
Abstract
Adipose tissue regulates systemic energy metabolism through adipokine production as well as energy storage and energy supply to other organs in response to changes in energy status. Adipose tissue dysfunction is therefore thought to be a key contributor to the pathogenesis of a variety of metabolic disorders including nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). Given that insulin plays a central role in the regulation of many aspects of adipocyte function, insulin resistance in adipose tissue is implicated in the pathogenesis of metabolic disorders as a cause of adipose tissue dysfunction. The concept of metabolic dysfunction-associated fatty liver disease (MAFLD) has recently been proposed for liver disease associated with metabolic disorders in both obese and nonobese individuals, with insulin resistance in adipose tissue likely being an important factor in its pathogenesis. This review outlines the relation between insulin resistance in adipose tissue and metabolic disorders, with a focus on the physiological relevance and mechanism of action of 3′-phosphoinositide-dependent kinase 1 (PDK1), a key kinase in insulin signaling, and its downstream transcription factor FoxO1 in adipocytes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adipocytes store excess energy as triglyceride in individuals in the fed state, whereas they break down the stored triglyceride and supply the released fatty acids to other organs such as skeletal muscle and the liver in the fasted state. In addition, adipocytes secrete various adipokines such as adiponectin and leptin that regulate the interactions between adipose tissue and other organs and play a key role in the regulation of energy metabolism. Insulin is a major regulator of many aspects of adipocyte function [1, 2]. Adipose tissue dysfunction related to either quantitative or qualitative changes in this tissue can give rise to metabolic disorders, such as systemic insulin resistance, diabetes mellitus, and dyslipidemia. Furthermore, such adipose tissue dysfunction has recently been implicated in the pathogenesis of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). NAFLD and the more severe NASH are usually associated with obesity and related comorbidities, but they can also develop in the absence of obesity, in which case they are referred to as lean NAFLD/NASH. Although lean NAFLD/NASH has often been described in Asian populations, it has been diagnosed in 10% to 20% of nonobese white individuals [3]. Pathological factors underlying the development of NAFLD/NASH in lean individuals remain unclear, but adipose tissue dysfunction, hormonal changes, reduced muscle mass, and gut dysbiosis in addition to genetic factors might play a role [3,4,5,6,7]. The concept of metabolic dysfunction–associated fatty liver disease (MAFLD) has recently been proposed for both obese and nonobese (normal weight or underweight) individuals [8]. Insulin resistance in adipose tissue that leads to adipose tissue dysfunction might contribute to the pathogenesis of this condition. This review outlines the relation between adipose tissue insulin resistance and metabolic disorders, focusing on insight into the physiological relevance and mechanism of action of the PDK1–FoxO1 signaling axis obtained from studies of adipocyte-specific knockout mice.
Insulin signaling and its physiological role in adipose tissue
The binding of insulin to the insulin receptor increases the tyrosine kinase activity of the receptor and the consequent phosphorylation of tyrosine residues of insulin receptor substrate (IRS). Tyrosine-phosphorylated IRS then binds to and activates phosphatidylinositol 3-kinase, which results in the production of phosphatidylinositol 3,4,5-trisphosphate (PIP3) at the plasma membrane. 3′-Phosphoinositide–dependent kinase 1 (PDK1) is a protein kinase with an NH2-terminal kinase domain and a COOH-terminal pleckstrin homology (PH) domain, and becomes localized to and activated at the plasma membrane by binding of its PH domain to PIP3 [9]. PDK1 activates the protein kinase Akt by phosphorylating a threonine residue in its activation loop, and Akt then targets various effector molecules including forkhead box O1 (FoxO1), mechanistic target of rapamycin (mTOR), and glucose transporter 4 (GLUT4) [9]. PDK1 is thought to be the only physiological kinase responsible for threonine phosphorylation of Akt [10].
