
Abstract
Restoration of the American chestnut (Castanea dentata) is underway using backcross breeding that confers chestnut blight disease resistance from Asian chestnuts (most often Castanea mollissima) to the susceptible host. Successful restoration will depend on blight resistance and performance of hybrid seedlings, which can be impacted by below-ground fungal communities. We compared fungal communities in roots and rhizospheres (rhizobiomes) of nursery-grown, 1-year-old chestnut seedlings from different genetic families of American chestnut, Chinese chestnut, and hybrids from backcross breeding generations as well as those present in the nursery soil. We specifically focused on the ectomycorrhizal (EcM) fungi that may facilitate host performance in the nursery and aid in seedling establishment after outplanting. Seedling rhizobiomes and nursery soil communities were distinct and seedlings recruited heterogeneous communities from shared nursery soil. The rhizobiomes included EcM fungi as well as endophytes, putative pathogens, and likely saprobes, but their relative proportions varied widely within and among the chestnut families. Notably, hybrid seedlings that hosted few EcM fungi hosted a large proportion of potential pathogens and endophytes, with possible consequences in outplanting success. Our data show that chestnut seedlings recruit divergent rhizobiomes and depart nurseries with communities that may facilitate or compromise the seedling performance in the field.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Non-native forest pests and invasive pathogens have had catastrophic impacts on tree species around the globe (Boyd et al. 2013; Santini et al. 2013; Lovett et al. 2016). In the USA, one such pathogen is the chestnut blight fungus, ascomycete Cryphonectria parasitica (Murr.) Barr, that caused a disease that rapidly eliminated the American chestnut (Castanea dentata (Marsh.) Borkh.) as an upper canopy dominant throughout its native range in the first half of the twentieth century (Anagnostakis 1987). Chestnut blight causes necrotic cankers on the branch and trunk surfaces leading to girdling and eventual mortality in susceptible trees. To restore the chestnut to eastern North American forests, a backcross breeding approach has been developed in an attempt to confer blight resistant genes from Asian chestnut species, most often the Chinese chestnut (Castanea mollisima Blume), into the American chestnut using conventional breeding (Burnham et al. 1986; Anagnostakis 2012). Such breeding programs aim to generate progeny that exhibit American chestnut phenotypic form and growth characteristics as well as maintain durable blight resistance (Hebard 2001; Diskin et al. 2006; Sniezko 2006; Anagnostakis 2012).
These breeding programs currently produce material that is being field tested under various forest management conditions and is exhibiting low to intermediate resistance (Clark et al. 2014a; Clark et al. 2016; Steiner et al. 2016). Despite the importance of resistance breeding programs for restoring pathogen-decimated tree populations and species, sparse research is available to better understand factors, other than disease resistance, that will affect outplanting success of hybrid populations (Thompson et al. 2006; Seddon 2010; Jacobs 2013; Clark et al. 2014a, 2014b; Pinchot et al. 2017). Like many other temperate forest trees, American chestnut forms important ectomycorrhizal (EcM) mutualisms (Palmer et al. 2008; Bauman et al. 2017; Newhouse et al. 2018) that facilitate host nutrient uptake, improve host growth and performance, and may improve pathogen resistance/tolerance (Smith and Read 1997). Emerging evidence suggests that different host genotypes may recruit distinct fungal communities (e.g., Lamit et al. 2016; Perez-Izquierdo et al. 2017), resulting in potential breeding-associated effects on non-target fungal communities that may be consequential to the establishment and survival of juvenile plants.
Nursery-reared seedlings recruit diverse communities of rhizosphere fungi (fungal rhizobiomes, or fungi associated with roots and adhering soils) including pathogens, endophytes, and putative mutualists (Menkis et al. 2005, 2016; Stenström et al. 2014). These nursery-borne fungi may affect the seedling performance under the nursery conditions (Sinclair et al. 1982; Menkis et al. 2007) and can be particularly important for seedling establishment once outplanted to the field (Kropp and Langlois 1990; Lilja and Rikala 2000). Heavy pathogen loads at the nursery can remain asymptomatic—and therefore undetected—yet compromising the outplanting success (Lilja and Rikala 2000). Alternatively, mycorrhizal mutualists that establish in the nursery may aid in survival after outplanting (Menkis et al. 2007). Furthermore, understanding the communities that may have been introduced into soil with the outplanted nursery stocks can later serve as a baseline for assessment of host performance and rhizobiome composition after outplanting.
Only a few studies have targeted the root-associated fungi of American chestnuts (Dulmer 2006; Bauman et al. 2013, 2017; D’Amico et al. 2015; Stephenson et al. 2017), although associating with compatible mutualists may be essential for restoration of threatened plants (Perry et al. 1987). As a result, restoration strategies already consider the importance of mycorrhizal inocula (Jacobs et al. 2013). Mycorrhizal seedlings generally outperform their nonmycorrhizal counterparts (Kropp and Langlois 1990; Quoreshi and Timmer 2000; Menkis et al. 2007), even though the early, nursery-recruited symbionts may be short-lived and rapidly outcompeted by naturally occurring fungi after outplanting (Menkis et al. 2007).
Fungi that colonize American chestnut include a number of common EcM taxa (Palmer et al. 2008; Dulmer et al. 2014; Stephenson et al. 2017) shared with other EcM hosts. The establishing seedlings likely benefit from sharing mycorrhizal partners with older overstory trees (Horton and van der Heijden 2008), as existing EcM networks are extensive and can expedite colonization (Dickie et al. 2002; Nara 2006a, 2006b), improve access to resources (Dickie et al. 2002, 2007), and minimize negative effects of root competition (Booth 2004). However, it remains largely unknown if chestnut blight breeding programs have adverse non-target effects on EcM colonization and diversity, i.e., does selection of blight resistant progeny alter the host compatibility with mycorrhizal partners (but see D’Amico et al. 2015).
In this study, we examined 1-year-old chestnut nursery seedlings for fungal rhizobiomes prior to outplanting to the field. We compared and contrasted six backcross hybrid, two American chestnut, and one Chinese chestnut families, and analyzed the nursery soil—that we considered the potential source inoculum. Similarly to D’Amico et al. (2015), we focused on EcM communities but surveyed them using high throughput sequencing to deeply dissect the rhizobiomes recruited from the nursery. We specifically aimed to address following research questions: (i) Does the nursery soil differ from the seedling host rhizobiomes in fungal richness, diversity and community composition? (ii) Do backcross hybrids and American and Chinese chestnut families differ in their rhizobiomes while growing in a presumably homogeneous nursery substrate? (iii) Finally, should the host rhizobiomes be distinct among species, breeding generations, or families, we also aimed to identify the distinguishing fungal taxa. The resultant data indicate substantial heterogeneity in the communities that are associated with the nursery-reared seedlings, raising thus questions about the need for mycorrhizal inoculation of high-value nursery stock prior to outplanting.
Materials and methods
Experimental material
We obtained nuts from two American Chestnuts trees (Pryor 043, Pryor182), one Chinese Chestnut tree (Princeton), five backcross hybrid trees from The American Chestnut Foundation (TACF), and one backcross hybrid tree from the Connecticut Agricultural Experimental Station (CAES) (Table 1). Hereafter, a “family” refers to progeny from a single open-pollinated orchard tree with a distinct lineage and limited pollen contamination from outside sources (Hebard 2006). The Chinese chestnut family was located on private property with limited pollen contamination (Paul Sisco, TACF, Asheville, NC, USA, personal communication) (Burnham et al. 1986). The TACF hybrids (D22, W3, W4, W5, and W6) are theoretically 94% C. dentata and 6% C. mollissima (Hebard 2006), whereas the CAES hybrid (4-75) is theoretically 90% American chestnut with remaining 10% a mix of Chinese chestnut, European chestnut (Castanea sativa), and Japanese chestnut (Castanea crenata) (Anagnostakis 2012).
