced during extended fasting durations [51]. As depicted in Fig. 1, augmenting ANGPTL4 expression triggers the activation of the peroxisome proliferator-activated receptor (PPAR) signaling pathway, subsequently suppressed upon refeeding [52]. When studying mice with obesity, diabetes, and atherosclerosis, researchers found that their ANGPTL4 expression was much lower. [53]. Increased serum triglyceride levels are caused by the N-terminal of ANGPTL4, which inhibits LPLs. At the same time, this protein hinders triglyceride clearance, further increasing blood lipid levels. In contrast to its effects on adipocytes, an augmentation in free fatty acid (FFA) concentration in the blood is observed, emanating from ectopic sources such as the liver or muscle cells [5, 54]. ANGPTL4 exhibits a glucose-lowering effect and enhances glucose processing capabilities, as evidenced by numerous studies. It may be possible to increase insulin sensitivity using this [4, 27, 55]. Human studies involving individuals with T2DM have reported a decrease in the serum concentration of ANGPTL4 [56]. Additionally, ANGPTL4 facilitates lipid accumulation in adipocytes, contributing to steatosis [57]. Conversely, the genetic inactivation of ANGPTL4 has shown promise in substantially reducing the risk of developing coronary artery disease and diabetes [58]. Is there a correlation between insulin resistance and serum concentrations of ANGPTL4 or not? The contradictory results of the research on this protein make it impossible to draw any firm conclusions. It seems that it has a dual role even in inflammation [58]. Stem cells have a wide range of applications in treating cardiovascular diseases (CVD). One example is the use of ANGPTL4 derived from mesenchymal stem cells, which have been found to suppress β1/αVβ3 integrin signaling by inhibiting NF-κB (Fig. 2). This contributes to the polarization of inflammatory macrophages as an anti-inflammatory molecule [33, 59]. Moreover, in other study involving mice, it was observed that ANGPTL4 levels increased in cases of acute lung injury. Furthermore, the reduction of this hepatokine led to an increase in sirtuin1 (SIRT1), which has anti-inflammatory and antioxidant properties [33].

The impact of hepatokines on insulin signaling pathways. GLUT4: Glucose transporter type 4, IRS: insulin receptor substrate, JAK2: Janus Kinase 2, IGF-1: Insulin-like Growth Factor 1, FGF-21: fibroblast growth factor 21, ANGPTL: Angiopoietin-like protein, SELENOP: Selenoprotein P, PPARs: Peroxisome proliferator-activated receptors, LCN13: Lipocalin 13, PI3K: Phosphoinositide 3-kinases, LECT2: Leukocyte cell-derived chemotaxin-2, FST: Follistatin, FSTL1: Follistatin-like protein 1, ACC: Acetyl-coA carboxylase, AMPK: Adenosine monophosphate-activated protein kinase, GDF-15: Growth differentiation factor 15, SMOC1: Secreted modular calcium-binding protein 1, mTORC1: Mammalian target of rapamycin complex 1

The impact of hepatokines on inflammation pathways. ANGPTL: Angiopoietin-like protein, TNF-α: Tumor necrosis factor α, FSTL1: Follistatin-like protein 1, P38 MAPK: P38 mitogen-activated protein kinase, ROS: Reactive oxygen species, LECT2: Leukocyte cell-derived chemotaxin-2, NF-κB: Nuclear factor-κB, JNK: Jun N-terminal kinase, GDF-15: Growth differentiation factor 15, IGF-1: Insulin-like Growth Factor 1
Previous research findings suggest that the levels of ANGPTL5 may be linked to inflammation in both obese individuals and those with type 2 diabetes. In addition, the rise in this hepatokine is correlated with level of hs-CRP [33, 60].
ANGPTL6, on the other hand, lacks a binding domain for LPLs, distinguishing it from the preceding two variants of ANGPTL [28]. Its liver expression is predominate, with little in other tissues [61]. This protein enhances AMPK and boosts insulin signaling in muscle (Fig. 1). Additionally, it suppresses gluconeogenesis by inhibiting the expression of the glucose-6-phosphatase (G6P) gene, leading to a catabolic shift in sugar metabolism [29, 62]. Fasting insulin and blood sugar levels are two indicators that are directly correlated with ANGPTL6 [63]. As well as, this hepatokine is involved in the inflammatory processes associated with metabolic syndromes. For instance, mice that were induced with psoriasis displayed elevated levels of epidermal ANGPTL6, resulting in more intense skin inflammation [33, 64].
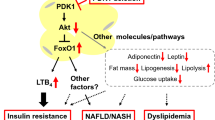
Regarding ANGPTL8, this protein possesses an LPL binding domain, albeit lacking a coiled-coil or fibrinogen C-terminal [28]. The ANGPTL8 protein is primarily secreted by adipose tissue and liver cells [65]. The inhibitory effect of this protein on LPL is greatly influenced by the concentration of ANPGTL3 and 4. It emerges as a potential biomarker for the identification and treatment of metabolic irregularities on a broad scale [4]. The impact of ANGPTL8 on both NAFLD and insulin resistance remains a subject of discourse. Arguments posit that ANGPTL8 may enhance insulin sensitivity by directly affecting Akt phosphorylation [66]. Decreasing ANGPTL8 levels is associated with a reduction in insulin resistance [67]. However, polymorphism of this protein is implicated in the development of NAFLD [68]. It has been reported that overexpression of ANGPTL8 improved insulin sensitivity and glucose tolerance and decreased fasting blood glucose levels in high-fat diet/streptozotocin-induced diabetic and db/db mice. In fact, ANGPTL8 promoted glucose metabolism via the inhibition of expression of gluconeogenesis-related genes [phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase catalytic subunit (G6PC)] by activating the AKT signaling pathway [69].
It is important to note that the final member of this family also plays a role in acute inflammatory conditions by acting as an anti-inflammatory agent. For instance, in individuals with acute infections or in laboratory mice with stimulated hepatocytes, ANGPTL8 controls the NF-κB signaling pathway by promoting the targeted autophagic breakdown of IKKγ [33, 70]. Zhang et al. [71] ‘s study expanded on the function of selective autophagy in fine-tuned inflammatory responses and proposed the ANGPTL8/p62-IKKγ axis as a negative feedback loop that controlled NF-κB activation.
Despite various potential explanations and connections between the members of this family and inflammatory pathways, the involvement of the ANGPTL family in inflammation remains uncertain and requires additional research.
Fibroblast growth factor 21 (FGF21)
Various tissues, such as the liver (hepatocytes), adipose tissue, pancreas, heart, and brain, release this protein [7, 72]. Fasting-induced metabolic changes in the liver, such as resistance to growth hormone (GH), fatty acid oxidation, ketogenesis, and gluconeogenesis, are tightly regulated by FGF21, as a physiological function [73]. Mice treated with FGF21 have improved insulin and leptin sensitivity, decreased hepatic steatosis, increased energy expenditure, and decreased sugar and alcohol consumption [4, 74, 75].
FGF21 primarily promotes liver fatty acid oxidation while blocking lipogenesis and gluconeogenesis [76]. Second, by reducing the severity of hyperlipidemia and hyperglycemia, FGF21 indirectly affects the liver. To do this, the following mechanisms are activated: mitochondrial activity is enhanced, fatty acid oxidation is facilitated, adipose tissue lipolysis is stimulated, energy is dissipated as heat, and glucose uptake and utilization are improved. Additionally, it influences several areas of the hypothalamus and hindbrain to encourage thermogenesis and energy expenditure as well as an aversion to sweets and alcohol intake [76]. Furthermore, there is a conjecture that FGF21 resistance in both rats and humans is attributed to obesity and insulin resistance [18]. Clinical trials involving FGF21 analogues indicate an elevation in high density cholesterol (HDL) and a reduction in plasma triglyceride concentrations, although they do not exert any discernible impact on insulin sensitivity or glucose levels [77].
The metabolic impacts attributed to FGF21 are acknowledged to arise from its interactions with various tissues [78]. FGF21 directly communicates with adipose tissue, enhancing insulin sensitivity [4]. Simultaneously, it stimulates the central nervous system to elevate calorie expenditure and facilitate weight loss [79, 80].
FGF21, recognized as an insulin-dependent hormone in humans [72], also plays a pivotal role in enhancing glucose balance by safeguarding β cells. FGF21 is believed to prevent β-cell dysfunction and cell death in vivo and in vitro through its inhibition of lipid accumulation in islet cells. As illustrated in Fig. 1, this likely occurs through the activation of the PPARδ/γ and AMPK-acetyl coenzyme A carboxylase (ACC) signaling pathways [81]. Beyond its reliance on insulin-dependent mechanisms for improving glucose homeostasis, FGF21 independently facilitates glucose uptake in adipocytes [72]. In addressing systemic insulin resistance, FGF21 demonstrates the capacity to reduce serum insulin levels and enhance insulin sensitivity [72]. In-depth investigations into the precise mechanisms underlying FGF21’s enhancement of hepatic insulin sensitivity involved administering FGF21 to mice. They discovered that FGF21 can enhance insulin sensitivity via blocking liver mTORC1 (Fig. 1). FGF21-deficient mice consistently exhibit heightened hepatic insulin resistance and activation of mTORC1 [82]. Furthermore, FGF21 targets subcutaneous fat tissue as a crucial component to control systemic insulin sensitivity. To prevent systemic insulin resistance in vivo, FGF21 can encourage the growth of subcutaneous fat and raise adiponectin levels in subcutaneous fat [72].
In summary, FGF21 provides protection against T2DM by improving glucose regulation [83]. Noteworthy is its demonstrated anti-inflammatory prowess [22]. Elevated FGF21 in insulin-sensitive obese individuals worsens the polarization of M2 macrophages and controls adiponectin levels in subcutaneous adipose tissue. As well as, elevating the concentration of this growth factor impacts the transforming growth factor-β (TGF-β) signaling pathway and inhibits adipogenesis by influencing the Smad3 pathway [84, 85]. As a result, because of its effects on hepatocytes, the brain-liver axis, and adiponectin synthesis in adipose tissue, FGF21 protects against hyperglycemia, dyslipidemia, non-alcoholic steatohepatitis (NASH), and other comorbidities [86]. FGF21 emerges as a promising therapeutic avenue for the management of T2DM and NAFLD [87].
Fetuin-A
Classified as α2-Heremans-Schmid-glycoprotein, Fetuin-A is a complex glycoprotein belonging to the cystatin protease inhibitor superfamily. Predominantly synthesized by hepatocytes and adipose tissue, this protein establishes a novel nexus linking obesity, inflammation, and insulin resistance [88, 89]. Fetuin-A, a plasma protein, regulates multiple physiological processes, including cellular protein metabolism, acute inflammatory responses, neutrophil and platelet release, lymphocyte activation, and binding to fatty acids, thyroid hormones, and calcium ions [89]. It serves as a natural antagonist to the insulin-stimulated insulin receptor tyrosine kinase, impeding the autophosphorylation of the insulin receptor and subsequent downstream signaling, as evidenced in laboratory investigations [90, 91]. Notably, as showed in Fig. 1, this hepatokine has been observed to bind to the receptor tyrosine kinase (RTK) at a distinct site from the insulin binding region; however, the precise domain of fetuin-A implicated in this interaction remains unclear [92]. Numerous investigations suggest a positive correlation between heightened levels of circulating fetuin-A and insulin resistance in human subjects [93]. Consequently, this protein emerges as a prospective independent risk factor in the predisposition to T2DM [94,95,96]. Through its modulation of adiponectin levels in lipid-induced inflamed adipocytes via the Wingless-related integration site (Wnt)-PPARγ pathway, Fetuin-A potentially instigates insulin resistance [88, 97]. This cascade of events underscores the plausible role of Fetuin-A in the pathogenesis of type 2 diabetes. Notably, there exists a consensus within the scientific community that NAFLD can be a precursor to insulin resistance and the subsequent onset of T2DM [18, 98]. Regardless of adiposity, Fetuin-A is elevated in NAFLD, suggesting a link between fatty liver and insulin resistance. [93, 99]. The pro-inflammatory or anti-inflammatory effects of this hepatokine are dependent on the particular clinical setting in which it is activated, thereby displaying dualistic inflammatory properties [89, 92, 100]. This molecule, renowned for its proinflammatory attributes, assumes a role in the development of insulin resistance. Additionally, Fetuin-A demonstrates neuroprotective properties and plays a significant part in mitigating inflammation in conditions such as sepsis and autoimmune diseases [100]. In instances of severe systemic inflammation, the protective role of Fetuin-A is ascribed to its capacity to impede the release of high mobility group box protein 1 (HMGB1), induced by pathogen-associated molecular patterns (PAMPs) [89]. As well as, Fig. 2 displays this hepatokine enhances the polarization of M1 macrophages and boosts the release of pro-inflammatory cytokines like interleukin-1β, Tumor Necrosis Factor α (TNF- α), and IL-6 [101].