Adipocyte-specific insulin receptor knockout (A-IRKO) mice generated with the use of a Cre recombinase transgene under the control of the adipocyte protein 2 (aP2) gene promoter manifest a reduced fat mass, increased insulin sensitivity and glucose tolerance, and longer life span compared with control mice [11, 12] (Table 1). In contrast, A-IRKO mice generated with a Cre transgene under the control of the adiponectin (Adipoq) gene promoter develop severe lipoatrophy and other metabolic disorders including insulin resistance, hyperglycemia, dyslipidemia, and NAFLD/NASH [13, 14] (Table 1). Potential problems with the aP2-Cre mouse model have been noted, including a possible allele-dependent reduction in recombination efficiency as well as Cre expression in cell types other than adipocytes [15]. The contrasting phenotypes of the two types of A-IRKO mice are therefore thought to be due to differences in the characteristics of the Cre transgenic mice. These knockout mice nevertheless revealed that insulin signaling in adipocytes plays a central role in the regulation of systemic insulin sensitivity, glucose and lipid metabolism, fat mass, and liver histology.
Physiological role of PDK1 in adipose tissue
To investigate signaling downstream of the insulin receptor related to insulin action in adipocytes, we generated adipocyte-specific PDK1 knockout (A-PDK1KO) mice with the use of the Adipoq-Cre transgene [16]. The regulation of lipid synthesis and lipolysis as well as of glucose uptake by insulin was markedly impaired in isolated adipocytes from A-PDK1KO mice. The plasma concentrations of adiponectin and leptin were also markedly decreased in these mice. In addition, A-PDK1KO mice manifested a reduced fat mass and reduced adipocyte diameter, reflecting dysregulation of lipid synthesis and lipolysis. Ablation of PDK1 in adipocytes thus resulted in extensive adipose tissue dysfunction. On the other hand, no obvious inflammation was apparent in adipose tissue of A-PDK1KO mice, as indicated by the absence of both increased macrophage infiltration and increased circulating concentrations of inflammatory cytokines and chemokines. A-PDK1KO mice showed hyperglycemia and dyslipidemia as well as systemic insulin resistance on a normal diet. Furthermore, they showed marked lipid accumulation in the liver by 10 weeks of age as well as, on further aging, histological changes to the liver—such as hepatocyte ballooning, inflammatory cell infiltration, and fibrosis—similar to those associated with human NASH. Consistent with these histological changes, gene expression related to lipogenesis, inflammation, and fibrosis was upregulated in the liver of A-PDK1KO mice. These phenotypes are thus highly similar to those of the A-IRKO mice generated with the Adipoq-Cre transgene described above (Table 1). Our observations therefore indicated that PDK1 signaling plays a central role in the metabolic effects of insulin in adipocytes, and that impaired insulin action as a result of PDK1 deficiency in adipocytes gives rise to NAFLD/NASH in addition to systemic insulin resistance and other metabolic disorders.
Physiological role of FoxO1 in adipose tissue
PDK1 signals via various effector molecules downstream of Akt by activating Akt. FoxO1 is a transcription factor that is highly expressed in insulin target tissues including adipose tissue and regulates the expression of various genes [17]. Activation of insulin signaling results in the phosphorylation of FoxO1 by Akt and its consequent inactivation through sequestration in the cytoplasm [17]. Insulin-dependent phosphorylation of FoxO1 was abolished in adipose tissue of A-PDK1KO mice, indicative of constitutive activation of FoxO1 in adipocytes. To clarify the relevance of FoxO1 to the metabolic disorders of A-PDK1KO mice, we generated adipocyte-specific PDK1/FoxO1 double-knockout (A-PDK1/FoxO1DKO) mice, which lack FoxO1 in addition to PDK1 specifically in adipocytes [16]. Compared with A-PDK1KO mice, A-PDK1/FoxO1DKO mice showed improved insulin sensitivity in the liver and skeletal muscle, resulting in amelioration of hyperglycemia. In addition, the NASH-like histology apparent in A-PDK1KO mice was markedly attenuated in A-PDK1/FoxO1DKO mice. On the other hand, the dyslipidemia, reduced plasma concentrations of adiponectin and leptin, and reduced fat mass observed in A-PDK1KO mice were not altered in A-PDK1/FoxO1DKO mice. These observations thus suggested that, of the various metabolic disorders caused by the loss of PDK1 in adipocytes, FoxO1 contributes to the pathogenesis of insulin resistance, hyperglycemia, and NAFLD/NASH, whereas other molecules or pathways may be responsible for the regulation of lipid metabolism, fat mass, and the production or secretion of adiponectin and leptin.