We sampled roots and soils within an operational nursery at the Indiana State Nursery in Vallonia, Indiana. The nursery beds were methyl bromide (CH3Br) fumigated before the nuts were sown by family in seed lots at the nursery on November 19, 2013. Likely as a result of the fumigation, we observed no fruiting bodies in the nursery beds at the time of sowing or at the time of seedling collection. At the nursery, each family seed lot was sown at a density of 65 nuts per m2, measured approximately 0.75 × 0.2 m, and was separated by 0.5 m of empty bed space. After sowing, the beds were open and arrival of inoculum was not controlled during the experiment. The seedlings were fertilized according to standard prescriptions to produce large, high-quality seedlings and irrigated as needed (cf. Kormanik et al. 1994). The fertilization regime ranged from roughly 5 g-N m−2 to an excess of 13 g-N m−2 applied every 2 weeks as NH4NO3 (Nov. 19, 5.2 g-N m−2; Dec. 3, 4.3 g-N m−2; Dec. 17, 4.5 g-N m−2; Dec. 31, 5.8 g-N m−2; Jan. 14, 8.2 g-N m−2; Jan. 28, 13.6 g-N m−2; Feb. 11, 13.6 g-N m−2) for a total of 55 g-N m−2 over the first 3 months in the nursery. Seedlings ranged from 13 to 213 cm in height averaging 99 cm at the time of preparation for outplanting. A machine lifter was used to undercut seedlings (25–30 cm) and loosen soil around the roots. Seedlings were manually removed from the nursery beds, roots packed in sphagnum moss as per the nursery standard operating protocol to minimize seedling desiccation during transport, and placed in poly-coated paper tree bags in cold storage until root sampling. We did not collect any root colonization data prior to outplanting. While we did not control for fungal inoculum in the sphagnum moss, we expect it to be uniform across the material, exposure short in duration, and an unlikely factor to explain divergence among the analyzed genetic families.
Our experiment included a total of four replicates of each of the nine chestnut families for a total of 36 root samples for the rhizobiome analyses. Seedlings were removed from the bags, rinsed free of sphagnum moss, and roots sampled with a pruner that removed approximately 20 g of secondary and feeder roots from each seedling. The pruner was sterilized in 20% solution of domestic bleach (0.534% sodium hypochlorite) between family samples. Tap roots were not sampled to avoid problems at planting to the field. The sampled roots were placed in Ziplock bags stored at 4 °C until shipped to Mississippi State University within 48 h.
To also assess the background soil inoculum in the nursery, we sampled four replicate bulk soil samples from Vallonia nursery immediately after lifting seedlings. Approximately 500 g of soil was collected for each sample within a 1 m by 1 m area by combining ten subsamples collected to a depth of 13 cm using a trowel. We sampled in the center of the beds to minimize potential edge effects. Sampling was conducted at four relatively equidistant locations to ensure adequate coverage of the nursery beds. The samples were refrigerated at 4 °C and shipped overnight on ice to Mississippi State University, where stored at − 80 °C until further processing. In total, our experimental design consisted of 36 root and 4 soil samples for a total of 40 samples.
DNA isolation and PCR
For each root and soil sample, genomic DNA was extracted from three subsamples of either roots or soil (~ 0.25 g fresh weight) using the PowerSoil DNA Isolation kit (MoBio Laboratories, Inc., Carlsbad, CA) as per the manufacturer’s protocol. The three replicate subsamples were pooled into one, DNA quantified with Nanodrop 2000 Spectophotometer (Thermo Scientific Waltham, MA), and DNA adjusted to 2 ng/μl. We also included a negative control, in which the tissue or soil was omitted. The control yielded minimal DNA; the elute was used as a negative control in the PCRs and included in the subsequent sequencing library.
For fungal community analyses, we targeted the Internal Transcribed Spacer region 2 (ITS2) that has been proposed as the universal metabarcode marker (Schoch et al. 2012). We amplified the ITS2 region in a 2-step PCR with the forward primer fITS7 (5′ - GTGARTCATCGAATCTTTG - 3′; Ihrmark et al. 2012) and the reverse primer ITS4 (5′ - TCCTCCGCTTATTGATATGC - 3′; White et al. 1990). All PCR reactions were carried out in duplicate 50 μl volumes with the following concentrations and volumes: 20 ng of template DNA (10 μl), 200 μM dNTPs (5 μl of 2 mM dNTP stock), 1 μM of forward and reverse primer (5 μl of each 10 μM primer stock), 10 μl of Phusion 5× HF buffer containing 7.5 mM MgCl2 for a final 1.5 mM MgCl2 concentration, 14.5 μl of molecular grade DEPC-treated water, and 1 unit (0.5 μl) of Phusion Green Hot Start II High-Fidelity DNA polymerase (Thermo Scientific, Pittsburgh, USA). The cycle conditions for the primary PCRs included an initial 30s denaturing at 98 °C, followed by 30 cycles of 10s denaturing at 98 °C, 10s annealing at 56 °C, 1 min extension at 72 °C, and final 5-min extension at 72 °C. Resulting duplicate amplicons were combined and purified using Sera-Mag SpeedBead Carboxylate-Modified Magnetic Particles (GE Healthcare, Little Chalfont Buckinghamshire, UK) in a 96-well SPRI plate format to remove excess primers and residual contaminants from the samples. We utilized a clean-up protocol identical to that provided by AgentCourt AMPure XP (Backman Coulter, Indianapolis, IN) but replaced the magnetic bead solution with Sera-Mag SpeedBead solution. To avoid the potential erroneous assignment of samples to experimental units (Carlsen et al. 2012), a secondary 5-cycle PCR included unique 12 bp barcodes appended to both forward and reverse primers under the same conditions as above, followed by a second magnetic Sera-Mag SpeedBead clean-up.
Illumina MiSeq library preparation
Purified amplicons were quantified using the ND2000 and 200 ng of each sample was pooled for sequencing. The negative extraction control did not yield comparable DNA concentration and the entire volume of the cleaned amplicon was included into the sequencing pool. Illumina specific primers and adapters were ligated to the amplicons using a NEBNext® DNA MasterMix for Illumina kit (New England Biolabs Inc., Ipswich, MA, USA) at the Integrated Genomics Facility at Kansas State University (Manhattan, KS, USA). The library was sequenced using paired-end MiSeq Reagent Kit v3 (Illumina, San Diego, CA, USA) with 2 × 300 cycles. The resulting raw paired-end sequence data are available at the Sequence Read Archive (SRA) at the National Center for Biotechnology Information (NCBI) under BioProject PRJNA432081, BioSamples SRS2903649-2903688.