Follistatin (FST)
This glucosylated plasma protein can bind and neutralize TGF-β family members [102]. Nowadays, 5 types of this family are identified [103]. The gene responsible for its expression is discernible in diverse tissues, including the pituitary gland, placenta, ovary, testis, brain, and skeletal muscle [104, 105]. In the human system, the predominant source of circulating FST is the hepatocytes, with its production and release being augmented in response to an elevated ratio of glucagon to insulin [106]. FST primarily operates through autocrine and paracrine signaling pathways [107, 108]. The manifold physiological roles ascribed to FST encompass the regulation of follicle-stimulating hormone (FSH) production in the pituitary, facilitation of ovarian follicle maturation, control of spermatogenesis, maintenance of liver homeostasis, facilitation of wound repair, and responsiveness to inflammatory stimuli [107]. Emergent evidence suggests a potential involvement of this hepatokine in insulin resistance and mild inflammation. However, it appears that plasma FST levels exhibit a closer association with metabolic disruptions than with low-grade inflammation [104].
Individuals with T2DM show a moderate increase in plasma FST levels, yet further investigations are imperative to elucidate whether the elevated plasma levels constitutes a causative factor or a consequence of metabolic irregularities in T2DM [104, 109]. A recent study has presented findings indicating that heightened circulating FST levels are linked to an increased susceptibility to developing type 2 diabetes, owing to their contribution to insulin resistance in adipose tissue [105]. Here, FST secretion by the liver causes insulin resistance and white adipose tissue fat breakdown. Increased levels of glycerol and non-esterified fatty acids in the bloodstream cause the liver to produce glucose uncontrollably, which ultimately results in glucose intolerance [105, 110]. Follistatin possesses the potential to influence insulin resistance and inflammation by virtue of its interaction with members of the TGF-β family. Thus, Fig. 2 presents the expression of FSTL1 is related to the activity of the NF-κB signaling pathway. Also, FSTL1 leads to the expression of various chemokines and inflammatory cytokines that are related to the NF-kB signaling pathway. Among them, CCL-2/MCP-1, TNF-α, CXCL8/IL-8, IL-1β and IL-6 can be mentioned [103]. Moreover, FSTL is attributed to its recognized capability to bind and counteract both myostatin and activin A within the circulatory system [111]. Notably, activin A, emanating from epicardial adipose tissue (in addition to liver) in individuals with T2DM, impedes insulin function by instigating the production of miR-143 in cardiomyocytes. This specific miRNA, in turn, suppresses the Akt pathway by diminishing the levels of oxysterol-binding protein–related protein 8 (ORP8), a recently identified regulator of insulin activity (Fig. 1) [112]. Myostatin, commonly acknowledged for its potent control over muscle growth and size, is implicated in metabolic processes. Although research indicates a potential correlation between myostatin and insulin resistance, the intricate details of this association remain under investigation [113]. Overall, Serum levels of FST are generally higher in those with T2DM. The underlying process, on the other hand, is still a mystery.
Leukocyte cell-derived chemotaxin-2 (LECT2)
This protein exhibits hormone-like properties and was initially identified as a chemokine influencing the regulation of neutrophil movement [114]. Subsequently, it garnered further characterization as chondromodulin II (CHM2), attributed to its capacity to stimulate proteoglycan synthesis by chondrocytes and facilitate cartilage growth [115]. Substantial progress has been achieved in comprehending its multifaceted roles, spanning liver regeneration, immune system modulation, bone development, neuronal growth, glucose regulation, metabolic syndrome, cancer, and amyloidosis [116]. While hepatocytes constitute the primary source of production and release of LECT2 into the bloodstream, its presence is also discernible in diverse cell types, encompassing vascular endothelial cells, smooth muscle cells, cerebral nerve cells, and adipocytes [117, 118]. LECT2, identified as a hepatokine, has been the subject of research establishing a direct correlation between hepatic LECT2 mRNA levels and body mass index (BMI). This association implies that elevated mRNA levels of hepatic LECT2 are associated with the severity of obesity in humans, potentially leading to insulin resistance in skeletal muscle [119, 120]. Glucose and insulin loading tests on Lect2–/– mice revealed reduced blood glucose levels following glucose or insulin administration. Furthermore, Lect2–/– mice exhibited heightened insulin-stimulated Akt phosphorylation specifically in skeletal muscle, with no discernible impact in the liver or adipose tissue. These observations suggest that the absence of the hepatokine LECT2 enhances insulin sensitivity in rodent skeletal muscle [119]. Moreover, as displayed in Fig. 1, the overproduction of LECT2 in the liver may contribute to JNK phosphorylation, resulting in insulin resistance in the skeletal muscle of obese individuals. However, the precise mechanism underlying how LECT2 facilitates JNK phosphorylation remains incompletely elucidated [120]. Additionally, LECT2 is involved in the body’s inflammation response by attaching to cell surface receptors like CD209a, Met, and Tie1. Additionally, in NAFLD, this hepatokine enhances the polarization of M2 macrophages and influences liver inflammation by impacting the JNK signaling pathway (Fig. 2) [121]. In summary, LECT2 emerges as a causative factor in inducing insulin resistance in skeletal muscles.
Hepassocin
Hepassocin, also recognized as hepatocyte-derived fibrinogen-related protein 1 (HFREP1) or fibrinogen-like protein 1, stands out as a specialized factor exerting mitogenic effects, particularly fostering the proliferation of liver cells (in an autocrine manner) [122,123,124]. Its primary source of secretion is the liver, but noteworthy findings indicate its expression within brown adipose tissues as well. Following liver damage, some signals stimulate an upregulation of its expression in brown adipose tissues [125]. While an investigation observed elevated levels of hepassocin in overweight or obese individuals compared to those with normal weight, the precise nature of the association between hepassocin and obesity remains elusive [126, 127]. As outlined in a study, this hepatokine assumes a pivotal role in the development of NAFLD, contributing to hepatic lipid accumulation through the activation of an ERK1/2-dependent mechanism [128]. Conversely, a reduction in hepassocin levels correlates with heightened insulin sensitivity, achieved through the modulation of ERK1/2 activity within the liver. This implies that elevated hepassocin concentrations may escalate the susceptibility to insulin resistance and diabetes [129]. Furthermore, hepassocin instigates insulin resistance in skeletal muscle cells through a 396 EGFR/JNK-mediated mechanism [130]. Nevertheless, conflicting perspectives arise from certain studies, challenging the earlier postulates. One investigation suggests a potentially beneficial impact of hepassocin on liver fat accumulation, coupled with a partial mitigation of hepatic apoptosis, fibrosis, and inflammation through the inhibition of oxidative stress. If the liver is injured, an increase in hepassocin in the liver can protect the liver against inflammation, steatosis, fibrosis and cell death. Therefore, the production of inflammatory cytokines such as IL-1β, IL-6, and TNFα increased in the mice where the expression of hepasosin was knocked down (Fig. 2) [121]. This, in turn, proved instrumental in preventing the development of steatohepatitis in mice [131].
Secreted modular calcium-binding protein 1 (SMOC1)
SMOC-1, secreted by hepatocytes, encompasses one N-terminal follistatin-like (FS), one extracellular calcium (EC), two thyroglobulin-like (TY) domains, and a distinctive domain lacking known homologs. Its expression is widespread across diverse tissues, frequently positioning it in proximity to the cell’s basement membrane [132, 133]. This hepatokine demonstrates interaction capabilities with laminins [132], C-reactive protein (CRP), fibulin-1, vitronectin [134], transglutaminase 2 [135], and tenascin-C [132, 136]. These interactions substantiate its involvement in integrin-matrix interactions and cell adhesion [137]. Through an acute intraperitoneal injection in mice, SMOC1 demonstrated the capacity to enhance insulin sensitivity and regulate glucose without influencing insulin secretion. The mechanism underlying its favorable glycemic effects involves the inhibition of adenosine 3′,5′-cyclic monophosphate (cAMP)–cAMP-dependent protein kinase (PKA)–cAMP response element–binding protein (CREB) signaling in the liver. Figure 1 depicts, this, in turn, led to a downregulation of gluconeogenic gene expression and a suppression of hepatic glucose production [138]. Additionally, individuals characterized by obesity and insulin resistance exhibited lower circulating levels of SMOC1, with a discernible association with both hepatic and systemic insulin sensitivity [138]. Overall, SMOC1 seems to manifest beneficial effects in the context of T2DM. In addition, SMOC1 is involved in the inflammatory processes of kidney diseases. A prior study showed that interleukin 1 beta reduces the expression of SMOC1 in animal mesangial cells by stimulating the production of nitric oxide (NO). Consequently, blocking the function of nitric oxide synthases results in an elevation of SMOC1 expression, exacerbating inflammation and fibrin deposition in the animal’s glomerulus. Furthermore, decreasing SMOC1 expression reduces the activity of the TGF-β signaling pathway, subsequently leading to a decline in the activity and expression of the Smad gene [139].
Insulin-like growth factor 1 (IGF-1)
This hepatokine, referred to as somatomedin C, is a hormone exhibiting structural similarities to insulin and holds pivotal significance in the growth processes during childhood. The liver (hepatocytes) serves as the primary site for IGF-1 synthesis, a response elicited by the stimulation of GH. The majority of IGF-1 is bound to one of six binding proteins (IGF-BP) [140]. IGF-1 exerts a comprehensive influence on systemic body growth, displaying growth-promoting effects across diverse cell types such as skeletal muscle, cartilage, bone, liver, kidney, nerve, skin, hematopoietic, and lung cells. In addition to its insulin-like effects, IGF-1 plays a regulatory role in cellular DNA synthesis [141]. GH engages with the cell-surface GH receptor (GHR), a homodimeric transmembrane protein. As showed in Fig. 1, this interaction initiates signal transduction by recruiting and activating cytosolic Janus Kinase 2 (JAK2) [142].
In individuals with T2DM, IGFBP1 exhibits an elevation and possesses the capacity to counteract insulin’s hypoglycemic effects by inhibiting IGF1 [143]. There exists compelling evidence indicating that insulin resistance, an increased susceptibility to metabolic syndrome and cardiovascular disease, along with diminished plasma levels of IGF1, collectively serve as prognostic indicators for the onset of type 2 diabetes [144,145,146]. Furthermore, certain studies propose that the modulation of IGF1 significantly influences glucose metabolism in mice subjected to a regular or high-fat diet. For instance, the systemic overexpression of IGFBP1 led to heightened plasma insulin levels, effectively mitigating the hypoglycemic impact of endogenous IGF1. However, this intervention also induced a reduction in insulin-stimulated glucose transport and glycogen synthesis in skeletal muscle, indicative of a subtle degree of insulin resistance [147]. Investigations have also unveiled a reduction in IGF levels among individuals afflicted with NAFLD and obesity [148]. A noteworthy observation arises from mice lacking Stat5a/b specifically in the central nervous system, denoted as Stat5NKO mice. These mice exhibit pronounced obesity, accompanied by insulin resistance, hyperphagia, hyperleptinemia, and a diminished capacity to respond thermally to cold stimuli [149]. In the realm of energy homeostasis regulation, a prominent player is UCP1 within Brown Adipose Tissue (BAT) activity. Comparative analyses between BAT from AAV-Igf1 mice (subjected to adeno-associated virus (AAV)-mediated delivery of IGF-I cDNA) and control mice (those injected with empty AAV) elucidated a marked augmentation in UCP1 expression, as discerned through Western blotting assays [150].