Mechanism of systemic insulin resistance determined by the PDK1–FoxO1 axis in adipose tissue
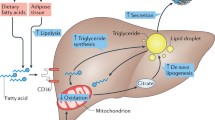
With the use of liquid chromatography and tandem mass spectrometry, we found that the concentration of leukotriene B4 (LTB4) in plasma as well as in adipose tissue was increased in A-PDK1KO mice and that this increase was reversed in A-PDK1/FoxO1DKO mice [16]. LTB4 is a lipid mediator generated from arachidonic acid derived from membrane phospholipids, and the first step in its biosynthesis is catalyzed by 5-lipoxygenase (5-LO) [18, 19]. Consistent with this increase in LTB4 levels, the expression of 5-LO in adipose tissue was found to be increased in A-PDK1KO mice, and again, this increase was normalized in A-PDK1/FoxO1DKO mice. In vitro experiments with isolated and cultured adipocytes revealed that insulin inhibits the production of LTB4 by down-regulating the expression of 5-LO, and that this action of insulin is mediated by the PDK1-FoxO1 pathway. LTB4 has been implicated in the pathogenesis of inflammatory diseases, such as bronchial asthma, rheumatoid arthritis, and atherosclerosis [20], and its plasma concentration has been found to be increased in individuals with metabolic syndrome [21]. Studies with mice have also shown that LTB4 is upregulated in adipose tissue of obese animals and that it contributes to the pathogenesis of systemic insulin resistance [22,23,24]. Consistent with these various observations, we found that pharmacological or genetic inhibition of the 5-LO–LTB4 pathway ameliorated insulin resistance in A-PDK1KO mice [16]. Insulin signaling via the PDK1–FoxO1 axis in adipose tissue of mice thus controls systemic insulin sensitivity through regulation of LTB4 production (Fig. 1). We also showed that the plasma LTB4 concentration in human subjects was positively correlated with homeostasis model assessment-insulin resistance (HOMA-IR) and blood insulin concentration, suggesting that LTB4 might also play a role in the pathogenesis of insulin resistance in humans [16].

Model for metabolic regulation by the PDK1–FoxO1 axis and other downstream pathways and factors in adipocytes. Red arrows and boxes indicate changes induced by loss of PDK1
Mechanism of NAFLD/NASH determined by the PDK1–FoxO1 axis in adipose tissue
About 60% of triglyceride that accumulates in the liver of individuals with NAFLD is derived from fatty acids produced by lipolysis in adipose tissue [25]. Insulin resistance in adipose tissue is therefore likely to promote hepatic fat accumulation associated with NAFLD/NASH by increasing the transfer of free fatty acids from adipose tissue to the liver as a result of impaired lipid synthesis and increased lipolysis. The plasma concentration of free fatty acids after feeding is higher in A-PDK1KO mice than in control animals, suggesting that an increased flux of free fatty acids from adipose tissue to the liver contributes to the pathogenesis of NAFLD/NASH in these mice. Whereas the NAFLD/NASH phenotype is attenuated in A-PDK1/FoxO1DKO mice, however, these mice show a similar increase in the plasma concentration of free fatty acids after feeding as do A-PDK1KO mice, suggesting that FoxO1 in adipocytes contributes to the pathogenesis of NAFLD/NASH through a mechanism independent of the regulation of free fatty acid flux to the liver. Given that genetic or pharmacological inhibition of the 5-LO–LTB4 pathway ameliorated systemic insulin resistance but not NAFLD/NASH in A-PDK1KO mice, the PDK1–FoxO1 axis in adipocytes may regulate the interaction between adipose tissue and the liver via an unidentified mechanism independent of LTB4 (Fig. 1).