Sequence data analyses
The sequence data were analyzed using the bioinformatics software mothur (v. 1.38, Schloss et al. 2009). After contig construction, the library contained 4,500,599 sequences. Contigs were screened to remove any that contained ambiguous bases, a disagreement in primer or barcode sequence, or a homopolymer of 8 bp or longer. The remaining 4,470,285 sequences were truncated to 236 bp to facilitate pre-clustering (Huse et al. 2008) and subsequent clustering using VSEARCH (Rognes et al. 2016) both of which require aligned sequences or sequences of equal length. Near identical sequences (up to 2 nucleotide differences) were pre-clustered to reduce sequencing bias (Huse et al. 2008) and screened for potential chimeras (UCHIME; Edgar et al. 2011). After the removal of the presumed chimeric sequences, the sequence data were clustered into operational taxonomic units (OTUs) at 97% similarity using VSEARCH (Rognes et al. 2016). Rare OTUs represented by 10 or fewer sequences were removed, as they may represent PCR and/or MiSeq artifacts (Brown et al. 2015; Oliver et al. 2015). The negative control yielded a small number of sequences, which were culled during the data processing. OTUs were assigned to taxon affinities using the Naïve Bayesian Classifier (Wang et al. 2007) and the UNITE taxonomy reference (http://unite.ut.ee/repository.php). The final data set contained 872 OTUs across the 40 samples representing chestnut roots and nursery soils. We subsampled the data to an equal 10,000 sequences per sample and iteratively calculated Good’s coverage, observed (SObs) and extrapolated (Chao1, Boneh) richness, diversity (Shannon’s H′), and evenness (Shannon’s EH) using mothur (v. 1.38, Schloss et al. 2009).
Statistical analyses
Estimators for coverage, richness (Sobs), extrapolative richness (ChaoI, Boneh), diversity (H′), and evenness (EH) data were non-normal and heteroscedastic. These data were accordingly natural log transformed (ln; Sobs, ChaoI, Boneh, H′) or arc sine square root-transformed (EH). To test for differences in richness (Sobs), extrapolative richness (ChaoI, Boneh), diversity (H′), and evenness (EH) between the roots and soils, we compared means using Dunnett’s test (Dunnett 1955). In this test, each richness, diversity, and evenness estimator for each family was contrasted against those in the nursery bulk soil using JMP (version 10.0.0). To compare richness, diversity, and evenness among the chestnut families, we used one-way ANOVAs followed by Tukey’s HSD to test for differences among all possible comparisons. These analyses excluded the soil samples to better focus on the differences among the families.
To visualize the fungal community composition, we calculated pairwise Bray–Curtis distance matrices and visualized the community composition using non-metric multidimensional scaling (NMS) in PC-ORD (version 6.19). Consistently with the analyses of the richness and diversity estimators, we conducted these analyses in two steps. First, we analyzed NMS ordination that included the nursery soil samples as well as the samples representing the nine chestnut families. In these analyses, a three-dimensional ordination (k = 3) provided an optimal solution and represented 86.4% of the variation with a stress of 0.13, separating the soils and a majority of the chestnut family rhizobiomes; these analyses are provided as a supplement (Supplemental Fig. S1a–c). Second, we reanalyzed the data after omitting the soil samples to focus only on the nine chestnut families. In these latter analyses, a three-dimensional ordination (k = 3) again provided an optimal solution and represented 85.6% of the variation with a stress of 0.14. The community data across the different treatments were compared using permutation-based MANOVA (PerMANOVA; Anderson 2001). In addition to these analyses that included the entire data matrix for the nine families, we analyzed a dataset that included only the 90 core OTUs present in at least half of the samples (see Unterseher et al. 2011 for core taxon analyses) and compared these communities using PerMANOVA. Similarly to the broader data matrix, a three-dimensional solution was optimal, representing 83.0% of the variation with a stress of 0.14.
To identify OTUs that were disproportionally enriched under one treatment condition over others, we performed Indicator Taxon Analyses (Dufrene and Legendre 1997) in PC-ORD. These analyses identified a large number of potential indicators (P < 0.05)—171 indicators in total, 26 for chestnut family rhizobiomes and 145 for soils (Supplemental Table S1). Indicator analyses that excluded the soils identified a total of 86 indicators (P < 0.05) across the nine families (Supplemental Table S2). As a result of the large number of potential indicator taxa and to better focus on the commonly occurring indicators, we present and discuss the indicator analyses only for the reduced core taxa that occurred in at least half of the 36 rhizobiome samples. However, we present the full indicator taxon lists for the complete datasets as supplements (Supplemental Tables S1 and S2).
To assign the detected OTUs to potential ecological functions and ecological guilds, we used FUNGuild (Nguyen et al. 2016) as described in Veach et al. (2017). We were specifically interested in the EcM fungi and those that were present when EcM fungi were in low abundance in our root samples. We used Dunnett’s test to compare the mean abundance of EcM and putative plant pathogens in the roots and soil. We also compared the FUNGuild-assigned functional roles across the nine breeding lines using one-way ANOVA followed by Tukey’s HSD post hoc tests. These analyses were first conducted using only the high probability FUNGuild assignments, followed by a separate analysis considering all assignments.
Results
General community description
Of the 872 OTUs in total, 330 (37.8%) OTUs occurred in soil, 617 (70.8%) in chestnut rhizobiomes, 177 (20.3%) in both soil and chestnut family rhizobiomes, and 153 (17.5%) OTUs were unique to soil and 440 (50.5%) were unique to chestnut rhizobiomes. The large proportion of OTUs observed only in the rhizobiomes is likely a result of nine times greater sampling of the rhizobiomes than of the nursery soils. Similarly to the small proportion of OTUs that were observed in both soils and rhizobiomes (20.3%), only 56 (9.1%) of the OTUs that occurred in rhizobiomes were present in all sampled chestnut families and 303 (49.1%) were shared by two or more sampled families. Taken together, these data indicate a great heterogeneity in the fungal communities in the nursery soils and in chestnut family rhizobiomes after 1 year in the nursery.
Overall, Ascomycota (242,468 sequences, 60.6%; 419 OTUs) and Basidiomycota (140,163 sequences, 35.0%; 223 OTUs) dominated the fungal communities, with only a small proportion of the data representing basal taxa formerly assigned to Zygomycota (10,744 sequences, 2.7%; 43 OTUs) or Chytridiomycota (570 sequences, 0.14%; 25 OTUs). Additionally, a small proportion of the data remained unclassified beyond kingdom Fungi (5981 sequences, 1.5%; 69 OTUs). Interestingly, approximately 95% of these unclassified sequences were found in the nursery soils (5707 sequences), whereas only few occurred in the rhizobiomes (274 sequences, 4.5%). The soil and rhizobiome communities were distinct (Supplemental Fig. S1a–c). Of the taxa that were assigned to a genus level, soils were dominated by Podospora (2311 sequences, 5.8%), Chaetomium (1962 sequences, 4.9%), and Mortierella (1342 sequences, 3.4%), whereas the rhizobiomes were dominated by Guehomyces (23,455 sequence, 6.5%), Aureobasidium (17,534 sequences, 4.9%), and Fusarium (12,480 sequences, 3.5%).