Robust neuroanatomical and functional evidence substantiates the involvement of the sympathetic nervous system (SNS) in the innervation and regulation of both WAT and BAT, orchestrating processes essential for thermoregulation and the generation of beige adipocytes [151]. The pivotal protein UCP1 in BAT, crucial for initiating thermogenesis in response to dietary and cold stimuli, is subject to control by SNS activity [152]. Intriguingly, the aforementioned findings suggest that the central activity of IGF1 can enhance central sympathetic outflow, consequently influencing UCP1 expression [150]. Beyond its role in improving insulin sensitivity and glucose tolerance, as well as fostering energy expenditure through thermogenesis, central IGF1 is implicated in the regulation of appetite [150]. Notably, administration of IGF1 has been associated with a boosted insulin sensitivity and lowered blood glucose levels in both individuals with T2DM and those without diabetes [150, 153, 154]. Moreover, Obese individuals have enlarged fat cells that, in collaboration with immune cells, elevate the production of pro-inflammatory cytokines like IL-6 and TNF-α. In such cases, the rise in serum IGF1 levels can serve as an anti-inflammatory agent and help regulate the levels of inflammatory factors (Fig. 2) [155].
Growth differentiation factor 15 (GDF15)
The protein secreted by hepatocytes, also known as macrophage inhibitory cytokine-1 (MIC-1), is a constituent of the TGF-β superfamily [9, 156]. In physiological condition, its regulatory influence extends to hematopoietic growth, energy equilibrium, adipose tissue metabolism, somatic growth, bone remodeling, and responsiveness to stress signals [9]. GDF15 assumes a multifaceted role in various pathological conditions, encompassing cancer, cardiometabolic disorders, etc. [9]. While GDF15 is typically absent in reproductive organs, it can be induced in numerous cell types under stress conditions [156]. A critical aspect of its operation is the regulation of energy balance, which is accomplished by increasing energy expenditure and decreasing body weight gain [4].
In mice with MIC-1/GDF15-induced anorexia/cachexia syndrome, glucose, insulin, and IGF-1 levels were lower [157]. In the context of NAFLD and other chronic liver disorders, there is a proportional elevation of GDF15 levels corresponding to disease severity and the presence of steatotic hepatitis. The augmentation of GDF15, either through overexpression or the introduction of recombinant GDF15, proves efficacious in ameliorating NAFLD and mitigating obesity [158]. In the milieu of obesity, the activation of p53 in adipose tissue contributes to the secretion of pro-inflammatory cytokines, insulin resistance, and the onset of diabetes. Concurrently, it mediates the expression of GDF15 within adipose tissue [159]. Also, in cases of insulin resistance or obesity, the integrative stress pathway and mitochondrial unfolded protein response (UPR) become active, resulting in the attachment of CHOP and ATF4 to the promoter region, this in turn triggers the transcription of GDF15 [160]. As well as, GDF-15 is intricately involved in lipid catabolism, heat generation, and oxidative metabolism. Its presence demonstrates a preventive effect against obesity and insulin resistance arising from dietary or genetic factors. In humans, there is a direct correlation between the expression of the GDF15 gene and the expression of genes associated with inflammation. It is also plausible that GDF15 may be linked to chronic low-grade inflammation, although the precise molecular mechanism for this connection has not yet been determined. When obese mice were given GDF15 antibody, there was an increase in inflammatory markers such as IL-6, CD-68, Tnf, monocyte chemoattractant protein-1 (Mcp1), and F480 in adipose tissue (Fig. 2). However, more research is needed to fully understand the relationship between GDF15 and inflammatory factors. Consequently, GDF15 emerges as a promising therapeutic target for addressing obesity and insulin resistance [161].
Lipocalin-13 (LCN13)
The lipocalin family encompasses a diverse array of small secreted proteins, typically comprising 160–180 amino acids and featuring an N-terminal signal peptide. LCN13 is produced in various tissues such as the hepatocytes, epididymis, pancreas, and skeletal muscle, and it is released into the blood circulation [162]. The distinctive biological activity exhibited by LCN family members is intricately linked to their capacity to selectively bind to diminutive hydrophobic molecules, including phospholipids, fatty acids, steroids, retinol, and pheromones. Research has illuminated the expansive role of LCN family members in governing chemical communication, reproduction, immunological responses, and the progression of cancer in rats. Recent revelations underscore the newfound significance of LCNs in modulating food metabolism and insulin sensitivity among individuals grappling with obesity. Among these, LCN13 stands out, being released into the circulation of mice and expressed by various organs such as the liver, pancreas, epididymis, and skeletal muscle [163]. Notably, LCN13 levels exhibit an inverse correlation with obesity, and interventions involving LCN13 in mice afflicted with hereditary or diet-induced obesity have demonstrated a reversal of LCN13 deficiency. This reversal, in turn, leads to improvements in insulin resistance, glucose intolerance, hepatic steatosis, hyperglycemia, and hyperinsulinemia. Mechanistically, LCN13 directly impedes hepatic lipogenesis and gluconeogenesis in hepatocytes, while concurrently promoting fatty acid β-oxidation. LCN13 also enhances adipocytes’ responsiveness to insulin. A thorough exploration of the underlying processes contributing to LCN13’s anti-diabetic and anti-steatotic effects is currently a subject of discourse [163].
Concerning glucose metabolism, the expression and secretion patterns of LCN13 in mice exhibit variability in response to alterations in metabolic conditions. There is an inconsistency in LCN13 expression, which diminishes in the context of obesity. Mice subject to genetic (db/db) or high-fat diet (HFD) conditions display a substantial reduction in circulating LCN13 levels, contributing to the progression of obesity. Remarkably, hepatic LCN13 operates in an autocrine/paracrine manner, exerting inhibitory effects on glucose synthesis within the liver. Even in the absence of insulin, LCN13 facilitates glucose absorption in adipocytes while concurrently suppressing glucose synthesis and the expression of key gluconeogenic genes, such as phosphoenolpyruvate carboxykinase and glucose-6-phosphatase, in primary hepatocytes. These observations suggest an alternate, insulin-independent mechanism through which LCN13 modulates glucose metabolism [164].
Shifting focus to lipid metabolism, there is a plausible mechanism suggesting that LCN13 plays a preventive role in obesity-related hepatic steatosis. This preventive action is primarily achieved through the enhancement of fatty acid β-oxidation and the concurrent reduction of lipogenesis within the liver (Fig. 1). LCN13 and its analogs exhibit therapeutic potential for the treatment of T2DM and NAFLD, owing to their dual properties of anti-steatosis and anti-diabetic effects [163]. It is hypothesized that LCN13 orchestrates lipid metabolism by upregulating the expression of carnitine palmitoyltransferase-1α (CPT1α) in the liver, while concurrently downregulating the expression of key lipogenic genes such as PPARγ and carbohydrate response element binding protein (ChREBP) (Fig. 1). This dual regulation serves to mitigate lipogenesis and augment fatty acid β-oxidation [165]. In the context of obese mice, LCN13 demonstrates a notable reduction in hepatic steatosis without a concomitant impact on adiposity, suggesting a preferential inhibition of lipogenesis and promotion of fatty acid oxidation specifically within hepatocytes, as opposed to adipocytes. Additionally, LCN13 directly facilitates glucose absorption by adipocytes through pathways that are both insulin-dependent and insulin-independent [164]. Although the level of LCN13 expression through the effect of Akt S473 phosphorylation and Glut4 expression can affect the insulin signaling pathway in the liver and skeletal muscles, its effect on the expression of inflammatory cytokines has not been observed [166].
Tsukushi (TSK)
TSK, secreted by hepatocytes, stands as an atypical member within the small leucine-rich proteoglycan family, exerting regulatory influence over developmental processes in diverse organisms [167]. TSK has been associated with the process of neurogenesis, the formation of the anterior commissure, the development of the eyes and inner ear, as well as bone growth and maintaining liver homeostasis [168]. This hepatokine, induced by both obesity and exposure to cold conditions, emerges as a significant player in the orchestration of physiological responses. The prevalence of liver steatosis, commonly associated with obesity and acute cold exposure, exhibits a high correlation with circulating TSK levels in various mouse models. Intriguingly, both inflammation and endoplasmic reticulum stress, implicated in excessive hepatic lipid accumulation, serve as stimulants for the expression of TSK in the liver [169]. A study by Wang et al. has demonstrated that the deletion of TSK amplifies sympathetic innervation and thermogenesis in BAT, contributing to a protective effect against diet-induced obesity and an enhancement in glucose regulation in mice [170]. TSK deficiency also heightens adrenergic action and augments thermogenic stimulation in brown fat by promoting the phosphorylation of hormone-sensitive lipase (HSL) and Protein Kinase A (PKA) substrates [171].
The interplay between Tsk and inflammation has been elucidated, with Lipopolysaccharide (LPS), a component of gram-negative bacteria, demonstrating an ability to elevate TSK levels in mice. This observation suggests a direct link between inflammation and the induction of Tsk expression [172]. Notably, deficiency in TSK has been associated with an increase in protein levels of UCP1 [169]. Liver cells respond to proinflammatory cytokines, such as TNF-α, IL-1β, and IFN-γ, collectively promoting the transcription of TSK [169]. The initial surge in TSK levels may serve a protective role in stressed liver cells by mitigating cholesterol influx into the liver. However, the prolonged elevation of TSK observed in conditions like obesity and NAFLD raises concerns about its potential contribution to atherosclerosis, potentially attributed to the chronic constriction of reverse cholesterol transport [172].
Adropin
Hepatocytes and brain are two of the many tissues that express adropin, a small peptide with 76 amino acids. Its presence has been noted in proteins of the pancreas, heart, blood vessels, and kidneys as well [173,174,175]. This peptide exhibits a decrease in response to heightened lipid availability in the liver. Furthermore, individuals with obesity and T2DM manifest a decline in serum adropin levels [5]. Mouse hepatic steatosis reduces energy homeostasis-associated gene (ENHO) gene expression, which encodes adropin [176]. Conversely, in hepatic steatosis induced by a lipid-rich diet, an upregulation of adropin expression is observed [177]. An intriguing observation reveals a positive correlation between adropin and phosphoenolpyruvate carboxykinase 1 (PCK1), a pivotal regulatory point in gluconeogenesis [178]. In addition, Adropin is used as a therapeutic stimulus to increase glucose tolerance and insulin sensitivity in rats induced by high-fat diet [179]. Furthermore, aside from its involvement in the body’s metabolic processes, adropin also has a function in reducing inflammatory factors in diabetes. Adropin is able to prevent the attachment of THP1 monocytes to Human Umbilical Vein Endothelial Cells (HUVEC) cells by affecting the TNF-α signaling pathway, thus exhibiting its anti-inflammatory properties (Fig. 2) [180].
Activins
Activins, members of the TGFβ superfamily, constitute a diverse group of proteins with multifaceted effects on various tissues. Traditionally perceived as heterogeneous, these proteins consist of multiple βA and βB subunits interconnected by disulfide bridges. Their pivotal role in the formation of gonadal follicles was initially discerned. Recent investigations have unveiled two additional subunits, namely βE and βC, characterized by sequences akin to their classical counterparts. Intriguingly, despite their initial association with gonads, it has been established that the hepatocytes serves as the primary source of activins [181, 182]. This protein instigates a cascade of signaling events by binding to the Activin A receptors type I (ActRI) and type II (ActRII), characterized as serine-threonine kinases. These signals subsequently culminate in the activation of genes with diverse functionalities [183].