Adipose tissue insulin resistance and NAFLD/NASH in humans
The adipose tissue insulin resistance index (Adipo-IR), calculated as the product of the fasting plasma concentrations of insulin and free fatty acids, is used as an indicator of insulin resistance in adipose tissue in humans [26], whereas the euglycemic–hyperinsulinemic clamp test allows for a more accurate assessment of insulin sensitivity in adipose tissue. Given that lipolysis in adipose tissue is suppressed by insulin, the presence of insulin resistance in adipose tissue reduces the rate of suppression of the circulating free fatty acid concentration during clamping. The extent of insulin resistance in adipose tissue as determined by the euglycemic–hyperinsulinemic clamp test was found to be higher in NAFLD patients without diabetes, dyslipidemia, or hypertension than in healthy individuals matched for body mass index (BMI) and body fat mass [27]. Studies of diabetic and nondiabetic men also found that insulin resistance in adipose tissue correlated with hepatic fat accumulation in a manner independent of BMI and visceral fat mass [28, 29]. Moreover, adipose tissue insulin resistance was shown to correlate with hepatic fat content as well as with metabolic abnormalities in a manner independent of body fat mass even in nonobese healthy Japanese men with a BMI of < 25 kg/m2 [30, 31]. Given that insulin resistance in adipose tissue is thought to contribute to the pathogenesis of hepatic fat accumulation independently of body weight and body fat mass, it is likely also an important contributor to the pathogenesis of MAFLD.
Adipose tissue insulin resistance was found to be higher in individuals with NAFLD than in those with a normal liver, and NAFLD patients with a higher adipose tissue insulin resistance also had higher circulating levels of liver transaminases and more pronounced liver fibrosis [32]. Insulin resistance in adipose tissue was also found to worsen with progression of NAFLD/NASH [33]. Adipose tissue insulin resistance has thus been implicated in the pathogenesis of NASH as well as NAFLD in humans. Although adipose tissue insulin resistance is thought to give rise to hepatic fat accumulation as a result of an increased flux of free fatty acids from adipose tissue to the liver, it remains unknown whether and how such resistance directly affects the pathogenesis of hepatic inflammation and fibrosis.
Concluding remarks
This review has described the relevance of adipose tissue insulin resistance to systemic insulin resistance and NAFLD/NASH, with a focus on our previous study of A-PDK1KO and A-PDK1/FoxO1DKO mice. PDK1 plays a central role in the metabolic effects of insulin, and impairment of PDK1 signaling in adipose tissue gives rise to NAFLD/NASH in addition to metabolic disorders including systemic insulin resistance, hyperglycemia, and dyslipidemia. Elucidation of the importance and mechanisms of signaling downstream of PDK1 in adipocytes should provide further insight into the pathogenesis of metabolic diseases including NAFLD/NASH as well as inform the development of new therapeutic strategies.
References
Santoro A, McGraw TE, Kahn BB. Insulin action in adipocytes, adipose remodeling, and systemic effects. Cell Metab. 2021;33:748–57.
White MF, Kahn CR. Insulin action at a molecular level–100 years of progress. Mol Metab. 2021;52: 101304.
Younes R, Bugianesi E. NASH in lean individuals. Semin Liver Dis. 2019;39:86–95.
Maier S, Wieland A, Cree-Green M, Nadeau K, Sullivan S, et al. Lean NAFLD: an underrecognized and challenging disorder in medicine. Rev Endocr Metab Disord. 2021;22:351–66.
Francque S, Wong VW. NAFLD in lean individuals: not a benign disease. Gut. 2022;71:234–6.
Younes R, Govaere O, Petta S, Miele L, Tiniakos D, et al. Caucasian lean subjects with non-alcoholic fatty liver disease share long-term prognosis of non-lean: time for reappraisal of BMI-driven approach? Gut. 2022;71:382–90.
Luukkonen PK, Qadri S, Ahlholm N, Porthan K, Männistö V, et al. Distinct contributions of metabolic dysfunction and genetic risk factors in the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2022;76:526–35.
Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73:202–9.
Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22.
Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014;6: a009191.
Blüher M, Michael MD, Peroni OD, Ueki K, Carter N, et al. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell. 2002;3:25–38.
Blüher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–4.
Boucher J, Softic S, El Ouaamari A, Krumpoch MT, Kleinridders A, et al. Differential roles of insulin and IGF-1 receptors in adipose tissue development and function. Diabetes. 2016;65:2201–13.