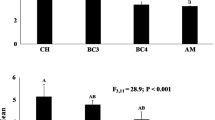
Of the 872 OTUs, 462 (52.9%) received a FUNGuild assignment (Fig. 1), representing 155,942 sequences (39.0% of total). Only a total of 72 OTUs (37,744 sequences, 9.4% of all sequences and 8.3% of all OTUs) received an assignment with a “high probability.” Among all OTUs that were assigned to a guild, “undefined saprotroph” OTUs were overwhelmingly most common (168, 36.4% of OTUs with a guild assignment; 16.3% of sequences representing those OTUs), followed by “fungal parasite-undefined saprotroph” OTUs (39, 8.4%; 1.9%) and “plant pathogen” OTUs (38, 8.2%; 7.8%). Although only relatively few OTUs were assigned to “ectomycorrhizal” guild, these OTUs (12, 2.6%) represented the second largest sequence count among the assigned sequences (12.2%) after the “saprotroph” OTUs (Fig. 1). These EcM included OTUs assigned to Ceratobasidium (1 OTU), Chloridium (1 OTU), Hebeloma (1 OTU), Laccaria (2 OTU), Pisolithus (2 OTUs), Tomentella (1 OTU), Scleroderma (3 OTUs), and Sphaerosporella (1 OTU). The relative abundances of EcM fungi were variable in soils (4.1 ± 3.3%) and ranged widely among the chestnut family rhizobiomes (from 0.04 ± 0.03% in Princeton to 17.4 ± 10.3% in D22; Fig. 1). Yet, none of the families differed in the sequence abundance of EcM fungi from the nursery soil (Dunnett’s test; P ≥ 0.05), likely due to large variability. When the rhizobiomes were compared, some chestnut families differed from one another (one-way ANOVA: F8,27 = 3.33, P = 0.0089): the family with the greatest proportion of EcM fungi (D22, 17.4 ± 10.3%) had a greater abundance of EcM fungi than the Chinese chestnut (Princeton, 0.04 ± 0.03%) and two of the other hybrids (W3, 0.08 ± 0.09% and W6, 0.12 ± 0.18%) with low EcM abundances (Tukey’s HSD, P < 0.05). Interestingly, in the near absence of EcM fungi, the Chinese chestnut family had a high abundance of putative plant pathogens (9.8% ± 11.3%; Fig. 1) representing a variety of taxa assigned to Xylariales (e.g., genus Monographella), Ophiostomatales (e.g., genus Ophiostoma), Pleosporales (e.g., genus Leptosphaeria), and Hypocreales (e.g., Clonostachys and Fusarium teleomorph Gibberella) among others. In contrast, the W3 hybrid had a large proportion of sequences assigned to a combined guild that contained plant pathogens, soil, and wood saprotrophs (13.7 ± 25.9%; Mixed 2 in Fig. 1) representing exclusively OTUs assigned to genus Fusarium, whereas hybrid W6 had a large proportion of putative fungal endophytes (14.7 ± 28.0%; Fig. 1) representing common root-associated taxa (e.g., Cadophora, Capronia, Leptodontidium, Phialocephala, and Trichoderma). These analyses highlight (1) the low and variable presence of EcM taxa in 1-year old seedlings in the nursery and (2) the heterogeneity among the fungal functional groups associated with their roots.

Assignment of the sequence data to the most commonly encountered fungal ecological guilds using FUNGuild (Nguyen et al. 2016). OTUs with no assignment were omitted for better visualization. Guilds listed here are as follows: 1, undefined saprotroph; 2, ectomycorrhizal; 3, mixed 1: animal pathogen-endophyte-epiphyte-plant pathogen; 4, mixed 2: plant pathogen-soil saprotroph-wood saprotroph; 5, plant pathogen; 6, mixed 3: animal endosymbiont-undefined saprotroph; 7, mixed 4: animal pathogen-endophyte-epiphyte-undefined saprotroph; 8, mixed 5: animal pathogen-endophyte-plant pathogen-wood saprotroph; 9, endophyte; 10, other guilds
Richness and diversity
Our coverage estimators (Supplemental Fig. S2a) indicate rather complete sampling of the fungal communities in both roots (99.5 ± 0.1%) and nursery soils (99.2 ± 0.1%). Although generally high, coverage was higher in all rhizobiomes than in the sampled nursery soils (Dunnett’s test, P < 0.05). The fungal community richness differed between the nursery soil and some chestnut roots (Supplemental Fig. S2b), four of the nine chestnut families had lower richness than soil (W3, D22, 4-75, and Pryor 043; Dunnett’s test, P < 0.05). Extrapolative richness estimators corroborated: ChaoI estimates were commonly lower in the rhizobiomes than in the soil (Dunnett’s test, P < 0.05) except for the Chinese chestnut family (Princeton; Dunnett’s test, P = 0.08) and one hybrid (W5; Dunnett’s test, P = 0.07) that did not differ (Supplemental Fig. S2c); and, Boneh estimators for the potential number of additional OTUs that would have been detected if sampling had been complete were consistently lower (Dunnett’s test, P < 0.05) except for the Chinese chestnut family that did not differ from the nursery soil (Princeton; Dunnett’s test, P = 0.07) (Supplemental Fig. S2d). In contrast to richness, diversity and evenness estimators between the chestnut rhizobiomes and the nursery soil did not differ (Supplemental Figs. S2e, f; Dunnett’s test; P > 0.34). Further analyses that omitted the soils and compared only the nine families, indicated no differences in coverage (one-way ANOVA: F8,27 = 1.16, P = 0.36), richness (one-way ANOVA: F8,27 = 1.34, P = 0.27), extrapolative richness–ChaoI (one-way ANOVA: F8,27 = 1.17, P = 0.35) and Boneh (one-way ANOVA: F8,27 = 1.94, P = 0.10), diversity (one-way ANOVA: F8,27 = 0.82, P = 0.59), or evenness (one-way ANOVA: F8,27 = 0.78, P = 0.63) (Supplemental Fig. S2).
Community composition
We first visualized and tested for differences in fungal community composition using the complete dataset including both the nursery soil and all rhizobiome samples (Supplemental Fig. S1a–c). These analyses indicated distinct rhizobiome and nursery soil communities (PerMANOVA; F9,30 = 3.08, P = 0.001). Further analyses that excluded soils distinguished fungal communities among the chestnut family rhizobiomes (Supplemental Fig. S3a–c; F8,27 = 2.98, P = 0.001). Subsequent pairwise comparisons indicated that fungal communities in the roots of most chestnut families differed from each other, with only a few exceptions (Table 2): only six of the possible 36 pairwise comparisons suggested non-distinct communities. The NMS ordinations suggest that the American and Chinese chestnut families—as well as their hybrids—recruited distinct fungal communities from nursery soil (Supplemental Fig. S3a–c).
We performed ordination analyses using only core OTUs (Fig. 2). Again, despite the limited replication and great within-treatment variability, the communities differed among the chestnut families (PerMANOVA; F8,27 = 2.93, P = 0.001). Pairwise comparisons show that these communities differed in 31 of the 36 possible comparisons (Supplemental Table S3). These analyses suggest that it is not only the peripheral members that distinguish the fungal communities but that core components are also recruited differently.

Non-metric multidimensional scaling (NMS) ordination of the core fungal communities that include only those 90 OTUs that occurred at least in half of the 36 chestnut rhizobiome samples. The ordination was optimally resolved on three axes that represent 15.1%, 31.6%, and 36.3% of the variability, for a total of 83.0% with stress 0.14. The permutation-based MANOVA indicated that fungal communities differ among the nine analyzed chestnut rhizobiomes (F8,27 = 2.93, P < 0.001). Shown are Axis 2 and Axis 3 that represent 67.9% of the variation. The ordination distinguishes most chestnut families (Supplemental Table S3)
Indicator taxa
To identify taxa that were disproportionately represented, we used indicator taxon analyses (Supplemental Tables S1 and S2). The analyses that focused on the core rhizobiomes identified a total of 30 indicator OTUs (Table 3). Among these, OTUs representing common soil-inhabiting genera—Cryptococcus (5 OTUs), Rhodotorula (4 OTUs), and Mortierella (2 OTUs)—were most abundant, but included putative pathogens exemplified by an OTU assigned to genus Ophiostoma and mycorrhizal symbionts exemplified by an OTU assigned to genus Tomentella. Interestingly, the three pure species families accounted for more than half of the indicator OTUs (Pryor 182, 11; Pryor 43, 2; and Princeton, 4), whereas—in general—the hybrid families had few indicator OTUs (e.g., D22, W3, W4, and W5 had only one indicator each; Table 3).