In 2023, Liu and colleagues uncovered that the expression of Activin A in the liver led to a decrease in inflammation, expansion of hematopoietic stem cells, reduction of liver steatosis, decreased fat accumulation and lower levels of circulating cholesterol. These findings suggested that these effects collectively contribute to the observed protection against atherosclerosis and metabolic related diseases [184]. Moreover, in murine models, the analysis of activin E mRNA levels within hepatocytes’ cytosol suggests a plausible involvement in both exacerbating insulin resistance associated with obesity and facilitating thermogenesis in brown adipose tissue [185]. In a study, it has been shown that overexpression of hepatic activin E led to the activation of thermogenesis by upregulating Ucp1 in both white and brown adipose tissues. Furthermore, hepatic activin E-transgenic mice showed enhanced insulin sensitivity. Additionally, experiments conducted in vitro indicated that activin E directly promoted the expression of Ucp1 and FGF21, potentially through the mediation of transforming growth factor-b or activin type I receptors [186]. Within this gene cohort, UCP1 holds significance, encoding uncoupler proteins that induce the leakage of hydrogen ions into the mitochondrial space, consequently generating heat within the targeted cell and tissue. Another noteworthy gene in this category is FGF21, contributing to the growth and specialization of brown adipocytes [187]. During fasting, the protein level of FGF21 is elevated, with its suppression attributed to the absence of an insulin receptor [188]. In contrast, there is an observed elevation in the level of activin E in mice experiencing obesity due to a high-fat diet [6].
Additionally, a prior study explored the inflammatory function of Activin in umbilical vein endothelial cells. As depicted in Fig. 2, the findings indicated that this hepatokine impacts the expression of chemokines and inflammatory cytokines by influencing NF-κB and MAPK signaling pathways. As a result, it exerts an anti-inflammatory effect against TNF-α in these cells [189].
Selenoproteins
Selenocysteine, an amino acid implicated in oxidation-reduction processes, is found in selenoprotein P [190, 191], which is primarily secreted from the hepatocytes in mammals, selenoprotein P may also be released into the bloodstream by the testis and brain tissue [192]. In addition, Selenoprotein S is involved in different metabolic conditions in the body, including insulin resistance, diabetes, and obesity. When selenoprotein S is not expressed in liver cells, it can worsen obesity and lead to reduced insulin sensitivity and glucose tolerance [193]. Human studies suggest a positive correlation between SELENOP and NAFLD [194]. The expression of this protein escalates in response to endoplasmic reticulum stress and the activation of the JNK protein. Furthermore, SELENOP synthesis is triggered by the activation of AMPK, which can be achieved by the use of anti-inflammatory or insulin-sensitizing medications (Fig. 1) [195, 196]. In the context of T2DM, there is an observed elevation in the mRNA level of SELENOP. In-vitro investigations indicate that SELENOP dysregulates glucose metabolism [197, 198]. While selenoprotein-S is involved in controlling the release of inflammatory cytokines, selenoprotein-P functions as a regulator of homeostasis and a transporter of Se from the liver to other tissues in the body [199, 200].
Hepcidin
Hepcidin, the primary regulator of iron homeostasis in the body, is a polypeptide hormone that primarily originates from liver hepatocytes. Hepcidin acts as an iron regulator by affecting the transfer of iron to the plasma through the action of ferroportin, reducing the amount of iron absorption from the diet and increasing cellular iron storage [201, 202]. Iron metabolism is one of the factors that influences the functioning of the endocrine system. Ferroptosis, a form of iron-dependent cell death, leads to insulin resistance in fat, liver, and muscle cells, and also reduces insulin secretion in pancreatic β cells [203]. Dysregulation of hepcidin leads to various metabolic diseases such as obesity, type 2 diabetes, and insulin resistance. Additionally, the plasma concentration of hepcidin changes in acute and chronic inflammatory conditions, and by affecting macrophages, it leads to the release of inflammatory cytokines [201, 202].
Iron metabolism plays a crucial role in blood glucose homeostasis through its effects on fat metabolism, insulin secretion, and liver metabolism [203]. Uncontrolled iron metabolism in adipose tissue and increased liver ferroptosis can lead to the development of diabetes and insulin resistance, respectively [203]. Previous studies have shown that individuals with T2DM tend to have higher levels of hepcidin compared to those without the condition [201, 204]. Additionally, inactivation of the hepcidin-coding gene in liver cells results in the activation of Akt and glycogen synthase kinase-3β (GSK3β) in the insulin signaling pathway [205]. Protein kinase Akt, by activating GSK3β and increasing the expression of GLUT4, leads to enhanced glucose uptake by cells and a reduction in insulin resistance [206, 207]. Increased iron levels can also lead to elevated blood glucose, fasting insulin, and hepatic ferroptosis. Through the JAK2/STAT3 signaling pathway, this can then result in the activation of SOCS-1 and the development of insulin resistance [208, 209]. Considering that hepcidin is an acute-phase reactant, the inflammation associated with T2DM can lead to increased secretion of IL-6 and activin-B (Act-B), which in turn stimulate the synthesis of hepcidin in the liver via the STAT3 signaling pathway [204].
Lipopolysaccharide binding protein
Lipopolysaccharide binding protein (LBP) is an acute-phase protein that plays a crucial role in the lipopolysaccharide (LPS) transport cascade. LBP is primarily produced by hepatocytes and, to a lesser extent, in non-hepatic tissues such as intestinal epithelial cells, lungs, and gum tissue, also, it is present in low amounts in the blood circulation. During the acute-phase response to infection and inflammation, LBP’s concentration increases 10 to 20 times compared to its baseline level [210, 211].
Lipopolysaccharide is a pathogenic factor found in the outer membrane of Gram-negative bacteria, commonly known as endotoxin [211]. Since lipopolysaccharide primarily exerts its endotoxic effects by activating the innate immune system, it is not inherently toxic [212]. However, the entry and increased level of lipopolysaccharide in the bloodstream can lead to a condition called endotoxemia, which can occur due to unhealthy eating habits or digestive disorders (derived from the intestinal microbiome). This endotoxemia can cause disorders such as metabolic endotoxemia (even in healthy individuals) and chronically contribute to inflammation-related disorders, including cardiovascular diseases, liver diseases, insulin resistance, metabolic syndromes, obesity, and diabetes [213].
LBP plays a crucial role in the LPS-to-TLR4 signaling cascade. LBP can form high-affinity complexes with LPS and then bind to CD14. LPS interacts with the LRR13-LRR15 region of TLR4, gets transferred from CD14 to MD2, and leads to signal generation, thereby greatly enhancing the host’s response to extracellular LPS [214]. Activation of the TLR4 pathway triggers the production of pro-inflammatory cytokines, such as TNF-α and IL-6. These pro-inflammatory cytokines can contribute to insulin resistance by interfering with insulin signaling pathways [215].
LBP is involved in the metabolism of lipids, particularly triglycerides and high-density lipoprotein (HDL) cholesterol. Clinical studies on metabolic endotoxemia in obese patients have shown that the serum level of LPS increases with a high-fat diet. As a result, obesity is considered a state of low-grade chronic inflammation [216, 217]. The signaling cascade initiated by the binding of LBP to LPS leads to an inflammatory response and modulation of lipid metabolism. These changes in lipid metabolism can affect insulin sensitivity and contribute to insulin resistance [87, 210, 218].
Conclusion
Recent investigations posit that the disorder in hepatokines significantly contributes to the genesis of insulin resistance and inflammation culminating in the onset of T2DM. In the context of this study, we scrutinized various hepatokines and explored their impacts on insulin resistance and inflammation. It is evident that the involvement of hepatokines stems from their modulation of key metabolic mechanisms, including lipogenesis, lipolysis, and the transfer of lipoproteins, insulin-dependent sugar metabolism and the modulation of inflammatory pathways. Briefly, fetuin-A facilitates the development of lipid-induced insulin resistance through its role as an endogenous ligand for TLR-4. FGF21 mitigates inflammation in diabetes by impeding the nuclear translocation of NF-κB in adipocytes and adipose tissue under insulin-resistant conditions, concurrently augmenting glucose metabolism. Moreover, the overproduction of LECT2 in the liver may contribute to JNK phosphorylation, resulting in insulin resistance in the skeletal muscle of obese individuals. ANGPTL6 enhances AMPK and boosts insulin signaling in muscle. Additionally, it suppresses gluconeogenesis by inhibiting the expression of the G6P gene. Furthermore, ANGPTL8 may enhance insulin sensitivity by directly affecting Akt phosphorylation. More importantly, Follistatin possesses the potential to influence insulin resistance and inflammation by virtue of its interaction with members of the TGF-β family. Also, Hepassocin assumes a pivotal role in the development of NAFLD, contributing to hepatic lipid accumulation through the activation of an ERK1/2-dependent mechanism. Furthermore, hepassocin instigates insulin resistance in skeletal muscle cells through a 396 EGFR/JNK-mediated mechanism. SMOC1 demonstrated the capacity to enhance insulin sensitivity and regulate glucose, as well. The mechanism underlying its favorable glycemic effects involves the inhibition of adenosine 3′,5′-cyclic monophosphate (cAMP)–cAMP-dependent protein kinase (PKA)–cAMP response element–binding protein (CREB) signaling in the liver. This, in turn, led to a downregulation of gluconeogenic gene expression and a suppression of hepatic glucose production. Intriguingly, the central activity of IGF1 can enhance central sympathetic outflow, consequently influencing UCP1 expression. Beyond its role in improving insulin sensitivity and glucose tolerance, as well as fostering energy expenditure through thermogenesis, central IGF1 is implicated in the regulation of appetite. In addition, LCN13 orchestrates lipid metabolism by upregulating the expression of CPT1α in the liver, while concurrently downregulating the expression of key lipogenic genes such as PPARγ and ChREBP. Liver cells respond to proinflammatory cytokines, such as TNF-α, IL-1β, and IFN-γ, collectively promoting the transcription of TSK. The initial surge in TSK levels may serve a protective role in stressed liver cells by mitigating cholesterol influx into the liver. An intriguing observation reveals a positive correlation between adropin and phosphoenolpyruvate carboxykinase 1 (PCK1), a pivotal regulatory point in gluconeogenesis. The prospect is optimistic that harnessing the potential of hepatokines and manipulating the pertinent pathways through the use of agonists and antagonists could pave the way for enhancing human health and advancing the treatment of T2DM and fatty liver disease. In addition, research in the field of hepatokines may help for the pathomechanism-based clustering of insulin resistance in some diseases to better implement precision medicine in clinical practice [18]. However, more complementary studies are needed in this regard.
Data availability
Not Applicable.