Softic S, Boucher J, Solheim MH, Fujisaka S, Hearing MF, et al. Lipodystrophy due to adipose tissue-specific insulin receptor knockout results in progressive NAFLD. Diabetes. 2016;65:2187–200.
Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62:864–74.
Hosooka T, Hosokawa Y, Matsugi K, Shinohara M, Senga Y, et al. The PDK1-FoxO1 signaling in adipocytes controls systemic insulin sensitivity through the 5-lipoxygenase-leukotriene B4 axis. Proc Natl Acad Sci USA. 2020;117:11674–84.
Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005;16:183–9.
Samuelsson B, Dahlén SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–6.
Serhan CN, Haeggstrom JZ, Leslie CC. Lipid mediator networks in cell signaling: update and impact of cytokines. FASEB J. 1996;10:1147–58.
Toda A, Yokomizo T, Shimizu T. Leukotriene B4 receptors. Prostagland Other Lipid Mediat. 2002;68–69:575–85.
Novgorodtseva TP, Karaman YK, Zhukova NV, Lobanova EG, Antonyuk MV, et al. Composition of fatty acids in plasma and erythrocytes and eicosanoids level in patients with metabolic syndrome. Lipids Health Dis. 2011;10:82.
Mothe-Satney I, Filloux C, Amghar H, Pons C, Bourlier V, et al. Adipocytes secrete leukotrienes: contribution to obesity-associated inflammation and insulin resistance in mice. Diabetes. 2012;61:2311–9.
Spite M, Hellmann J, Tang Y, Mathis SP, Kosuri M, et al. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J Immunol. 2011;187:1942–9.
Li P, Oh DY, Bandyopadhyay G, Lagakos W, Talukdar S, et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med. 2015;21:239–47.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51.
Ter Horst KW, van Galen KA, Gilijamse PW, Hartstra AV, de Groot PF, et al. Methods for quantifying adipose tissue insulin resistance in overweight/obese humans. Int J Obes (Lond). 2017;41:1288–94.
Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, et al. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia. 2005;48:634–42.
Kotronen A, Juurinen L, Tiikkainen M, Vehkavaara S, Yki-Järvinen H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology. 2008;135:122–30.
Kotronen A, Seppälä-Lindroos A, Bergholm R, Yki-Järvinen H. Tissue specificity of insulin resistance in humans: fat in the liver rather than muscle is associated with features of the metabolic syndrome. Diabetologia. 2008;51:130–8.
Sugimoto D, Tamura Y, Takeno K, Kaga H, Someya Y, et al. Clinical features of nonobese, apparently healthy, Japanese men with reduced adipose tissue insulin sensitivity. J Clin Endocrinol Metab. 2019;104:2325–33.
Kiya M, Tamura Y, Takeno K, Someya Y, Kakehi S, et al. Adipose insulin resistance and decreased adiponectin are correlated with metabolic abnormalities in nonobese men. J Clin Endocrinol Metab. 2021;106:e2228–38.
Lomonaco R, Ortiz-Lopez C, Orsak B, Webb A, Hardies J, et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:1389–97.
Musso G, Cassader M, De Michieli F, Rosina F, Orlandi F, et al. Nonalcoholic steatohepatitis versus steatosis: adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology. 2012;56:933–42.
Acknowledgements
We thank our present and former colleagues at the University of Shizuoka and Kobe University Graduate School of Medicine as well as all collaborators for contributions to the research described in this review. This study was supported in part by Japan Society for the Promotion of Science KAKENHI grants 15K09389, 18K08478, and 21H03355, as well as by grants from Yamaguchi Endocrine Research Foundation, Suzuken Memorial Foundation, Hyogo Science and Technology Association, Astellas Foundation for Research on Metabolic Disorders, and Japan Diabetes Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Imi, Y., Ogawa, W. & Hosooka, T. Insulin resistance in adipose tissue and metabolic diseases. Diabetol Int 14, 119–124 (2023). https://doi.org/10.1007/s13340-022-00616-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13340-022-00616-8