Discussion
We analyzed chestnut hybrid and parent family rhizobiomes from an operational nursery to evaluate their rhizobiomes prior to outplanting. As a result of the use of high-value nursery stock from an operational nursery, our within-treatment replication was low and variation high. Yet, these data show (i) that chestnut seedling rhizobiomes are distinct from those in nursery inoculum; (ii) that chestnuts of different species and hybrid families recruit distinct rhizobiomes from the nursery inoculum with no discernable pattern conforming to the backcross breeding program; and (iii) that the rhizobiomes include fungi ranging from saprobes and mycorrhizal mutualists to root-associated endophytes and plant pathogens. Importantly, although our data poorly permit decoupling effects of the genotypic background and the possible founder/priority effects (see Kennedy et al. 2009; Fukami et al. 2010) or spatial heterogeneity in the nursery soils, they highlight the distinct and heterogeneous rhizobiomes and distinguish the loads of putative pathogens and benign endophytes when a substantial EcM component is absent. Taken together, these findings indicate diverse fungal rhizobiomes that have the potential to either burden (see Lilja and Rikala 2000) or aid (see Menkis et al. 2007) post-transplanting performance and successful establishment.
We broadly dissected fungal rhizobiomes present within an operational nursery. The data—for OTUs with functional assignments—included a continuum of potential functions but were dominated by diverse OTUs assigned to putative saprotrophic guilds, or guilds that combined saprotrophs and plant pathogens. Although OTUs assigned to plant pathogens (38 OTUs) were more diverse than those assigned to EcM (12 OTUs), the latter were more abundant based on sequence counts. The EcM fungi can positively impact tree seedling growth in forest nurseries (Sinclair et al. 1982; Menkis et al. 2007) and improve transplanting success (Perry et al. 1987; Kropp and Langlois 1990; Ortega et al. 2004; Menkis et al. 2007). In contrast, in their absence, the nursery-grown seedlings may be more susceptible to environmental stressors, such as root-borne pathogens, drought, and/or toxic metals (Morin et al. 1999; Lilja and Rikala 2000; van Tichelen et al. 2001; Ortega et al. 2004). Our data indicate that, although the EcM inhabit both nursery soil and the chestnut rhizobiomes therein, the EcM communities include only a limited number of taxa likely as a result of the fumigation and fertilization treatments as per standard operating protocols. Our estimates of low EcM richness agree with others focusing on EcM in forest nurseries. For example, Menkis et al. (2005), studying Lithuanian forest nurseries, observed 21 EcM taxa in Pinus sylvestris and 13 in Picea abies.
Most EcM fungi have broad host ranges (Molina et al. 1992), although host ranges may vary from a few hosts to wide generality (Molina and Horton 2012). Recent studies have concluded that genotypic differences among conspecific hosts may select for distinct EcM (Korkama et al. 2006; Velmala et al. 2013; Lamit et al. 2016) or fungal rhizobiomes (Perez-Izquierdo et al. 2017). Although our data suggest differences among the chestnut families, we observed no clear trends supporting that breeding for pathogen resistance in American chestnut may alter its compatibility with EcM fungi. Our results contrast those of D’Amico et al. (2015), who bioassayed root tips to compare EcM colonization and communities of a C. dentata wild-type, transgenic resistance line, American-Chinese hybrids, or other Fagaceae. However, the hybrids that were included in our experiment did not consistently possess less diverse or less abundant EcM than their parents suggesting no strong negative selection against EcM partners as a result of the breeding focusing on disease resistance.
The EcM communities that we observed are not unique compared to other nurseries or studies that target American chestnut. We observed EcM taxa previously detected in nursery soils or even considered adapted to nursery conditions (Laccaria and Hebeloma (Mikola 1970; Stenström and Ek 1990; Henrion et al. 1994; Menkis et al. 2005, 2016; Menkis and Vasaitis 2011), Pisolithus (Marx 1977), Tomentella (Menkis et al. 2005)). Similarly, previous studies on American chestnut using either laboratory or field bioassays (Palmer et al. 2008; Dulmer et al. 2014; Stephenson et al. 2017; Bauman et al. 2018) reported Laccaria, Tomentella, and Scleroderma that we also observed in our nursery-reared seedlings. Another study (Bauman et al. 2013), focusing on EcM of backcrossed bare-root hybrid and seed-initiated seedlings (inoculated with Pisolithus tinctorius) in a coal mine restoration project using select soil preparation practices in central Ohio, observed genera detected in the current study including Hebeloma, Laccaria, Pisolithus, Scleroderma, and Tomentella. In sum, our data indicate that nursery soil inocula include EcM taxa that nursery-reared seedlings likely encounter at their outplanting sites.
In addition to the low EcM richness, our data were characterized by the highly variable EcM occurrence as indicated by the large within and between treatment variability. It is of note that we did not estimate colonization rates microscopically, but rely exclusively on sequence yields and relative abundances. Some sampled chestnut rhizobiomes yielded very few EcM sequences. These data agree with previous observations from forest nurseries. Menkis et al. (2016), analyzing container-grown conifer forest nursery seedlings in Sweden, concluded that the communities “were largely composed of saprotrophic, mycorrhizal and endophytic fungi, while pathogens were relatively rare.” Although OTUs assigned to “plant pathogens” using FUNGuild (Nguyen et al. 2016) were definitely present in our data, they were not abundant across all soil and many rhizobiome samples. Even so, the sparse presence of EcM fungi seemed to permit pathogen establishment as indicated by the Chinese parent line Princeton. This may have substantial consequences for the seedling establishment success. Saunders et al. (1992) estimated that a quarter of post-planting seedling mortality could be a result of pathogens that originated from the nursery, and Lilja and Rikala (2000) concluded that a majority of Rhizoctonia-inoculated seedlings failed to survive in the field. Although the pathogens can negatively impact stand regeneration, they occurred heterogeneously in our experiment: even when pathogens were present, their occurrence was highly variable. In sum, our data highlight the continuum of fungi ranging from pathogens to mycorrhizal symbionts that occupy the rhizobiomes as well as the stochasticity in their occurrence.
Similarly to previous reports (Kernaghan et al. 2003; Menkis et al. 2005, 2016; Stenström et al. 2014), our data show that putative endophytes commonly occupy nursery-grown seedling rhizobiomes—particularly when EcM OTUs are few. In our study, the hybrid line W6 serves as an example: with its low EcM abundance, endophytes were a large component in the rhizobiome. Interestingly, OTUs from chestnut rhizobiomes represented nursery-borne endophytes commonly detected in both Europe (Menkis et al. 2005; Stenström et al. 2014) and North America (Kernaghan et al. 2003): Cadophora, Leptodontidium, Meliniomyces, and Phialocephala. There is a considerable debate about the function of these root-associated fungi. Meta-analyses disagree and suggest that the endophyte effects on the hosts are either primarily negative (Alberton et al. 2010; Mayerhofer et al. 2013) or, if positive, may depend on availability or form of nitrogen (Newsham 2011). Mandyam and Jumpponen (2015) combined and analyzed a large number of small endophyte inoculation experiments. They highlighted context dependencies perhaps best explained by interactions specific to host genotypes and fungal strains. Our data focusing on chestnut families highlight that the endophytes can be abundant in the rhizobiomes and may thus impact the seedling outplanting performance, albeit perhaps in an unpredictable manner.