Abbreviations
- NAFLD:
-
Non-alcoholic fatty liver disease
- T2DM:
-
Type 2 diabetes mellitus
- TLR-4:
-
Toll-like receptor 4
- FGF-21:
-
Fibroblast growth factor 21
- ANGPTLs:
-
Angiopoietin-like proteins
- GDF15:
-
Growth/Differentiation Factor-15
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- GLUT4:
-
Glucose transporter type 4
- MAPKs:
-
Mitogen-activated protein kinases
- JNK:
-
c-Jun N-terminal kinases
- ERK5:
-
Extracellular signal-regulated kinases 5
- IRS1:
-
insulin receptor substrate 1
- mTOR:
-
Mammalian target of rapamycin
- Fet A:
-
Fetuin A
- NF-κB:
-
Nuclear factor-κB
- WAT:
-
white adipose tissue
- LPLs:
-
Lipoprotein lipase gene
- FAK:
-
Focal adhesion kinase
- IL-6:
-
Interleukin 6
- ICAM-1:
-
Intercellular adhesion molecule 1
- VLDLs:
-
Very-low-density lipoproteins
- LXR:
-
Liver X receptor
- PPAR:
-
Peroxisome proliferator-activated receptor
- FFA:
-
Free fatty acid
- CVD:
-
Cardiovascular diseases
- G6P:
-
glucose-6-phosphatase
- GH:
-
Growth hormone
- HDL:
-
High-density lipoprotein
- ACC:
-
AMPK-acetyl coenzyme A carboxylase
- NASH:
-
non-alcoholic steatohepatitis
- MIC-1:
-
Macrophage inhibitory cytokine-1
- GDNF:
-
Glial cell-derived neurotrophic factor
- UPR:
-
unfolded protein response
- MCP-1:
-
Monocyte chemoattractant protein-1
- RTK:
-
Receptor tyrosine kinase
- HMGB1:
-
High mobility group box protein 1
- PAMPs:
-
Pathogen-associated molecular patterns
- TGF-β:
-
Transforming growth factor-β
- IGF-1:
-
Insulin-like growth factor 1
- FSH:
-
Follicle-stimulating hormone
- TNF-α:
-
Tumor Necrosis Factorα
- CHM2:
-
Chondromodulin II
- BMI:
-
Body mass index
- HFREP1:
-
Hepatocyte-derived fibrinogen-related protein 1
- IL:
-
Interleukin
- EC:
-
Extracellular calcium
- TY:
-
Thyroglobulin-like
- CRP:
-
C-reactive protein
- NO:
-
Nitric oxide
- GHR:
-
GH receptor
- JAK2:
-
Janus Kinase 2
- BAT:
-
Brown Adipose Tissue
- SNS:
-
Sympathetic nervous system
- HFD:
-
High-fat diet
- CPT1α:
-
Carnitine palmitoyltransferase-1α
- ChREBP:
-
carbohydrate response element binding protein
- TSK:
-
Tsukushi
- LCN-13:
-
Lipocalin-13
- HSL:
-
Hormone-sensitive lipase
- PKA:
-
Protein Kinase A
- HUVEC:
-
Human Umbilical Vein Endothelial CellsActR: Activin A receptors, PCK1:Phosphoenolpyruvate carboxykinase 1
- LBP:
-
Lipopolysaccharide binding protein
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- LPS:
-
Lipopolysaccharide
- G6PC:
-
Glucose-6-phosphatase catalytic subunit
References
Meigs JB (2003) Epidemiology of the insulin resistance syndrome. Curr Diab Rep 3(1):73–79
Roden M, Shulman GI (2019) The integrative biology of type 2 diabetes. Nature 576(7785):51–60
Lebovitz HE (2001) Insulin resistance: definition and consequences. Exp Clin Endocrinol Diabetes 109(Suppl 2):S135–S148
Jensen-Cody SO, Potthoff MJ (2021) Hepatokines and metabolism: deciphering communication from the liver. Mol Metab 44:101138
Watt MJ, Miotto PM, De Nardo W, Montgomery MK (2019) The liver as an endocrine organ-linking NAFLD and insulin resistance. Endocr Rev 40(5):1367–1393
Adam RC, Pryce DS, Lee JS, Zhao Y, Mintah IJ, Min S et al (2023) Activin E-ACVR1C cross talk controls energy storage via suppression of adipose lipolysis in mice. Proc Natl Acad Sci U S A 120(32):e2309967120
Kliewer SA, Mangelsdorf DJ (2019) A dozen years of discovery: insights into the physiology and pharmacology of FGF21. Cell Metabol 29(2):246–253
Min L, Xiang J, Wang B, Ye C, Su X (2023) Novel insights of ANGPTL-3 on modulating cholesterol efflux Capacity Induced by HDL particle. Curr Mol Med
Siddiqui JA, Pothuraju R, Khan P, Sharma G, Muniyan S, Seshacharyulu P et al (2022) Pathophysiological role of growth differentiation factor 15 (GDF15) in obesity, cancer, and cachexia. Cytokine Growth Factor Rev 64:71–83
Govender N, Khaliq OP, Moodley J, Naicker T (2021) Insulin resistance in COVID-19 and diabetes. Prim Care Diabetes 15(4):629–634
Pei J, Wang B, Wang D (2022) Current studies on molecular mechanisms of insulin resistance. Journal of Diabetes Research. ;2022
Lee S-H, Park S-Y, Choi CS (2022) Insulin resistance: from mechanisms to therapeutic strategies. Diabetes Metabolism J 46(1):15–37
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414(6865):799–806
Czech MP (2017) Insulin action and resistance in obesity and type 2 diabetes. Nat Med 23(7):804–814
Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S et al (2012) Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med 18(8):1279–1285
Stefan N, Häring H-U (2013) Circulating fetuin-A and free fatty acids interact to predict insulin resistance in humans. Nat Med 19(4):394–395
Shulman GI (2014) Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 371(12):1131–1141
Stefan N, Schick F, Birkenfeld AL, Häring H-U, White MF (2023) The role of hepatokines in NAFLD. Cell Metabol 35(2):236–252
Cayatte AJ, Kumbla L, Subbiah MT (1990) Marked acceleration of exogenous fatty acid incorporation into cellular triglycerides by fetuin. J Biol Chem 265(10):5883–5888
Chattopadhyay D, Das S, Guria S, Basu S, Mukherjee S (2021) Fetuin-A regulates adipose tissue macrophage content and activation in insulin resistant mice through MCP-1 and iNOS: involvement of IFNγ-JAK2-STAT1 pathway. Biochem J 478(22):4027–4043
Afrisham R, Sadegh-Nejadi S, Meshkani R, Emamgholipour S, Paknejad M (2020) Effect of circulating exosomes derived from normal-weight and obese women on gluconeogenesis, glycogenesis, lipogenesis and secretion of FGF21 and fetuin A in HepG2 cells. Diabetol Metab Syndr 12:1–11
Wang N, Xu TY, Zhang X, Li JY, Wang YX, Guo XC et al (2018) Improving hyperglycemic effect of FGF-21 is associated with alleviating inflammatory state in diabetes. Int Immunopharmacol 56:301–309
Rui L (2014) Energy metabolism in the liver. Compr Physiol 4(1):177–197
Azimifar SB, Nagaraj N, Cox J, Mann M (2014) Cell-type-resolved quantitative proteomics of murine liver. Cell Metab 20(6):1076–1087
Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R et al (2014) A draft map of the human proteome. Nature 509(7502):575–581
Choi KM (2016) The impact of Organokines on insulin resistance, inflammation, and atherosclerosis. Endocrinol Metab (Seoul) 31(1):1–6
Meex RCR, Watt MJ (2017) Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol 13(9):509–520
Carbone C, Piro G, Merz V, Simionato F, Santoro R, Zecchetto C et al (2018) Angiopoietin-like proteins in angiogenesis, inflammation and Cancer. Int J Mol Sci. ;19(2)
Hato T, Tabata M, Oike Y (2008) The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc Med 18(1):6–14
Wu SA, Kersten S, Qi L (2021) Lipoprotein Lipase and its regulators: an Unfolding Story. Trends Endocrinol Metab 32(1):48–61
Wang H, Eckel RH (2009) Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab 297(2):E271–E288
Yang J, Song QY, Niu SX, Chen HJ, Petersen RB, Zhang Y et al (2022) Emerging roles of angiopoietin-like proteins in inflammation: mechanisms and potential as pharmacological targets. J Cell Physiol 237(1):98–117
Yang J, Song Qy N, Sx C, Hj, Petersen RB, Zhang Y et al (2022) Emerging roles of angiopoietin-like proteins in inflammation: mechanisms and potential as pharmacological targets. J Cell Physiol 237(1):98–117
Carbone C, Piro G, Merz V, Simionato F, Santoro R, Zecchetto C et al (2018) Angiopoietin-like proteins in angiogenesis, inflammation and cancer. Int J Mol Sci 19(2):431
Yan Q, Jiang L, Liu M, Yu D, Zhang Y, Li Y et al (2017) ANGPTL1 interacts with integrin α1β1 to suppress HCC angiogenesis and metastasis by inhibiting JAK2/STAT3 signaling. Cancer Res 77(21):5831–5845
Chen HA, Kuo TC, Tseng CF, Ma JT, Yang ST, Yen CJ et al (2016) Angiopoietin-like protein 1 antagonizes MET receptor activity to repress sorafenib resistance and cancer stemness in hepatocellular carcinoma. Hepatology 64(5):1637–1651
Tabata M, Kadomatsu T, Fukuhara S, Miyata K, Ito Y, Endo M et al (2009) Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metabol 10(3):178–188
Horio E, Kadomatsu T, Miyata K, Arai Y, Hosokawa K, Doi Y et al (2014) Role of endothelial cell–derived Angptl2 in vascular inflammation leading to endothelial dysfunction and atherosclerosis progression. Arteriosclerosis, thrombosis, and vascular biology. 34(4):790–800
Farhat N, Thorin-Trescases N, Mamarbachi M, Villeneuve L, Yu C, Martel C et al (2013) Angiopoietin‐like 2 promotes atherogenesis in mice. J Am Heart Association 2(3):e000201
Nakamura M, Yamada K (1967) Studies on a diabetic (KK) strain of the mouse. Diabetologia 3(2):212–221
Koishi R, Ando Y, Ono M, Shimamura M, Yasumo H, Fujiwara T et al (2002) Angptl3 regulates lipid metabolism in mice. Nat Genet 30(2):151–157
Shimizugawa T, Ono M, Shimamura M, Yoshida K, Ando Y, Koishi R et al (2002) ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem 277(37):33742–33748
Minicocci I, Montali A, Robciuc MR, Quagliarini F, Censi V, Labbadia G et al (2012) Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab 97(7):E1266–E1275
Wang Y, McNutt MC, Banfi S, Levin MG, Holland WL, Gusarova V et al (2015) Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc Natl Acad Sci U S A 112(37):11630–11635
Ono M, Shimizugawa T, Shimamura M, Yoshida K, Noji-Sakikawa C, Ando Y et al (2003) Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J Biol Chem 278(43):41804–41809
Kaplan R, Zhang T, Hernandez M, Gan FX, Wright SD, Waters MG et al (2003) Regulation of the angiopoietin-like protein 3 gene by LXR. J Lipid Res 44(1):136–143
Shimamura M, Matsuda M, Ando Y, Koishi R, Yasumo H, Furukawa H et al (2004) Leptin and insulin down-regulate angiopoietin-like protein 3, a plasma triglyceride-increasing factor. Biochem Biophys Res Commun 322(3):1080–1085
Tikka A, Soronen J, Laurila PP, Metso J, Ehnholm C, Jauhiainen M (2014) Silencing of ANGPTL 3 (angiopoietin-like protein 3) in human hepatocytes results in decreased expression of gluconeogenic genes and reduced triacylglycerol-rich VLDL secretion upon insulin stimulation. Biosci Rep 34(6):e00160
Wang C, Tong Y, Wen Y, Cai J, Guo H, Huang L et al (2018) Hepatocellular carcinoma-associated protein TD26 interacts and enhances sterol regulatory element‐binding protein 1 activity to promote tumor cell proliferation and growth. Hepatology 68(5):1833–1850
Biterova E, Esmaeeli M, Alanen HI, Saaranen M, Ruddock LW (2018) Structures of Angptl3 and Angptl4, modulators of triglyceride levels and coronary artery disease. Sci Rep 8(1):6752
Koliwad SK, Gray NE, Wang JC (2012) Angiopoietin-like 4 (Angptl4): a glucocorticoid-dependent gatekeeper of fatty acid flux during fasting. Adipocyte 1(3):182–187