Many comparisons between the chestnut families and nursery soil were consistent with expectations. Soils often had greater fungal richness than the rhizobiomes, whereas these estimates did not differ among the chestnut families. Similarly to the richness estimates, the soil and rhizobiome communities were compositionally distinct. Interestingly, our ordination analyses highlighted also distinct fungal communities in the chestnut rhizobiomes—even when only the so-called core OTUs were included. Our subsequent indicator taxon analyses identified a total of 30 indicator OTUs—many of which assigned to common soil-inhabiting taxa: Cryptococcus, Rhodotorula, and Mortierella. Half of the observed indicator taxa were associated with two of the three parental lines, whereas the hybrid lines had few indicator OTUs. It remains unclear whether or not such parent-hybrid line distinctions suggest a limited ability of the hybrid families to recruit rhizobiomes. This is particularly the case since our data provided no evidence for less diverse fungal communities in these hybrids bred for greater disease resistance.
We dissected the rhizobiomes of chestnut families bred for resistance against the devastating pathogen to evaluate the potential non-target effects. Our data unequivocally show that chestnut families depart the nurseries with distinct fungal loads that may be consequential for their outplanting success. While some seedlings and families may host a large EcM component, others may be burdened by large proportion of potential antagonists or endophytes whose host interactions are unpredictable. Follow-up studies of the seedlings whose rhizobiomes have been pre-screened may provide a means to evaluate whether or not the nursery-recruited rhizobiomes differently determine the establishment success after transplanting.
References
Alberton O, Kuyper TW, Summerbell RC (2010) Dark septate root endophytic fungi increase growth of Scots pine seedlings under elevated CO2 through enhanced nitrogen use efficiency. Plant Soil 328:459–470
Anagnostakis SL (1987) Chestnut blight: the classical problem of an introduced pathogen. Mycologia 79:23–37
Anagnostakis SL (2012) Chestnut breeding in the United States for disease and insect resistance. Plant Dis 96:1392–1403
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46
Bauman JM, Keiffer CH, Hiremath S, McCarthy BC (2013) Soil preparation methods promoting ectomycorrhizal colonization and American chestnut Castanea dentata establishment in coal mine restoration. J Appl Ecol 50:721–729
Bauman JM, Adamson J, Brisbin R, Cline ET, Keiffer CH (2017) Soil metals and ectomycorrnizal fungi associated with American chestnut hybrids as reclamation trees on formerly coal mined land. Int J Agronomy Article ID 9731212:1–12
Bauman JM, Francino S, Santas A (2018) Interactions between ectomycorrhizal fungi and chestnut blight (Cryphonectria parasitica) on American chestnut (Castanea dentata) under in coal mine restoration. AIMS Microbiol 4:104–122
Booth MG (2004) Mycorrhizal networks mediate overstorey–understory competition in a temperate forest. Ecol Lett 7:538–546
Boyd IL, Freer-Smith PH, Gilligan CA, Godfray HC (2013) The consequence of tree pests and diseases for ecosystem services. Science 342:1235773
Brown SP, Veach AM, Grond K, Lickteig SK, Lothamer K, Oliver AK, Rigdon-Huss AR, Jumpponen A (2015) Scraping the bottom of the barrel: are rare high throughput sequences artifacts? Fungal Ecol 13:221–225
Burnham CR, Rutter PA, French DW (1986) Breeding blight-resistant chestnuts. Plant Breed Rev 4:347–397
Carlsen T, Aas AB, Lindner D, Vrålstad T, Schumacher T, Kauserud H (2012) Don’t make mista(g)kes: is tag switching an overlooked source of error in amplicon pyrosequencing studies? Fungal Ecol 5:747–749
Clark SL, Schlarbaum SE, Pinchot CC, Anagnostakis SL, Saunders MR, Thomas-Van Gundy M, Schaberg PG, McKenna J, Bard J, Berrang P, Casey DM, Casey CE, Crane B, Jackson B, Kochenderfer J, Lewis R, MacFarlane R, Makowski R, Miller M, Rodrigue J, Stelock J, Thornton C, Williamson T (2014a) Reintroduction of American chestnut in the National Forest System. J Forestry 112:501–512
Clark SL, Schlarbaum SE, Saxton AM, Hebard FV (2014b) The first research plantings of third-generation, third-backcross American chestnut (Castanea dentata) in the Southeastern United States. In: Double ML, MacDonald WL (eds) Proceedings of the fifth international chestnut symposium, ISHS. Acta Hortic 1019:39–44
Clark SL, Schlarbaum SE, Saxton AM, Hebard FV (2016) Establishment of American chestnuts (Castanea dentata) bred for blight (Cryphonectria parasitica) resistance: influence of breeding and nursery grading. New For 47:243–270
D’Amico KM, Horton TR, Maynard CA, Stehman SV, Oakes AD, Powell WA (2015) Comparisons of ectomycorrhizal colonization of transgenic American chestnut with those of wild type, conventionally bred hybrid, and related Fagaceae species. Appl Environ Microbiol 81:100–108
Dickie IA, Koide RT, Steiner KC (2002) Influences of established trees on mycorrhizas, nutrition, and growth of Quercus rubra seedlings. Ecol Monogr 72:505–521
Dickie IA, Montgomery RA, Reich PB, Schnitzer SA (2007) Physiological and phenological responses of oak seedlings to oak forest soil in the absence of trees. Tree Physiol 27:133–140
Diskin M, Steiner KC, Hebard FV (2006) Recovery of American chestnut characteristics following hybridization and backcross breeding to restore blight-ravaged Castanea dentata. For Ecol Manag 223:439–447
Dufrene M, Legendre P (1997) Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol Monogr 67:345–366
Dulmer KM (2006) Mycorrhizal associations of American chestnut seedlings: a lab and field bioassay. M.Sc. Thesis. State University of New York, Syracuse, New York
Dulmer KM, LeLuc SD, Horton TR (2014) Ectomycorrhizal inoculum potential of northeastern US forest soils for American chestnut restoration: results from field and laboratory bioassays. Mycorrhiza 24:65–74
Dunnett CW (1955) A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc 50:1096–1121
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Fukami T, Dickie IA, Wilkie JP, Paulus BC, Park D, Roberts A, Buchanan P, Allen RB (2010) Assembly history dictates ecosystem functioning: evidence from wood decomposer communities. Ecol Lett 13:675–684
Hebard FV (2001) Backcross breeding program produces blight-resistant American chestnuts. Ecol Restor 19:252–254
Hebard FV (2006) The backcross breeding program of the American chestnut foundation. In: Steiner KC, Carlson JE (eds) Restoration of American chestnut to forest lands. Proceedings of a conference and workshop. National Park Service, Washington, pp 61–77
Henrion B, Di Battista C, Bouchard D, Vairelles D, Thompson BD, Le Tacon F, Martin F (1994) Monitoring the persistence of Laccaria bicolor as an ectomycorrhizal symbiont of nursery-grown Douglas fir by PCR of the RDNA intergenic spacer. Mol Ecol 3:571–580
Horton TR, van der Heijden MGA (2008) The role of symbiosis in seedling establishment and survival. In: Leck M, Parker VT, Simpson RL (eds) Seedling ecology and evolution. Cambridge University Press, Cambridge, pp 150–171
Huse SM, Dethlefsen L, Huber JA, Welch MD, Relman DA, Sogin ML (2008) Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet 4:e1000255