La Paglia L, Listì A, Caruso S, Amodeo V, Passiglia F, Bazan V et al (2017) Potential role of ANGPTL4 in the Cross talk between metabolism and Cancer through PPAR signaling pathway. PPAR Res 2017:8187235
Singh AK, Chaube B, Zhang X, Sun J, Citrin KM, Canfrán-Duque A et al (2021) Hepatocyte-specific suppression of ANGPTL4 improves obesity-associated diabetes and mitigates atherosclerosis in mice. J Clin Invest. ;131(17)
Yoshida K, Shimizugawa T, Ono M, Furukawa H (2002) Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J Lipid Res 43(11):1770–1772
Wang Y, Liu LM, Wei L, Ye WW, Meng XY, Chen F et al (2016) Angiopoietin-like protein 4 improves glucose tolerance and insulin resistance but induces liver steatosis in high-fat-diet mice. Mol Med Rep 14(4):3293–3300
Xu A, Lam MC, Chan KW, Wang Y, Zhang J, Hoo RL et al (2005) Angiopoietin-like protein 4 decreases blood glucose and improves glucose tolerance but induces hyperlipidemia and hepatic steatosis in mice. Proc Natl Acad Sci U S A 102(17):6086–6091
Altun Ö, Dikker O, Arman Y, Ugurlukisi B, Kutlu O, Ozgun Cil E et al (2018) Serum angiopoietin-like peptide 4 levels in patients with hepatic steatosis. Cytokine 111:496–499
Zuo Y, He Z, Chen Y, Dai L (2023) Dual role of ANGPTL4 in inflammation. Inflamm Res 72(6):1303–1313
Im Cho D, Kang H-j, Jeon JH, Eom GH, Cho HH, Kim MR et al (2019) Antiinflammatory activity of ANGPTL4 facilitates macrophage polarization to induce cardiac repair. JCI Insight. ;4(16)
Alghanim G, Qaddoumi MG, Alhasawi N, Cherian P, Al-Khairi I, Nizam R et al (2019) Higher levels of ANGPTL5 in the circulation of subjects with obesity and type 2 diabetes are associated with insulin resistance. Front Endocrinol 10:461900
Fan KC, Wu HT, Wei JN, Chuang LM, Hsu CY, Yen IW et al (2020) Serum angiopoietin-like protein 6, risk of type 2 diabetes, and response to hyperglycemia: a prospective cohort study. J Clin Endocrinol Metab. ;105(5)
Kitazawa M, Ohizumi Y, Oike Y, Hishinuma T, Hashimoto S (2007) Angiopoietin-related growth factor suppresses gluconeogenesis through the Akt/forkhead box class O1-dependent pathway in hepatocytes. J Pharmacol Exp Ther 323(3):787–793
Erkan G, Muratoglu S, Ercin U, Bilgihan A (2018) Angiopoietin-like protein 2 and angiopoietin-like protein 6 levels in patients with nonalcoholic fatty liver disease. Arch Med Sci 14(4):781–787
Tanigawa H, Miyata K, Tian Z, Aoi J, Kadomatsu T, Fukushima S et al (2016) Upregulation of ANGPTL6 in mouse keratinocytes enhances susceptibility to psoriasis. Sci Rep 6(1):34690
Zhang L, Shannon CE, Bakewell TM, Abdul-Ghani MA, Fourcaudot M, Norton L (2020) Regulation of ANGPTL8 in liver and adipose tissue by nutritional and hormonal signals and its effect on glucose homeostasis in mice. Am J Physiol Endocrinol Metab 318(5):E613–e24
Zhang Z, Wu H, Dai L, Yuan Y, Zhu Y, Ma Z et al (2020) ANGPTL8 enhances insulin sensitivity by directly activating insulin-mediated AKT phosphorylation. Gene 749:144707
Bai Y, Du Q, Zhang L, Li L, Wang N, Wu B et al (2021) Silencing of ANGPTL8 alleviates insulin resistance in trophoblast cells. Front Endocrinol (Lausanne) 12:635321
Saghafi S, Chamani E, Salmani F, Fadaei R, Shafiei E, Moradi N et al (2023) Genetic predisposition to nonalcoholic fatty liver disease: insights from ANGPTL8 gene variants in Iranian adults. Lipids Health Dis 22(1):147
Zhao Z, Deng X, Jia J, Zhao L, Wang C, Cai Z et al (2022) Angiopoietin-like protein 8 (betatrophin) inhibits hepatic gluconeogenesis through PI3K/Akt signaling pathway in diabetic mice. Metabolism 126:154921
Zhang Y, Zheng L, Huang K (2018) A new way to regulate inflammation: selective autophagic degradation of IKKγ mediated by ANGPTL8. Cell Stress 2(3):66
Zhang Y, Guo X, Yan W, Chen Y, Ke M, Cheng C et al (2017) ANGPTL8 negatively regulates NF-κB activation by facilitating selective autophagic degradation of IKKγ. Nat Commun 8(1):2164
Tan H, Yue T, Chen Z, Wu W, Xu S, Weng J (2023) Targeting FGF21 in cardiovascular and metabolic diseases: from mechanism to medicine. Int J Biol Sci 19(1):66–88
Ge X, Wang Y, Lam KS, Xu A (2012) Metabolic actions of FGF21: molecular mechanisms and therapeutic implications. Acta Pharm Sinica B 2(4):350–357
von Holstein-Rathlou S, BonDurant LD, Peltekian L, Naber MC, Yin TC, Claflin KE et al (2016) FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet taste preference by the liver. Cell Metab 23(2):335–343
Hotta Y, Nakamura H, Konishi M, Murata Y, Takagi H, Matsumura S et al (2009) Fibroblast growth factor 21 regulates Lipolysis in White Adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology 150(10):4625–4633
Lu W, Li X, Luo Y (2021) FGF21 in obesity and cancer: new insights. Cancer Lett 499:5–13
Kliewer SA, Mangelsdorf DJ (2019) A Dozen years of Discovery: insights into the physiology and pharmacology of FGF21. Cell Metab 29(2):246–253
BonDurant LD, Potthoff MJ (2018) Fibroblast growth factor 21: a Versatile Regulator of metabolic homeostasis. Annu Rev Nutr 38(1):173–196
BonDurant LD, Ameka M, Naber MC, Markan KR, Idiga SO, Acevedo MR et al (2017) FGF21 regulates metabolism through adipose-dependent and -independent mechanisms. Cell Metab 25(4):935–44e4
Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K et al (2014) FGF21 acts centrally to induce sympathetic nerve activity, energy expenditure, and weight loss. Cell Metab 20(4):670–677
Xie T, So WY, Li XY, Leung PS (2019) Fibroblast growth factor 21 protects against lipotoxicity-induced pancreatic β-cell dysfunction via regulation of AMPK signaling and lipid metabolism. Clin Sci (Lond) 133(19):2029–2044
Gong Q, Hu Z, Zhang F, Cui A, Chen X, Jiang H et al (2016) Fibroblast growth factor 21 improves hepatic insulin sensitivity by inhibiting mammalian target of rapamycin complex 1 in mice. Hepatology 64(2):425–438
Goto T, Hirata M, Aoki Y, Iwase M, Takahashi H, Kim M et al (2017) The hepatokine FGF21 is crucial for peroxisome proliferator-activated receptor-α agonist-induced amelioration of metabolic disorders in obese mice. J Biol Chem 292(22):9175–9190
Al-Mansoori L, Al-Jaber H, Prince MS, Elrayess MA (2022) Role of inflammatory cytokines, growth factors and adipokines in adipogenesis and insulin resistance. Inflammation. :1–14
ZhuGe D-L, Javaid HMA, Sahar NE, Zhao Y-Z, Huh JY (2020) Fibroblast growth factor 2 exacerbates inflammation in adipocytes through NLRP3 inflammasome activation. Arch Pharm Res 43:1311–1324
Jin L, Yang R, Geng L, Xu A (2023) Fibroblast growth factor-based pharmacotherapies for the treatment of obesity-related metabolic complications. Annu Rev Pharmacol Toxicol 63:359–382
Jensen-Cody SO, Potthoff MJ (2021) Hepatokines and metabolism: deciphering communication from the liver. Mol Metabolism 44:101138
Agarwal S, Chattopadhyay M, Mukherjee S, Dasgupta S, Mukhopadhyay S, Bhattacharya S (2017) Fetuin-A downregulates adiponectin through Wnt-PPARγ pathway in lipid induced inflamed adipocyte. Biochim Biophys Acta Mol Basis Dis 1863(1):174–181
Komsa-Penkova RS, Golemanov GM, Radionova ZV, Tonchev PT, Iliev SD, Penkov VV (2017) Fetuin-A–alpha2-heremans-schmid glycoprotein: from structure to a novel marker of chronic diseases part 1. Fetuin-A as a calcium chaperone and inflammatory marker. J Biomedical Clin Res 10(2):90–97
Mathews ST, Chellam N, Srinivas PR, Cintron VJ, Leon MA, Goustin AS et al (2000) Alpha2-HSG, a specific inhibitor of insulin receptor autophosphorylation, interacts with the insulin receptor. Mol Cell Endocrinol 164(1–2):87–98
Auberger P, Falquerho L, Contreres JO, Pages G, Le Cam G, Rossi B et al (1989) Characterization of a natural inhibitor of the insulin receptor tyrosine kinase: cDNA cloning, purification, and anti-mitogenic activity. Cell 58(4):631–640
Chekol Abebe E, Tilahun Muche Z, Behaile TMA, Mengie Ayele T, Mekonnen Agidew M, Teshome Azezew M et al (2022) The structure, biosynthesis, and biological roles of fetuin-A: a review. Front Cell Dev Biol 10:945287
Stefan N, Hennige AM, Staiger H, Machann J, Schick F, Kröber SM et al (2006) Alpha2-Heremans-Schmid glycoprotein/fetuin-A is associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care 29(4):853–857
Stefan N, Fritsche A, Weikert C, Boeing H, Joost H-G, Häring H-U et al (2008) Plasma fetuin-A levels and the risk of type 2 diabetes. Diabetes 57(10):2762–2767
Kroeger J, Meidtner K, Stefan N, Guevara M, Kerrison ND, Ardanaz E et al (2018) Circulating fetuin-A and risk of type 2 diabetes: a mendelian randomization analysis. Diabetes 67(6):1200–1205
Ix JH, Wassel CL, Kanaya AM, Vittinghoff E, Johnson KC, Koster A et al (2008) Fetuin-A and incident diabetes mellitus in older persons. JAMA 300(2):182–188
Hennige AM, Staiger H, Wicke C, Machicao F, Fritsche A, Häring HU et al (2008) Fetuin-A induces cytokine expression and suppresses adiponectin production. PLoS ONE 3(3):e1765
Meex RC, Watt MJ (2017) Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Reviews Endocrinol 13(9):509–520
Pan X, Kaminga AC, Chen J, Luo M, Luo J (2020) Fetuin-A and Fetuin-B in non-alcoholic fatty liver disease: a Meta-analysis and Meta-regression. Int J Environ Res Public Health. ;17(8)
Mukhopadhyay S, Mondal SA, Kumar M, Dutta D (2014) Proinflammatory and antiinflammatory attributes of fetuin-a: a novel hepatokine modulating cardiovascular and glycemic outcomes in metabolic syndrome. Endocr Pract 20(12):1345–1351
Mukhuty A, Fouzder C, Kundu R (2021) Fetuin-A secretion from β-cells leads to accumulation of macrophages in islets, aggravates inflammation and impairs insulin secretion. J Cell Sci 134(21):jcs258507
Jones KL, Mansell A, Patella S, Scott BJ, Hedger MP, de Kretser DM et al (2007) Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc Natl Acad Sci U S A 104(41):16239–16244
Parfenova OK, Kukes VG, Grishin DV (2021) Follistatin-like proteins: structure, functions and biomedical importance. Biomedicines 9(8):999
Hansen J, Rinnov A, Krogh-Madsen R, Fischer CP, Andreasen AS, Berg RM et al (2013) Plasma follistatin is elevated in patients with type 2 diabetes: relationship to hyperglycemia, hyperinsulinemia, and systemic low-grade inflammation. Diabetes Metab Res Rev 29(6):463–472
Wu C, Borné Y, Gao R, López Rodriguez M, Roell WC, Wilson JM et al (2021) Elevated circulating follistatin associates with an increased risk of type 2 diabetes. Nat Commun 12(1):6486
Hansen JS, Rutti S, Arous C, Clemmesen JO, Secher NH, Drescher A et al (2016) Circulating follistatin is liver-derived and regulated by the glucagon-to-insulin ratio. J Clin Endocrinol Metab 101(2):550–560
Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M, Schneyer A (2006) Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 147(7):3586–3597
Welt C, Sidis Y, Keutmann H, Schneyer A (2002) Activins, inhibins, and follistatins: from endocrinology to signaling. A paradigm for the new millennium. Experimental Biology Med 227(9):724–752
Hansen JS, Plomgaard P (2016) Circulating follistatin in relation to energy metabolism. Mol Cell Endocrinol 433:87–93
Tao R, Stöhr O, Wang C, Qiu W, Copps KD, White MF (2023) Hepatic follistatin increases basal metabolic rate and attenuates diet-induced obesity during hepatic insulin resistance. Mol Metabolism 71:101703
Lee S-J, McPherron AC (2001) Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci 98(16):9306–9311
Blumensatt M, Greulich S, Herzfeld de Wiza D, Mueller H, Maxhera B, Rabelink MJ et al (2013) Activin a impairs insulin action in cardiomyocytes via up-regulation of miR-143. Cardiovasc Res 100(2):201–210
Yang J (2014) Enhanced skeletal muscle for effective glucose homeostasis. Prog Mol Biol Transl Sci 121:133–163
Yamagoe S, Yamakawa Y, Matsuo Y, Minowada J, Mizuno S, Suzuki K (1996) Purification and primary amino acid sequence of a novel neutrophil chemotactic factor LECT2. Immunol Lett 52(1):9–13
Hiraki Y, Inoue H, Kondo J, Kamizono A, Yoshitake Y, Shukunami C et al (1996) A novel growth-promoting factor derived from fetal bovine cartilage, chondromodulin II: purification and amino acid sequence. J Biol Chem 271(37):22657–22662
Slowik V, Apte U (2017) Leukocyte cell-derived Chemotaxin-2: it’s role in pathophysiology and future in Clinical Medicine. Clin Transl Sci 10(4):249–259
Nagal H, Hamada T, Uchida T, Yamagoe S, Suzuki K (1998) Systemic expression of a newly recognized protein, LECT2, in the human body. Pathol Int 48(11):882–886
Yamagoe S, Mizuno S, Suzuki K (1998) Molecular cloning of human and bovine LECT2 having a neutrophil chemotactic activity and its specific expression in the liver. Biochim Biophys Acta 1396(1):105–113
Misu H (2019) Identification of hepatokines involved in pathology of type 2 diabetes and obesity. Endocr J 66(8):659–662
Lan F, Misu H, Chikamoto K, Takayama H, Kikuchi A, Mohri K et al (2014) LECT2 functions as a hepatokine that links obesity to skeletal muscle insulin resistance. Diabetes 63(5):1649–1664
Zhu M-H, Liu Y-J, Yang G-J, Chen J (2023) The emerging roles of leukocyte cell-derived chemotaxin-2 in immune diseases: from mechanisms to therapeutic potential. Front Immunol 14:1158083
Hara H, Uchida S, Yoshimura H, Aoki M, Toyoda Y, Sakai Y et al (2000) Isolation and characterization of a novel liver-specific gene, hepassocin, upregulated during liver regeneration. Biochim Biophys Acta 1492(1):31–44
Hara H, Yoshimura H, Uchida S, Toyoda Y, Aoki M, Sakai Y et al (2001) Molecular cloning and functional expression analysis of a cDNA for human hepassocin, a liver-specific protein with hepatocyte mitogenic activity. Biochim Biophys Acta 1520(1):45–53
Cao MM, Xu WX, Li CY, Cao CZ, Wang ZD, Yao JW et al (2011) Hepassocin regulates cell proliferation of the human hepatic cells L02 and hepatocarcinoma cells through different mechanisms. J Cell Biochem 112(10):2882–2890
Demchev V, Malana G, Vangala D, Stoll J, Desai A, Kang HW et al (2013) Targeted deletion of fibrinogen like protein 1 reveals a novel role in energy substrate utilization. PLoS ONE 8(3):e58084
Huang RL, Li CH, Du YF, Cheng KP, Lin CH, Hu CY et al (2020) Discovery of a role of the novel hepatokine, hepassocin, in obesity. BioFactors 46(1):100–105
Wu HT, Chen SC, Fan KC, Kuo CH, Lin SY, Wang SH et al (2020) Targeting fibrinogen-like protein 1 is a novel therapeutic strategy to combat obesity. Faseb j 34(2):2958–2967
Wu HT, Lu FH, Ou HY, Su YC, Hung HC, Wu JS et al (2013) The role of hepassocin in the development of non-alcoholic fatty liver disease. J Hepatol 59(5):1065–1072
Wu HT, Ou HY, Hung HC, Su YC, Lu FH, Wu JS et al (2016) A novel hepatokine, HFREP1, plays a crucial role in the development of insulin resistance and type 2 diabetes. Diabetologia 59(8):1732–1742
Jung TW, Chung YH, Kim HC, Abd El-Aty AM, Jeong JH (2018) Hyperlipidemia-induced hepassocin in the liver contributes to insulin resistance in skeletal muscle. Mol Cell Endocrinol 470:26–33
Yang Y, Liu X, Chen H, Wang P, Yao S, Zhou B et al (2022) HPS protects the liver against steatosis, cell death, inflammation, and fibrosis in mice with steatohepatitis. Febs j 289(17):5279–5304
Vannahme C, Smyth N, Miosge N, Gösling S, Frie C, Paulsson M et al (2002) Characterization of SMOC-1, a novel modular calcium-binding protein in basement membranes. J Biol Chem 277(41):37977–37986
Bornstein P, Sage EH (2002) Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14(5):608–616
Novinec M, Kovacic L, Skrlj N, Turk V, Lenarcic B (2008) Recombinant human SMOCs produced by in vitro refolding: calcium-binding properties and interactions with serum proteins. Protein Expr Purif 62(1):75–82
Di Niro R, Sulic AM, Mignone F, D’Angelo S, Bordoni R, Iacono M et al (2010) Rapid interactome profiling by massive sequencing. Nucleic Acids Res 38(9):e110
Brellier F, Ruggiero S, Zwolanek D, Martina E, Hess D, Brown-Luedi M et al (2011) SMOC1 is a tenascin-C interacting protein over-expressed in brain tumors. Matrix Biol 30(3):225–233
Klemenčič M, Novinec M, Maier S, Hartmann U, Lenarčič B (2013) The heparin-binding activity of secreted modular calcium-binding protein 1 (SMOC-1) modulates its cell adhesion properties. PLoS ONE 8(2):e56839
Montgomery MK, Bayliss J, Devereux C, Bezawork-Geleta A, Roberts D, Huang C et al (2020) SMOC1 is a glucose-responsive hepatokine and therapeutic target for glycemic control. Sci Transl Med. ;12(559)
Gao Q, Mok H-P, Zhuang J (2019) Secreted modular calcium-binding proteins in pathophysiological processes and embryonic development. Chin Med J 132(20):2476–2484
Decourtye L, Mire E, Clemessy M, Heurtier V, Ledent T, Robinson IC et al (2017) IGF-1 induces GHRH Neuronal Axon Elongation during early postnatal life in mice. PLoS ONE 12(1):e0170083
Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL et al (2002) Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest 110(6):771–781
Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J et al (2003) Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med 349(12):1139–1147
Rajkumar K, Krsek M, Dheen ST, Murphy LJ (1996) Impaired glucose homeostasis in insulin-like growth factor binding protein-1 transgenic mice. J Clin Investig 98(8):1818–1825
Sandhu MS, Heald AH, Gibson JM, Cruickshank JK, Dunger DB, Wareham NJ (2002) Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: a prospective observational study. Lancet 359(9319):1740–1745
Succurro E, Andreozzi F, Marini MA, Lauro R, Hribal ML, Perticone F et al (2009) Low plasma insulin-like growth factor-1 levels are associated with reduced insulin sensitivity and increased insulin secretion in nondiabetic subjects. Nutr Metab Cardiovasc Dis 19(10):713–719
Boger RH, Frystyk J, Ledet T, Moller N, Flyvbjerg A, Orskov H (2003) Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease. Circulation 107(20):e193 author reply e
Rajkumar K, Krsek M, Dheen ST, Murphy LJ (1996) Impaired glucose homeostasis in insulin-like growth factor binding protein-1 transgenic mice. J Clin Invest 98(8):1818–1825
Bach MA, Shen-Orr Z, Lowe WL Jr., Roberts CT Jr., LeRoith D (1991) Insulin-like growth factor I mRNA levels are developmentally regulated in specific regions of the rat brain. Brain Res Mol Brain Res 10(1):43–48
Lee JY, Muenzberg H, Gavrilova O, Reed JA, Berryman D, Villanueva EC et al (2008) Loss of cytokine-STAT5 signaling in the CNS and pituitary gland alters energy balance and leads to obesity. PLoS ONE 3(2):e1639
Hong H, Cui ZZ, Zhu L, Fu SP, Rossi M, Cui YH et al (2017) Central IGF1 improves glucose tolerance and insulin sensitivity in mice. Nutr Diabetes 7(12):2
Nguyen NL, Barr CL, Ryu V, Cao Q, Xue B, Bartness TJ (2017) Separate and shared sympathetic outflow to white and brown fat coordinately regulates thermoregulation and beige adipocyte recruitment. Am J Physiol Regul Integr Comp Physiol 312(1):R132–r45
Bonet ML, Mercader J, Palou A (2017) A nutritional perspective on UCP1-dependent thermogenesis. Biochimie 134:99–117
Laron Z (1999) The essential role of IGF-I: lessons from the long-term study and treatment of children and adults with Laron syndrome. J Clin Endocrinol Metab 84(12):4397–4404
Laron Z, Avitzur Y, Klinger B (1995) Carbohydrate metabolism in primary growth hormone resistance (Laron syndrome) before and during insulin-like growth factor-I treatment. Metabolism 44(10 Suppl 4):113–118
Wang X, Sun H, Ma B, Gao J, Yin J, Qu S (2020) Insulin-like growth factor 1 related to chronic low-grade inflammation in patients with obesity and early change of its levels after laparoscopic sleeve gastrectomy. Obes Surg 30(9):3326–3332
Wischhusen J, Melero I, Fridman WH (2020) Growth/Differentiation Factor-15 (GDF-15): from biomarker to Novel Targetable Immune Checkpoint. Front Immunol 11:951
Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, Bauskin AR et al (2007) Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat Med 13(11):1333–1340
Breit SN, Brown DA, Tsai VW (2021) The GDF15-GFRAL pathway in Health and Metabolic Disease: friend or foe? Annu Rev Physiol 83:127–151
Kelly JA, Lucia MS, Lambert JR (2009) p53 controls prostate-derived factor/macrophage inhibitory cytokine/NSAID-activated gene expression in response to cell density, DNA damage and hypoxia through diverse mechanisms. Cancer Lett 277(1):38–47
Asrih M, Wei S, Nguyen TT, Yi HS, Ryu D, Gariani K (2023) Overview of growth differentiation factor 15 in metabolic syndrome. J Cell Mol Med 27(9):1157–1167
Chrysovergis K, Wang X, Kosak J, Lee SH, Kim JS, Foley JF et al (2014) NAG-1/GDF-15 prevents obesity by increasing thermogenesis, lipolysis and oxidative metabolism. Int J Obes (Lond) 38(12):1555–1564
Zhou Y, Rui L (2013) Lipocalin 13 regulation of glucose and lipid metabolism in obesity. Vitamins Horm 91:369–383
Zhou Y, Rui L (2013) Lipocalin 13 regulation of glucose and lipid metabolism in obesity. Vitam Horm 91:369–383
Cho KW, Zhou Y, Sheng L, Rui L (2011) Lipocalin-13 regulates glucose metabolism by both insulin-dependent and insulin-independent mechanisms. Mol Cell Biol 31(3):450–457
Sheng L, Cho KW, Zhou Y, Shen H, Rui L (2011) Lipocalin 13 protein protects against hepatic steatosis by both inhibiting lipogenesis and stimulating fatty acid β-oxidation. J Biol Chem 286(44):38128–38135
Ekim Üstünel B, Friedrich K, Maida A, Wang X, Krones-Herzig A, Seibert O et al (2016) Control of diabetic hyperglycaemia and insulin resistance through TSC22D4. Nat Commun 7(1):13267
Ahmad SAI, Anam MB, Ito N, Ohta K (2018) Involvement of Tsukushi in diverse developmental processes. J Cell Communication Signal 12(1):205–210
Istiaq A, Ohta K (2022) A review on Tsukushi: mammalian development, disorders, and therapy. J Cell Communication Signal 16(4):505–513
Mouchiroud M, Camiré É, Aldow M, Caron A, Jubinville É, Turcotte L et al (2019) The hepatokine Tsukushi is released in response to NAFLD and impacts cholesterol homeostasis. JCI Insight. ;4(15)
Wang Q, Sharma V, Shen H, Xiao Y, Zhu Q, Xiong X et al (2019) The hepatokine Tsukushi gates energy expenditure via brown fat sympathetic innervation. Nat Metab 1:251–260
Wang Q, Sharma VP, Shen H, Xiao Y, Zhu Q, Xiong X et al (2019) The hepatokine Tsukushi gates energy expenditure via brown fat sympathetic innervation. Nat Metab 1(2):251–260
Mouchiroud M, Camiré É, Aldow M, Caron A, Jubinville É, Turcotte L et al (2019) The Hepatokine TSK does not affect brown fat thermogenic capacity, body weight gain, and glucose homeostasis. Mol Metab 30:184–191
Aydin S (2014) Three new players in energy regulation: preptin, adropin and irisin. Peptides 56:94–110
Ali II, D’Souza C, Singh J, Adeghate E (2022) Adropin’s Role in Energy Homeostasis and Metabolic Disorders. Int J Mol Sci. ;23(15)
Kumar KG, Trevaskis JL, Lam DD, Sutton GM, Koza RA, Chouljenko VN et al (2008) Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab 8(6):468–481
Jasaszwili M, Billert M, Strowski MZ, Nowak KW, Skrzypski M (2020) Adropin as a Fat-Burning hormone with multiple functions-review of a Decade of Research. Molecules. ;25(3)
Ganesh Kumar K, Zhang J, Gao S, Rossi J, McGuinness OP, Halem HH et al (2012) Adropin deficiency is associated with increased adiposity and insulin resistance. Obes (Silver Spring) 20(7):1394–1402
Banerjee S, Ghoshal S, Stevens JR, McCommis KS, Gao S, Castro-Sepulveda M et al (2020) Hepatocyte expression of the micropeptide adropin regulates the liver fasting response and is enhanced by caloric restriction. J Biol Chem 295(40):13753–13768
Gao S, McMillan RP, Zhu Q, Lopaschuk GD, Hulver MW, Butler AA (2015) Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol Metab 4(4):310–324
Jasaszwili M, Billert M, Strowski MZ, Nowak KW, Skrzypski M (2020) Adropin as a fat-burning hormone with multiple functions—review of a decade of research. Molecules 25(3):549
Takanori Nakamura MA, Yuzuru EI, Takio K, Uchiyama H, Moriya N, Ariizumi T (1992) Takayuki Yashiro, Kishiko Sugino, Koiti Titani, and Hiromu Sugino. Isolation and Characterization of Native Activin B
Hashimoto O, Ushiro Y, Sekiyama K, Yamaguchi O, Yoshioka K, Mutoh K et al (2006) Impaired growth of pancreatic exocrine cells in transgenic mice expressing human activin betaE subunit. Biochem Biophys Res Commun 341(2):416–424
Abe Y, Minegishi T, Leung PC (2004) Activin receptor signaling. Growth Factors 22(2):105–110
Liu H, Hallauer Hastings M, Kitchen R, Xiao C, Baldovino Guerra JR, Kuznetsov A, Arteriosclerosis et al (2023) Thromb Vascular Biology 43(2):330–349
Hashimoto O, Tsuchida K, Ushiro Y, Hosoi Y, Hoshi N, Sugino H et al (2002) cDNA cloning and expression of human activin betaE subunit. Mol Cell Endocrinol 194(1–2):117–122
Hashimoto O, Funaba M, Sekiyama K, Doi S, Shindo D, Satoh R et al (2018) Activin E controls energy homeostasis in both brown and white adipose tissues as a hepatokine. Cell Rep 25(5):1193–1203
Sekiyama K, Ushiro Y, Kurisaki A, Funaba M, Hashimoto O (2019) Activin E enhances insulin sensitivity and thermogenesis by activating brown/beige adipocytes. J Vet Med Sci 81(5):646–652
Smati S, Régnier M, Fougeray T, Polizzi A, Fougerat A, Lasserre F et al (2020) Regulation of hepatokine gene expression in response to fasting and feeding: influence of PPAR-α and insulin-dependent signalling in hepatocytes. Diabetes Metab 46(2):129–136
Ko H, Il Kim Y, Ahn HJ (2022) Activin suppresses the inflammatory response of TNF-α-stimulated human umbilical vein endothelial cells. Die Pharmazie-An Int J Pharm Sci 77(5):152–156
Schmidt RL, Simonović M (2012) Synthesis and decoding of selenocysteine and human health. Croat Med J 53(6):535–550
Yu S-s, Du J-l (2017) Selenoprotein S: a therapeutic target for diabetes and macroangiopathy? Cardiovasc Diabetol 16(1):101
Arteel GE, Klotz L-O, Buchczyk DP, Sies H (2002) [11] - selenoprotein P. In: Sies H, Packer L (eds) Methods in Enzymology, vol 347. Academic, pp 121–125
Yu S-s, Du J-l (2024) Current views on selenoprotein S in the pathophysiological processes of diabetes-induced atherosclerosis: potential therapeutics and underlying biomarkers. Diabetol Metab Syndr 16(1):5
Polyzos SA, Kountouras J, Goulas A, Duntas L (2020) Selenium and selenoprotein P in nonalcoholic fatty liver disease. Horm (Athens) 19(1):61–72
Takayama H, Misu H, Iwama H, Chikamoto K, Saito Y, Murao K et al (2014) Metformin suppresses expression of the selenoprotein P gene via an AMP-activated kinase (AMPK)/FoxO3a pathway in H4IIEC3 hepatocytes. J Biol Chem 289(1):335–345
Ye R, Huang J, Wang Z, Chen Y, Dong Y (2022) The role and mechanism of essential selenoproteins for Homeostasis. Antioxid (Basel). ;11(5)
Misu H, Takamura T, Takayama H, Hayashi H, Matsuzawa-Nagata N, Kurita S et al (2010) A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab 12(5):483–495
Yang SJ, Hwang SY, Choi HY, Yoo HJ, Seo JA, Kim SG et al (2011) Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: implications for insulin resistance, inflammation, and atherosclerosis. J Clin Endocrinol Metab 96(8):E1325–E1329
Hariharan S, Dharmaraj S (2020) Selenium and selenoproteins: it’s role in regulation of inflammation. Inflammopharmacology 28:667–695
Polyzos SA, Kountouras J, Goulas A, Duntas L (2020) Selenium and selenoprotein P in nonalcoholic fatty liver disease. Hormones 19:61–72
Alfawaz HA, Alfaifi AA, Yakout SM, Khattak MNK, Alnaami AM, Al-Thayidi A et al (2022) Circulating hepcidin and its associations with low-grade inflammation and iron indices among arab adults with and without T2DM. Am J Translational Res 14(10):7520
Chambers K, Ashraf MA, Sharma S (2019) Physiology, hepcidin
Miao R, Fang X, Zhang Y, Wei J, Zhang Y, Tian J (2023) Iron metabolism and ferroptosis in type 2 diabetes mellitus and complications: mechanisms and therapeutic opportunities. Cell Death Dis 14(3):186
Maherdika M, Samsuria IK, Hendrianingtyas M, Widyastiti NS, Rahayu M (2022) The difference levels of hepcidin and interleukin-6 between obese and non-obese type 2 diabetes mellitus. Indonesian Biomedical J 14(1):98–103
Zhang X, Zhang L, Tan Y-m, Liu Y-p, Li J-j, Deng Q-m et al (2021) Hepcidin gene silencing ameliorated inflammation and insulin resistance in adipose tissue of db/db mice via inhibiting METs formation. Mol Immunol 133:110–121
Zhang Z, Liu H, Liu J (2019) Akt activation: a potential strategy to ameliorate insulin resistance. Diabetes Res Clin Pract 156:107092
Zhang X-g, Liu A-x, Zhang Y-x, Zhou M-y, Li X-y, Fu M-h et al (2022) A diarylheptanoid compound from alpinia officinarum hance ameliorates high glucose-induced insulin resistance by regulating pi3k/akt-nrf2-gsk3β signaling pathways in hepg2 cells. J Ethnopharmacol 295:115397
Mo M, Pan L, Deng L, Liang M, Xia N, Liang Y (2024) Iron Overload induces hepatic ferroptosis and insulin resistance by inhibiting the Jak2/stat3/slc7a11 signaling pathway. Cell Biochem Biophys. :1–16
Lu L, Ye X, Yao Q, Lu A, Zhao Z, Ding Y et al (2018) Egr2 enhances insulin resistance via JAK2/STAT3/SOCS-1 pathway in HepG2 cells treated with palmitate. Gen Comp Endocrinol 260:25–31
Page M, Kell D, Pretorius E (2022) The role of lipopolysaccharide-induced cell signalling in chronic inflammation. Chronic Stress (Thousand Oaks) 6:24705470221076390
Meng L, Song Z, Liu A, Dahmen U, Yang X, Fang H (2021) Effects of lipopolysaccharide-binding protein (LBP) single nucleotide polymorphism (SNP) in infections, inflammatory diseases, metabolic disorders and cancers. Front Immunol 12:681810
Page MJ, Kell DB, Pretorius E (2022) The role of lipopolysaccharide-induced cell signalling in chronic inflammation. Chronic Stress 6:24705470221076390
Pussinen PJ, Kopra E, Pietiäinen M, Lehto M, Zaric S, Paju S et al (2022) Periodontitis and cardiometabolic disorders: the role of lipopolysaccharide and endotoxemia. Periodontol 2000 89(1):19–40
Liu J, Kang R, Tang D (2024) Lipopolysaccharide delivery systems in innate immunity. Trends Immunol
Mazgaeen L, Gurung P (2020) Recent advances in lipopolysaccharide recognition systems. Int J Mol Sci 21(2):379
Frances L, Tavernier G, Viguerie N (2021) Adipose-derived lipid-binding proteins: the good, the bad and the metabolic diseases. Int J Mol Sci 22(19):10460
Won Y, Yang JI, Park S, Chun JS (2021) Lipopolysaccharide binding protein and CD14, cofactors of toll-like receptors, are essential for low‐Grade inflammation–Induced exacerbation of cartilage damage in mouse models of Posttraumatic Osteoarthritis. Arthritis Rheumatol 73(8):1451–1460
Wu H, Ballantyne CM (2020) Metabolic inflammation and insulin resistance in obesity. Circul Res 126(11):1549–1564
Acknowledgements
This project supported by the Doctoral Research Fund of Hubei University of Science and Technology, project No. Q201810. Therefore, we express our gratitude to them for their assistance.
Funding
This project supported by the Doctoral Research Fund of Hubei University of Science and Technology, project No. Q201810.
Author information
Authors and Affiliations
Contributions
RA and XM were involved in designing the study. Searching were conducted and results were interpreted by RA, YJ, MD and XM. The initial draft of the manuscript was written by VS, AA, NJ, FS and FA. The manuscript was then edited by RA and MD. All authors reviewed and endorsed the final version of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not Applicable.
Consent of publication
Not Applicable.
Competing Interests
Not Applicable.
Additional information
Managed by Massimo Federici.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Miao, X., Alidadipour, A., Saed, V. et al. Hepatokines: unveiling the molecular and cellular mechanisms connecting hepatic tissue to insulin resistance and inflammation. Acta Diabetol (2024). https://doi.org/10.1007/s00592-024-02335-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00592-024-02335-9