Ihrmark K, Bӧdeker ITM, Cruz-Martinez K, Friberg H, Kubartova A, Schenck J, Strid Y, Stenlid J, Brandstӧm-Durling M, Clemmensen KE, Lindhal BD (2012) New primers to amplify the fungal ITS2-region: evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol 82:666–677
Jacobs DF, Dalgleish HJ, Nelson CD (2013) A conceptual framework for restoration of threatened plants: the effective model of American chestnut (Castanea dentata) reintroduction. New Phytol 197:378–393
Kennedy PG, Peay KG, Bruns TD (2009) Root tip competition among ectomycorrhizal fungi: are priority effects a rule of an exception? Ecology 90:2098–2107
Kernaghan G, Sigler L, Khasa D (2003) Mycorrhizal and root endophytic fungi of containerized Picea glauca seedlings assessed by rDNA sequence analysis. Microb Ecol 45:128–136
Korkama T, Pakkanen A, Pennanen T (2006) Ectomycorrhizal community structure varies among Norway spruce (Picea abies) clones. New Phytol 171:815–824
Kormanik PP, Sung SS, Kormanik TL (1994) Toward a single nursery protocol for oak seedlings. 22nd Southern Forest Tree Improvement Conference
Kropp BR, Langlois CG (1990) Ectomycorrhizae in reforestation. Can J For Res 20:438–451
Lamit LJ, Holeski LM, Flores-Renteria L, Whitham TG, Gehring CA (2016) Tree genotype influences ectomycorrhizal community structure: ecological and evolutionary implications. Fungal Ecol 24:124–134
Lilja A, Rikala R (2000) Effect of uninucleate Rhizoctonia on the survival of outplanted Scots pine and Norway spruce seedlings. For Pathol 30:109–115
Lovett GM, Weiss M, Liebhold AM, Holmes TP, Leung B, Lambert KF, Orwig DA, Campbell FT, Rosenthal J, McCullough DG, Wildova R, Ayres MR, Canham CD, Foster DR, LaDeau SL, Weldy T (2016) Nonnative forest insects and pathogens in the United States: impacts and policy options. Ecol Appl 26:1437–1455
Mandyam KG, Jumpponen A (2015) Mutualism-parasitism paradigm synthesized from results of root-endophyte models. Front Microbiol 5:e776
Marx DH (1977) Tree host range and world distribution of the ectomycorrhizal fungus Pisolithus tinctorius. Can J Microbiol 23:217–223
Mayerhofer MS, Kernaghan G, Harper KA (2013) The effects of fungal root endophytes on plant growth: a meta-analysis. Mycorrhiza 23:119–128
Menkis A, Vasaitis R (2011) Fungi in roots of nursery grown Pinus sylvestris: ectomycorrhizal colonization, genetic diversity and spatial distribution. Microb Ecol 61:52–63
Menkis A, Vasiliauskas R, Taylor AFS, Stenlid J, Finlay R (2005) Fungal communities in mycorrhizal roots of conifer seedlings in forest nurseries under different cultivation systems, assessed by morphotyping, direct sequencing and mycelial isolation. Mycorrhiza 16:33–41
Menkis A, Vasiliauskas R, Taylor AFS, Stenlid J, Finlay R (2007) Afforestation of abandoned farmland with conifer seedlings inoculated with three ectomycorrhizal fungi—impact on plant performance and ectomycorrhizal community. Mycorrhiza 17:337–348
Menkis A, Burokiene D, Stenlid J, Stenström E (2016) High-throughput sequencing shows high fungal diversity and community segregation in the rhizospheres of container-grown conifer seedlings. Forests 7:44
Mikola P (1970) Mycorrhizal inoculation in afforestation. Int Rev Forest Res 3:123–196
Molina R, Horton TR (2012) Mycorrhiza specificity: its role in the development and function of common mycelial networks. In: Horton TR (ed) Mycorrhizal networks. Springer Verlag, Berlin Heidelberg, pp 1–39
Molina R, Massicotte H, Trappe JM (1992) Specificity phenomena in mycorrhizal symbioses: community-ecological consequences and practical implications. In: Allen MF (ed) Mycorrhizal functioning: an integrative plant-fungal process. Chapman and Hall, New York, pp 357–423
Morin C, Samson J, Dessureault M (1999) Protection of black spruce seedlings against Cylindrocladium root rot with ectomycorrhizal fungi. Can J Bot 77:169–174
Nara K (2006a) Ectomycorrhizal networks and seedling establishment during early primary succession. New Phytol 169:169–178
Nara K (2006b) Pioneer dwarf willow may facilitate tree succession by providing late colonizers with compatible ectomycorrhizal fungi in a primary successional volcanic desert. New Phytol 171:187–198
Newhouse AE, Oakes AD, Pilkey HC, Roden HE, Horton TR, Powell WA (2018) Transgenic American chestnuts do not inhibit germination of native seeds or colonization of mycorrhizal fungi. Front Plant Sci 9:1046
Newsham KK (2011) A meta-analysis of plant responses to dark septate root endophytes. New Phytol 144:517–524
Nguyen NH, Song Z, Bater S, Branco S, Tedersoo L, Menke J, Schilling JS, Kennedy PG (2016) FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol 20:241–248
Oliver AK, Brown SP, Callaham MA, Jumpponen A (2015) Polymerase matters: non-proofreading enzymes inflate community richness estimates by up to 15%. Fungal Ecol 15:86–89
Ortega U, Dunabeitia M, Menendez S, Gonzalez-Murua C, Majada J (2004) Effectiveness of mycorrhizal inoculation in the nursery on growth and water relations of Pinus radiata in different water regimes. Tree Physiol 24:65–73
Palmer J, Lindner D, Volk T (2008) Ectomycorrhizal characterization of an American chestnut (Castanea dentata)-dominated community in western Wisconsin. Mycorrhiza 19:27–36
Perez-Izquierdo L, Zabal-Aguirre M, Flores-Renteria D, Gonzales-Martinez SC, Buee M, Rincon A (2017) Functional outcomes of fungal community shifts driven by tree genotype and spatial-temporal factors in Mediterranean pine forests. Environ Microbiol 19:1639–1952
Perry AD, Molina R, Amaranthus PM (1987) Mycorrhizae, mycorrhizospheres, and reforestation: current knowledge and research needs. Can J For Res 17:929–940
Pinchot CC, Schlarbaum SE, Clark SL, Saxton AM, Sharp AM, Schweitzer CJ, Hebard FV (2017) Growth, survival, and competitive ability of chestnut (Castanea Mill.) seedlings planted across a gradient of light levels. New For 48:491–512
Quoreshi AM, Timmer VR (2000) Early outplanting performance of nutrient-loaded containerized black spruce seedlings inoculated with Laccaria bicolor: a bioassay study. Can J For Res 30:744–752
Rognes T, Flouri T, Nichols B, Quince C, Mahe F (2016) VSEARCH: a versatile open source tool for metagenomics. PEERJ 4:e2584
Santini A, Ghelardini L, De Pace C, Desprez-Loustau ML, Capretti P, Chandelier A, Cech T, Chira D, Diamandis S, Gaitniekis T, Hantula J, Holdenrieder O, Jankovsky L, Jung T, Jurc D, Kirisits T, Kunca A, Lygis V, Malecka M, Marcais B, Schmitz S, Schumacher J, Solheim H, Solla A, Szabo´ I, Tsopelas P, Vannini A, Vettraino AM, Webber J, Woodward S, Stenlid J (2013) Biogeographical patterns and determinants of invasion by forest pathogens in Europe. New Phytol 197:238–250
Saunders JE, Juzwik J, Hutchison R (1992) Outplanting survival of Cylindrocladium root-rot affected black spruce seedlings. Can J For Res 22:1204–1207
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Fungal Barcoding Consortium (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc Natl Acad Sci USA 109:6241–6246
Seddon PJ (2010) From reintroduction to assisted colonization: moving along the conservation translocation spectrum. Restor Ecol 18:796–802
Sinclair WA, Sylvia DM, Larsen AO (1982) Disease suppression and growth promotion in Douglas-fir seedlings by the ectomycorrhizal fungus Laccaria laccata. For Sci 28:191–201
Smith SE, Read DJ (1997) Mycorrhizal symbiosis, 2nd edn. Academy, London
Sniezko RA (2006) Resistance breeding against nonnative pathogens in forest trees—current successes in North America. Can J Plant Path 28:S270–S279
Steiner KC, Westbrook JW, Hebard FV, Georgia LL, Powell WA, Fitzsimmons SF (2016) Rescue of American chestnut with extraspecific genes following its destruction by a naturalized pathogen. New For 48:1–20
Stenström E, Ek M (1990) Field growth of Pinus sylvestris following nursery inoculation with mycorrhizal fungi. Can J For Res 20:914–918
Stenström E, Ndobe NE, Jonsson M, Stenlid J, Menkis A (2014) Root-associated fungi of healthy-looking Pinus sylvestris and Picea abies seedlings in Swedish forest nurseries. Scan J Forest Res 29:12–21
Stephenson SL, Ben Ali MHB, Rollins AW, Furches MS, Atherton KR (2017) Ectomycorrhizal fungi associated with American chestnut at a site in Tennessee, USA. Castanea 82:2–7
Thompson LM, Van Manen FT, Schlarbaum SE, DePoy M (2006) A spatial modeling approach to identify potential butternut restoration sites in Mammoth Cave National Park. Restor Ecol 14:289–296
Unterseher M, Dorman CF, Jumpponen A, Moora M, Öpik M, Tedersoo L, Schnittler M (2011) Species abundance distributions in fungal metagenomics—lessons learned from community ecology. Mol Ecol 20:275–285
van Tichelen KK, Colpaert JV, Vangronsveld J (2001) Ectomycorrhizal protection of Pinus sylvestris against copper toxicity. New Phytol 150:203–213
Veach, A.M, Stokes, C.E., Knoepp, J., Jumpponen, A., Baird, R. 2018. Fungal communities and functional guilds shift along an elevational gradient in the southern Appalachian Mountains. Microbial Ecology 76:156–168. https://doi.org/10.1007/s00248-017-1116-6
Velmala SM, Rajala T, Haapanen M, Taylor AFS, Pennanen T (2013) Genetic host-tree effects on the ectomycorrhizal community and root characteristics of Norway spruce. Mycorrhiza 23:21–33
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand MA, Sininsky JJ, White TF (eds) PCR protocols: a guide to methods and applications. Academic Press, New York, pp 315–322
Acknowledgments
The authors are grateful to Highlands Biological Station Grant-In-Aide in 2013 that provided travel support and to Cornelia Pinchot (USDA-FS) for collecting soil samples from nursery in Indiana. We also thank Alina Akhunova and the Kansas State University Integrated Genomics Facility (http://www.k-state.edu/igenomics/index.html) for library quality control, preparation, and Illumina MiSeq sequencing. This project was partly supported by the USDA-NIFA capacity program KS495.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplemental Table S1
Indicator taxon analyses (indicator values, mean ± St. Dev and associated P values) contrasting nursery soil and chestnut rhizosphere samples in the full dataset including all samples and OTUs. The Indicator Taxon Analyses (Dufrene and Legendre 1997) were performed in PC-ORD and indicate OTUs (taxa) that are disproportionately enriched under one treatment comparison compared to others. Column “Indicator” identifies if the OTU is an indicator (P < 0.05) for either soil or rhizosphere, or neither (Not Significant - NS). The analyses identify 171 indicators in total; 26 for chestnut rhizospheres and 145 for soils. The table also includes the taxon affinities for each of the observed OTUs (TXT 48 kb)
Supplemental Table S2
The 86 significant indicators identified in the indicator taxon analyses (indicator values, mean ± St. Dev and associated P values) of the rhizosphere samples representing different chestnut families in the full dataset including all samples and OTUs. The Indicator Taxon Analyses (Dufrene and Legendre 1997) were performed in PC-ORD and indicate OTUs (taxa) that are disproportionately enriched in one chestnut family in comparison to others. Column ‘Family’ represents chestnut family in which the OTU was most abundant and column ‘P Value’ identifies if the OTU is an indicator (P < 0.05). (TXT 9 kb)
Supplemental Table S3
Pairwise comparisons of the fungal communities associated with the rhizospheres of nine families. PerMANOVA analyses that included only core OTUs that occurred in at least half of the 36 rhizosphere samples and excluded soil samples distinguished fungal communities (F8,27 = 2.93, P = 0.001). These pairwise comparisons indicate that fungal communities of most families differ from each other. Shown are the t-test variables and associated P-values, those that do not differ are highlighted in bold. (DOCX 14 kb)
Supplemental Figure S1
Non-metric Multidimensional Scaling (NMS) ordination of fungal communities of the one-year-old American chestnut seedlings and the nursery soils in which they were grown. A three-dimensional ordination (k = 3) provided an optimal solution and represented 86.4% of the variation with a stress of 13.36, separating the soils and a majority of the chestnut breeding line rhizobiomes. a) Axis 1 and Axis 2 that represent 57.5% of the variation; b) Axis 1 and Axis 3 that represent 59.6% of the variation and distinguish nursery soils from the American chestnut rhizobiomes; and, c) Axis 2 and Axis 3 that represent 55.7% of the variation and also distinguish nursery soils from the American chestnut rhizobiomes. (PNG 179 kb)
Supplemental Figure S2
Richness and diversity estimators of American chestnut rhizobiomes and the nursery soils they were grown: a) coverage; b) observed richness (SObs); c) extrapolative ChaoI that estimates the total OTU number; d) extrapolative Boneh that estimates the number of additional OTUs that would have been observed had the sampling been complete; e) Shannon diversity (H′); and, f) evenness based on Shannon’s diversity (EH) of nursery soil and chestnut hybrid roots. The asterisks show the results of Dunnett’s tests, which compare means of each chestnut rhizobiome estimates against the nursery soil: *** – P < 0.001; ** – 0.001 ≤ P < 0.01; * – 0.01 ≤ P < 0.05. The data indicate greater coverage and lower richness (Observed and extrapolated) in the roots than in the soils, whereas diversity and evenness did not differ. Further, none of the estimators differed among the American chestnut rhizobiomes. (PNG 114 kb)
Supplemental Figure S3
Non-metric Multidimensional Scaling (NMS) ordination of fungal communities representing American chestnut rhizobiomes. The ordination was optimally resolved on three axes that represent 29.7%, 27.6%, and 28.5% of the variability, for a total of 85.6% with stress 0.14. Permutation-based MANOVA indicated that fungal communities differ among the nine analyzed chestnut rhizobiomes (F8,27 = 2.98, P < 0.001). a) Axis 1 and Axis 2 that represent 57.3% of the variation; b) Axis 1 and Axis 3 that represent 58.0% of the variation; and, c) Axis 2 and Axis 3 that represent 55.9% of the variation and also distinguish nursery soils from the American chestnut rhizobiomes. The ordination distinguishes most chestnut species and hybrid families (Table 2). (PNG 205 kb)
Rights and permissions
About this article

Cite this article
Reazin, C., Baird, R., Clark, S. et al. Chestnuts bred for blight resistance depart nursery with distinct fungal rhizobiomes. Mycorrhiza 29, 313–324 (2019). https://doi.org/10.1007/s00572-019-00897-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00572-019-00897-z